

Abstract
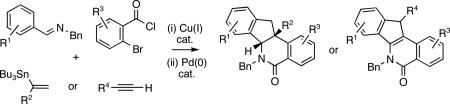
Copper-catalyzed coupling of imines, vinylstannanes or alkynes and o-bromoaroyl chlorides followed by Pd(0)-catalyzed annulations afforded indenoisoquinolines. Protocols requiring minimal purifications were developed, providing new methods for the construction of combinatorial libraries.
The prevalance of N-heterocycles among established pharmaceutical agents1 continues to inspire the development of new synthetic methods. We have been exploring a protocol based on an assembly of α-N-substituted amides followed by various intramolecular cyclizations,2 opening up access to combinatorial libraries of hexahydro-1H-isoindolones (Figure 1).3
Figure 1.

The general strategy
Herein, we describe a powerful novel combination of the Cu(I)-catalyzed three-component coupling and an intramolecular Pd(0)-catalyzed 1,2-bisarylation of an olefin or an alkyne in amides IV and V to deliver substituted indenoisoquinolines VI and VII (Figures 1 and 2). The protocol allows a rapid increase in molecular complexity in only two steps.
Figure 2.

Strategy toward indenoisoquinolines
Structurally related indenoisoquinolines have been shown to possess potent biological activities.4 Our protocol provides a more efficient alternative to the established preparations of indenoisoquinolines, particularly those substituted at the angular position or the benzylic carbon in the indene ring.5 The method reported herein opens up a modular access to indenoisoquinolines, and is well amenable to automation.
Initial studies were focused on extending the scope of the known Cu(I)-catalyzed coupling6 to o-bromoaroyl chlorides III as well as to 1,1-disubstituted vinylstannanes II (Figure 2). We were able to decrease the molar excess of stannane 3a,7 from 2.0 equiv to 1.5 equiv8 and realize the coupling to imine 1a and aroyl chloride 2a providing amide 4a in good yields (Scheme 1). An increase in the CuCl catalyst load improved the yield of amide 4a from 67% (with 10 mol % CuCl) and to 82% (with 20 mol % CuCl, Scheme 1).9 Next, the Pd-catalyzed cyclization of amide 4 a was explored, anticipating that an intramolecular Heck reaction would afford intermediate VIII poised for electrophilic arylation to yield dihydroindeno[1,2-c]isoquinolines VI (Figure 2).10 The treatment of amide 4a with Pd(OAc)2 (5 %) and NaOAc (1 equiv) afforded the indenoisoquinoline 5a in a 93% yield as a single diastereomer (Scheme 1).11
Scheme 1.

Protocol utilizing an isolated amide
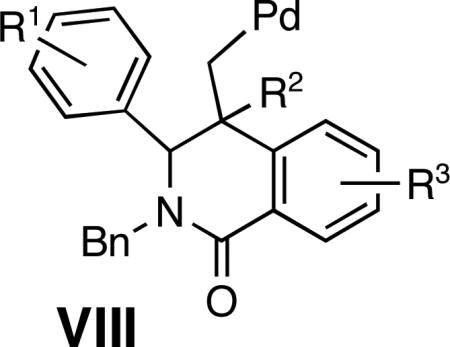
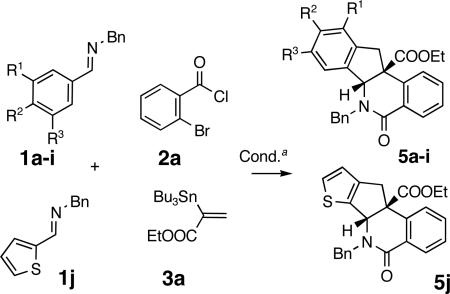
Aiming to establish a protocol amenable to automated synthesis, we sought to eliminate chromatographic purification of amide 4a. The addition of solid KF and small quantities of water, followed by filtration was employed to remove tin residues from the reaction mixtures. The resulting crude amide 4a was treated with Pd(OAc)2 catalyst under conditions reported in Scheme 1 to afford indenoisoquinoline 5a in 75% yield over two steps (entry 1, Table 1, Method A). A brief survey revealed that sodium acetate was the optimum base for the Pd-catalyzed cyclization.10 The replacement of NaOAc with Na2CO3/n-Bu4NCl applying modified Jeffery's conditions12 (compare Methods A and B, entries 1, 3 and 4, Table 1) resulted in a decrease in the reaction yields, particularly severe for the electronically deactivated imines 1c (R2 = H) and 1d (R2 = Cl) (entries 3 and 4, Method B, Table 1). Overall, the optimized sequential protocol afforded the corresponding indenoisoquinolines 5a-5e in 38-75% yields over two steps (entries 1-5, Table 1, Method A). The lower yields of the electronically deactivated chloro and ester-substituted indenoisoquinolines 5d-e are in agreement with the proposed involvement of electrophilic palladation in the key step, although the less facile iminolysis of the acyl chlorides may also be a contributing factor. The 3,4-disubstituted imines 1f and 1g afforded single regioisomers of heterocycles 5f (77%) and 5g (71%) arising from palladation at the least hindered position in the aromatic ring (entries 6 and 7, Table 1). A contiguous 1,2,3,4,5-substitution pattern was achieved in an activated imine yielding indenoisoquinoline 5h in 64% yield (entry 8, Table 1). Efficient preparation of indenoisoquinolines 5 i and 5 j demonstrated the compatibility of the method with heteroatoms other than oxygen (entries 9 and 10, Table 1).
Table 1.
Angularly Substituted Indenoisoquinolines
| |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | prdtb | yieldc (%) |
| 1 | H | OMe | H | 5a | 75 (71)d |
| 2 | H | Me | H | 5b | 69 |
| 3 | H | H | H | 5c | 67 (17)d |
| 4 | H | Cl | H | 5d | 49 (11)d |
| 5 | H | COOMe | H | 5e | 38 |
| 6 | H | OMe | OMe | 5f | 77 |
| 7 | H | -OCH2CH2O- | 5ge | 71 | |
| 8 | OMe | OMe | OMe | 5h | 64 |
| 9 | H | NMe2 | H | 5i | 59 |
| 10 | thiophene-2-yl | 5j | 74 | ||
aMethod A: was used for all the entries: (i) CuCl (20%), MeCN/CH2Cl2, 45 °C, 6 h, imine : acyl chloride : stannane = 1.0 : 1.2 : 1.5 (mol); (ii) aqueous KF, filtration, evaporation; (iii) Pd(OAc)2 (5%), NaOAc (1.0 equiv), DMF, 120 °C, 24 h.
With one exception, a single diastereomer was isolated.
Isolated yield of heterocycles 5 obtained via Method A calculated per imine as the limiting reagent.
Yield of heterocycle 5 obtained by Method B is given in parentheses. Method B: same as Method A, but substituting Na2CO3 (1.0 equiv)/n-Bu4NCl (1.0 equiv) for NaOAc.
Product was isolated as a 4 : 1 mixture of diastereomers (by 1H NMR), the major diastereomer is shown.
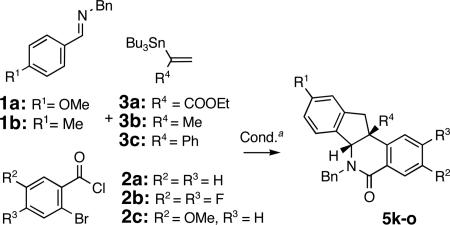
To expand the reaction scope, imines 1a and 1b were coupled to substituted aroyl chlorides 2b-c and vinylstannanes 3a and 3b-c13 bearing aliphatic (Me) and aromatic (Ph) substituents. Indenoisoquinolines 5k-o were obtained in good yields (59-76%) over two steps (Table 2). Heterocycles 5k-o were isolated as single diastereomers following chromatography and trituration of the crude products. The relative stereochemistry in heterocycles 5 (R4 = COOEt and Ph, Tables 1 and 2) was assigned based on analogy with indenoisoquinolines 5j and 5n, the structure of which was elucidated via single crystal X-ray crystallographic analyses. The relative stereochemistry in products 5 m and 5 o (R4 = Me) was assigned via spectroscopic methods.14
Table 2.
Diversification of Additional Building Blocks
| ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | R4 | prdtb | yieldc (%) |
| 1 | Me | OMe | H | COOEt | 5k | 62 |
| 2 | OMe | F | F | COOEt | 5l | 76 |
| 3 | OMe | H | H | Me | 5m | 68 |
| 4 | OMe | H | H | Ph | 5n | 59 |
| 5 | Me | H | H | Me | 5o | 54 |
aReaction conditions: (i) CuCl (20%), MeCN/ CH2Cl2, 45 °C, 6 h, imine : acyl chloride : stannane = 1.0 : 1.2 : 1.5 (mol); (ii) treatment with aqueous KF, filtration, evaporation; (iii) Pd(OAc)2 (5%), NaOAc (1.0 equiv), DMF, 120 °C, 24 h.
Single diastereomer was isolated.
Isolated yield of heterocycles 5 calculated per imine as the limiting reagent.
To access a distinct substitution pattern in the indenoisoquinolines, propargyl amide 7 a was prepared from imine 1a, aroyl chloride 2a and alkyne 6a in a good yield (54%) using conditions reported by Arndtsen15 (Scheme 2). We envisioned that Pd-catalyzed intramolecular bisfunctionalization of the alkyne16 would proceed via intermediate IX to afford indenoisoquinolines VII (Figure 2). Conceivably, a 1,3-shift of the allylic hydrogen in the intermediate I X would provide an organopalladium intermediate poised for the terminal electrophilic palladation.
Scheme 2.

Protocol utilizing an isolated propargyl amide
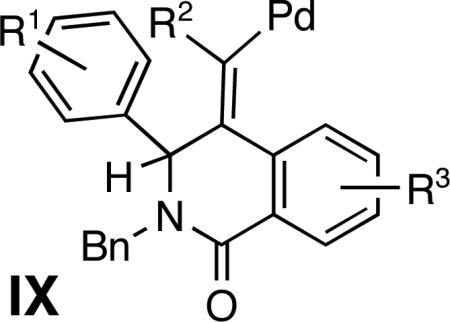
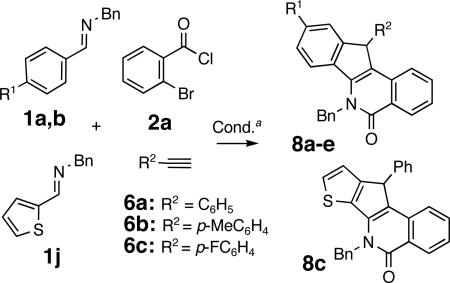
Indeed, the treatment of amide 7a with Pd(OAc)2 catalyst and Na2CO3/n-Bu4NCl additive mixture for a prolonged time period (36 h at 120 °C in DMF) afforded the corresponding indenoisoquinoline 8a in an excellent yield (91%) (Scheme 2). Ultimately, the isolation of amide 7a was avoided, limiting the purification of the crude reaction mixtures to the removal of excess alkyne via filtration through a short plug of silica. The resulting crude product was directly subjected to Pd-catalysis, affording indenoisoquinoline 8a in a good yield (66%) over two steps (entry 1, Table 2). This protocol was then applied to the coupling of imines 1a, 1b and 1j with aroyl chloride 2a and aryl acetylenes 6a-6c to provide indenoisoquinolines 8a-e in good yields (51-68%) over two steps (Table 2). Single crystal X-ray crystallographic studies on heterocycle 8c unequivocally established the structure, including the position of the double bond within the isoquinoline ring.17
The new synthetic protocol described here rapidly and efficiently assembles indenoisoquinolines with distinct substitution patterns from three simple building blocks. The modular strategy is particularly well suited for the construction of combinatorial libraries of indenoisoquinolines.
Supplementary Material
Table 3.
Indenoisoquinolines with a benzylic substituent
| ||||
|---|---|---|---|---|
| entry | R1 | R2 (aryl) | prdtb | yieldc (%) |
| 1 | OMe | C6H5- | 8a | 66 |
| 2 | Me | C6H5- | 8b | 51 |
| 3 | thiophene-2-yl | C6H5- | 8c | 68 |
| 4 | OMe | p-CH3C6H4- | 8d | 63 |
| 5 | OMe | p-FC6H4- | 8e | 57 |
aReaction conditions: (i) CuCl (20%), i-PrEt2N (1.5 equiv) MeCN, rt. 1 h, imine : acyl chloride : alkyne = 1.0 : 1.2 : 1.5 (mol); (ii) treatment with aqueous KF, filtration, evaporation; (iii) Pd(OAc)2 (5%), Na2CO3(1.0 equiv), n-Bu4NCl (1.0 equiv), DMF, 120 °C, 36 h.
Single diastereomer was isolated.
Isolated yield of heterocycles 8 calculated per imine as the limiting reagent.
Acknowledgment
Support for this work from the National Institutes of Health (KU Center for Methodology and Library Development, Grant No. P 50 GM069663) is gratefully acknowledged. We thank our colleague Dr. Victor W. Day for his assistance with the X-ray crystallographic analyses.
Footnotes
Supporting Information Available Description of the synthesis and characterization of all new compounds. and X-ray crystallographic analyses on compounds 5j, 5n and 8c. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Taylor JB, Triggle DJ, editors. Comprehensive Medicinal Chemistry II. Elsevier; Amsterdam, Boston: 2007. [Google Scholar]
- 2.Martin SF, Sunderhause JD, Dockendorff C. Org. Lett. 2007;9:4223. doi: 10.1021/ol7018357. [DOI] [PubMed] [Google Scholar]
- 3.a Zhang L, Lushington GH, Neuenswander B, Hershberger JC, Malinakova HC. J. Comb. Chem. 2008;10:285. doi: 10.1021/cc700151m. [DOI] [PubMed] [Google Scholar]; b Zhang L, Malinakova HC. J. Org. Chem. 2007;72:1484. doi: 10.1021/jo0621773. [DOI] [PubMed] [Google Scholar]
- 4.a Ryckebusch A, Garcin D, Lansiaux A, Goossens J-F, Baldeyrou B, Houssin R, Bailly C, Henichart J-P. J. Med. Chem. 2008;51:3617. doi: 10.1021/jm800017u. [DOI] [PubMed] [Google Scholar]; b Teicher BA. Biochem. Pharmacol. 2008;75:1262. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]; c Antony S, Agama KK, Miao Z-H, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Cancer. Res. 2007;67:10397. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 5.a D'Souza DM, Kiel A, Herten D-P, Rominger F, Müller TJJ. Chem. Eur. J. 2008;14:529. doi: 10.1002/chem.200700759. [DOI] [PubMed] [Google Scholar]; b Morrell A, Placzek M, Parmley S, Grella B, Antony S, Pommier Y, Cushman M. J. Med. Chem. 2007;50:4388. doi: 10.1021/jm070307+. [DOI] [PubMed] [Google Scholar]; c D'Souza DM, Rominger F, Müller TJJ. Angew. Chem. Int. Ed. 2005;44:153. doi: 10.1002/anie.200461489. [DOI] [PubMed] [Google Scholar]; d Xiao X, Miao Z-H, Antony S, Pommier Y, Cushman M. Bioorg. Med. Chem. Lett. 2005;15:2795. doi: 10.1016/j.bmcl.2005.03.101. [DOI] [PubMed] [Google Scholar]; e Jagtap PG, Baloglu E, Southan G, Williams W, Roy A, Nivorozhkin A, Landrau N, Desisto K, Saltzman AL, Szabo C. Org. Lett. 2005;7:1753. doi: 10.1021/ol050331m. [DOI] [PubMed] [Google Scholar]; f Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Bioorg. Med. Chem. 2004;12:5147. doi: 10.1016/j.bmc.2004.07.027. [DOI] [PubMed] [Google Scholar]; g Fox BM, Xiao X, Antony S, Kohlhagen G, Pommier Y, Staker B, Stewart L, Cushman M. J. Med. Chem. 2003;46:3275. doi: 10.1021/jm0300476. [DOI] [PubMed] [Google Scholar]; h Cho W-J, Park M-J, Imanishi T, Chung B-H. Chem. Pharm. Bull. 1999;47:900. doi: 10.1248/cpb.47.900. [DOI] [PubMed] [Google Scholar]
- 6.Black DA, Arndtsen BA. J. Org. Chem. 2005;70:5133. doi: 10.1021/jo0503557. [DOI] [PubMed] [Google Scholar]
- 7.For the preparation of 3a via hydrostannylation, see: Darwish A, Lang A, Kim T, Chong JM. Org. Lett. 2008;10:861. doi: 10.1021/ol702982x.
- 8.Compare to our original method reported in reference 3a.
- 9.Variations in the CuCl load and the excess of stannane 3a in reactions under the conditions described in Scheme 1 affected the yields of amide 4a: (i) 20 mol % CuCl, 2.0 equiv 3a gave 4a in 80% yield; 20 mol % CuCl, 1.0 equiv 3a gave 4a in 62% yield; (iii) 10 mol % CuCl, 1.5 equiv 3a gave 4a in 67% yield.
- 10.Zeni G, Larock RC. Chem. Rev. 2000;100:3009.Brown D, Grigg R, Sridharan V, Tambyrajah V. Tetrahedron Lett. 1995:8137. For a review on the synthesis of heterocycles via transition metal catalysis, see: D'Souza DM, Müller TJ. J. Chem. Soc. Rev. 2007;36:1095. doi: 10.1039/b608235c.
- 11.1H NMR analysis of the crude reaction mixtures indicated the presence of traces of a diastereomeric indenoisoquinoline. Isolation via chromatography followed by trituration from hexanes afforded a pure single diastereomer 5a. The relative stereochemistry was asssigned based on the comparison of the spectroscopic data with the spectroscopic data recorded for heterocycle 5j, the structure of which was established by X-ray crystallography (vide infra).
- 12.a Jeffery T. Tetrahedron Lett. 1985;26:2667. [Google Scholar]; b Jeffery T. J. Chem. Soc. Chem. Commun. 1984:1287. [Google Scholar]
- 13.For the preparation of the stannanes, see: Darwish A, Chang JM. J. Org. Chem. 2007;72:1507. doi: 10.1021/jo062145f.
- 14.The relative stereochemistry in heterocycle 5o was established via 1H NMR NOE study and the structure of 5 m was assigned accordingly. For details see the Supporting Information.
- 15.Black DA, Arndtsen BA. Org. Lett. 2004;6:1107. doi: 10.1021/ol036462+. [DOI] [PubMed] [Google Scholar]
- 16.Grigg R, Loganathan V, Sridharan V. Tetrahedron Lett. 1996;37:3399. [Google Scholar]
- 17.Structures of all the remaining indenoisoquinolines were assigned accordingly. The structure assignment is also supported by 2D NMR and NOE 1H NMR spectral studies performed on heterocycles 8c and 8d (see the Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.