

Abstract
Background. The majority of patients receiving the platinum-based chemotherapy drug oxaliplatin develop peripheral neurotoxicity. Because this neurotoxicity involves ROS production, we investigated the efficacy of mangafodipir, a molecule that has antioxidant properties and is approved for use as an MRI contrast enhancer.
Methods. The effects of mangafodipir were examined in mice following treatment with oxaliplatin. Neurotoxicity, axon myelination, and advanced oxidized protein products (AOPPs) were monitored. In addition, we enrolled 23 cancer patients with grade ≥2 oxaliplatin-induced neuropathy in a phase II study, with 22 patients receiving i.v. mangafodipir following oxaliplatin. Neuropathic effects were monitored for up to 8 cycles of oxaliplatin and mangafodipir.
Results. Mangafodipir prevented motor and sensory dysfunction and demyelinating lesion formation. In mice, serum AOPPs decreased after 4 weeks of mangafodipir treatment. In 77% of patients treated with oxaliplatin and mangafodipir, neuropathy improved or stabilized after 4 cycles. After 8 cycles, neurotoxicity was downgraded to grade ≥2 in 6 of 7 patients. Prior to enrollment, patients received an average of 880 ± 239 mg/m2 oxaliplatin. Patients treated with mangafodipir tolerated an additional dose of 458 ± 207 mg/m2 oxaliplatin despite preexisting neuropathy. Mangafodipir responders managed a cumulative dose of 1,426 ± 204 mg/m2 oxaliplatin. Serum AOPPs were lower in responders compared with those in nonresponders.
Conclusion. Our study suggests that mangafodipir can prevent and/or relieve oxaliplatin-induced neuropathy in cancer patients.
Trial registration. Clinicaltrials.gov NCT00727922.
Funding. Université Paris Descartes, Ministère de la Recherche et de l’Enseignement Supérieur, and Assistance Publique-Hôpitaux de Paris.
Introduction
The activity of platinum drugs is attributed to the formation of adducts with DNA that lead to cell death and intracellular generation of ROS following DNA and mitochondrial lesions (1). Oxaliplatin, a third-generation organoplatinum, also interacts with reduced glutathione (GSH), and the depletion of GSH is one of the pathways through which platinum-based cytotoxic drugs generate ROS in malignant cells (2).
Peripheral neuropathy occurs in 85%–95% of all patients exposed to oxaliplatin (3). This side effect may limit the dosage and/or duration of administration of the drug (4). The onset of acute neuropathy immediately follows administration of the drug, with paresthesias and possibly muscle cramps in the hands or feet generally reversible within hours or days. Electrophysiological studies have shown the hyperexcitability of peripheral nerves (5). The chronic form is characterized by the persistence of symptoms and distal sensory axonal degeneration, with myelin loss involving large fibers in the absence of a motor component (6). The occurrence of paresthesias and dysesthesias immediately after drug administration suggests a direct effect on the nerves. Several clinical and electrophysiological features of the acute form are similar to those of neuromyotonia, which is characterized by the impairment of cellular voltage-gated K+ channels (5). Although most ex vivo and in vivo investigations have confirmed an oxaliplatin-induced “channelopathy,” there is no evidence yet that a dysfunction of voltage-gated channels is the specific causal factor of the neuropathy. The acute effects of oxaliplatin can also be caused by the chelation of calcium by oxalate released from oxaliplatin and its effects on neural membranes and synapses (7). Chronic neuropathy results from the accumulation of platinum in dorsal root ganglion cells (8), and the repeated oxidative stress can be an important pathogenetic mechanism (9). Neither calcium or magnesium infusions, nor neuromodulatory agents have proved sufficiently effective to become routine treatments of acute or chronic neuropathy (10, 11). An alternative therapeutic approach is to target an upstream phenomenon, that is, the generation of oxidative stress by oxaliplatin. Indeed, in 1990, Mollman et al. observed that cisplatin-induced neuropathy is clinically indistinguishable from the neuropathy observed in patients with vitamin E deficiency (12). Since vitamin E is an antioxidant that maintains lipid-rich neural membranes, a molecule able to detoxify the ROS generated by oxaliplatin could prevent the drug’s neurotoxicity (13). A clinical trial conducted by Cascinu et al. showed limited efficacy of GSH infusions in oxaliplatin-treated patients (14). However, the efficacy was probably limited by the inability of GSH to cross cell membranes. This led us to try another molecule, mangafodipir.
Mangafodipir is a chelate of manganese and of the ligand fodipir, a vitamin B6 derivative. Vitamin B6 is known for its ability to maintain normal neurological functions and for its neuroprotective activity (15). In addition, given that mangafodipir is endowed with SOD-, catalase-, and glutathione reductase–like (GR-like) properties, it can target multiple steps of the ROS cascade by detoxifying superoxide anions and hydrogen peroxide and by restoring GSH (16, 17). Moreover, this molecule is known to cross the cell membrane, and we have already shown its ability to improve the therapeutic index of oxaliplatin (18).
We first designed a mouse model of oxaliplatin-induced neuropathy and tested the preventive and curative effects of mangafodipir on the clinical and electrophysiological signs and oxidative stress. Given the positive results obtained in the animal model, a phase II clinical trial was designed to include patients with cancer and oxaliplatin-induced neuropathy.
Results
Mangafodipir prevented oxaliplatin-induced locomotor disturbances in mice with oxaliplatin-induced neuropathy.
We studied locomotor dysfunction in mice using the rotarod test. After 8 weeks, mice injected weekly with oxaliplatin (10 mg/kg; n = 8) displayed a decreased latency to fall (514 ± 120 seconds) versus mice receiving vehicle alone (n = 8, 1,073 ± 106 seconds; P = 0.006). In contrast, injections of oxaliplatin plus mangafodipir had no effect on the latency to fall (at 8 weeks: 961 ± 137 seconds with oxaliplatin plus mangafodipir versus 1,073 ± 106 seconds with vehicle; P = 0.505 and 1,217 ± 201 seconds with mangafodipir alone; P = 0.598) (Figure 1A).
Figure 1. In vivo study of oxaliplatin-induced neurotoxicity.

Experimental mice received oxaliplatin (10 mg/kg) weekly and mangafodipir (10 mg/kg) 3 times a week for 8 weeks. Evaluation of hyperalgesia required two 5-day cycles of daily oxaliplatin (3 mg/kg). Control mice received either oxaliplatin or vehicle alone. Locomotor disturbances, cold hypoesthesia, nociception, or cold hyperalgesia were evaluated using rotarod testing, cold plate testing at +4°C, von Frey tests, or cold plate testing at –4°C, respectively (A–D). MnTBAP (10 mg/kg) or vitamin B6 (10 mg/kg), two components of mangafodipir, were substituted for mangafodipir, and mice were tested under the same experimental conditions (E–H). Data are presented as the means ± SEM of 8 different mice under each condition. *P < 0.05 and **P < 0.01 versus vehicle.
Mangafodipir prevented oxaliplatin-induced cold hypoesthesia.
We studied cold hypoesthesia in mice injected weekly with 10 mg/kg of oxaliplatin on a cold plate set at +4°C ± 0.2°C. By 8 weeks, oxaliplatin increased the latency to escape evaluated on a cold plate (n = 8, 108 ± 11 seconds) versus vehicle alone (16 ± 3 seconds; P = 0.002) or mangafodipir alone (21 ± 3 seconds, P = 0.006). We found that mangafodipir abrogated the alterations induced by oxaliplatin (43 ± 5 seconds with oxaliplatin plus mangafodipir versus 108 ± 11 seconds with oxaliplatin alone, P = 0.005) (Figure 1B).
Mangafodipir prevented oxaliplatin-induced nociception.
A von Frey test at 8 weeks showed that the threshold of withdrawal of the touched paw was higher in mice treated with oxaliplatin (n = 8, 8.2 ± 0.5 g) than in mice treated with vehicle alone (1.1 ± 0.2 g; P = 0.002) (Figure 1C). Mangafodipir corrected the hypoesthesia observed in the von Frey test (1.36 ± 0.25 g with oxaliplatin plus mangafodipir versus 8.2 ± 0.5 g with oxaliplatin alone, P = 0.002) (Figure 1C).
Mangafodipir prevented oxaliplatin-induced cold hyperalgesia.
Cold hyperalgesia was studied on a cold plate set at –4°C ± 0.2°C in mice treated daily with 3 mg/kg of oxaliplatin. After 5 days of treatment (oxaliplatin cycle 1), the mean number of brisk lifts of either hind paw was higher in mice that were injected daily with oxaliplatin (n = 10, 20 ± 1.4 paw lifts) than in mice injected with vehicle alone (n = 10, 11 ± 1.1 paw lifts; P = 0.0003). We observed the same changes after the second cycle of treatment. The association of mangafodipir with oxaliplatin prevented an increase in the number of paw lifts (n = 10, 12 ± 1.0 after the first cycle and 13 ± 1.2 after the second cycle; P = 0.90 and P = 0.97 versus vehicle) (Figure 1D).
The preventive effects of mangafodipir on oxaliplatin-induced neuropathy are mediated by its antioxidant properties.
We explored separately the activities of the two components of mangafodipir using MnTBAP, a chelate of manganese with superoxide dismutase and catalase activities, and vitamin B6 (Figure 1, E–H). As we had already observed, after 8 weeks, oxaliplatin (10 mg/kg, once a week) induced locomotor disturbances (latency to fall: 514 ± 120 seconds), cold hypoesthesia (latency to escape: 108 ± 11 seconds), and increased the threshold of paw withdrawal (8.20 ± 0.5 g). After 8 weeks, we observed that the antioxidant MnTBAP (10 mg/kg, 3 times per week) in addition to oxaliplatin prevented locomotor disturbances (latency to fall: 961 ± 137 seconds; P = 0.032 versus oxaliplatin alone), cold hypoesthesia (latency to escape: 43 ± 5 seconds; P = 0.00058 versus oxaliplatin alone), and an increase in the paw withdrawal threshold (1.36 ± 0.5 g; P < 0.0001 versus oxaliplatin alone). We found that vitamin B6 had no effect on the neuropathy.
Mangafodipir prevented oxaliplatin-induced neuromuscular hyperexcitability in mice.
We studied the multimodal excitability of the neuromuscular system to characterize the effects of oxaliplatin and mangafodipir. We performed excitability tests (stimulus-response, strength-duration, and current-threshold relationships, as well as the threshold electrotonus and recovery cycle) on the compound muscle action potential (CMAP) recorded in the plantar muscles of mice injected with vehicle, oxaliplatin, mangafodipir, or oxaliplatin plus mangafodipir for 2 and 4 weeks (Table 1 and Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI68730DS1). Off-line treatment of CMAP recordings from mice injected over a 2-week period with either oxaliplatin or mangafodipir, or oxaliplatin plus mangafodipir revealed no markedly abnormal excitability waveform features compared with those of vehicle-injected mice. We confirmed this finding by analyzing the data gained from the five excitability tests (Table 1).
Table 1.
Excitability parameters derived from plantar muscle recordings of mice injected for 15 and 30 days (means ± SEM, n = 8 mice in each group) with vehicle, oxaliplatin, mangafodipir, or mangafodipir plus oxaliplatin
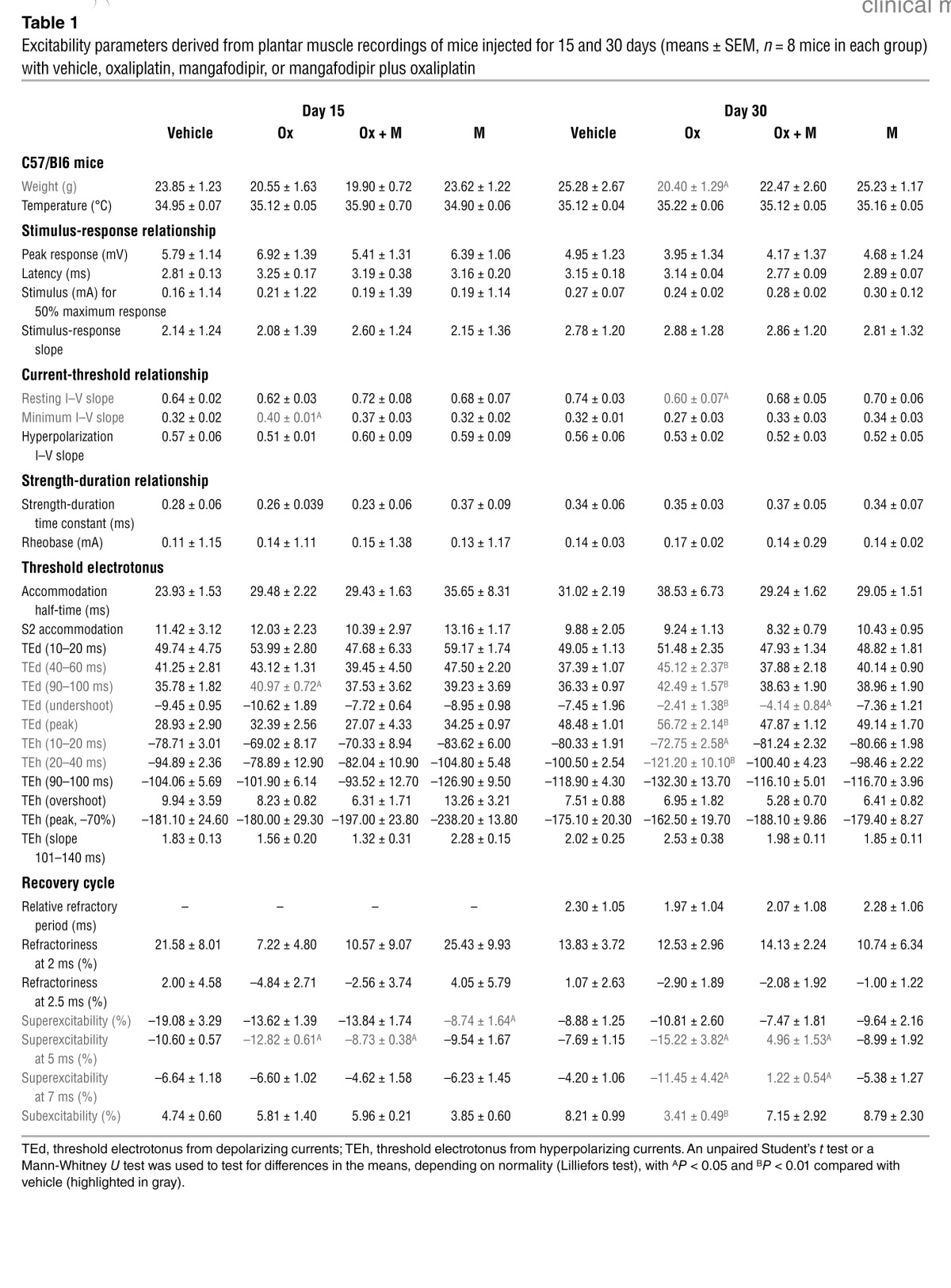
In contrast, the recordings made after 4 weeks of oxaliplatin injections showed alterations in neuromuscular excitability and lower body weight compared with the vehicle-injected animals. These alterations consisted of (a) a reduction in the resting slope (Table 1 and Supplemental Figure 1A), suggesting that fewer ion channels were open at the resting membrane potential; (b) an increase in threshold changes in response to both depolarizing and hyperpolarizing currents (Table 1 and Supplemental Figure 1C), likely caused by a reduced potassium conductance; and (c) greater superexcitability and lower subexcitability (Table 1 and Supplemental Figure 1D), indicative of potassium channel dysfunction. Consistent with membrane hyperexcitability, we did not detect these modifications by 4 weeks in the recordings from mice injected with oxaliplatin plus mangafodipir or with mangafodipir alone.
Morphological characterization of mangafodipir effects on oxaliplatin-induced neuropathy in mice.
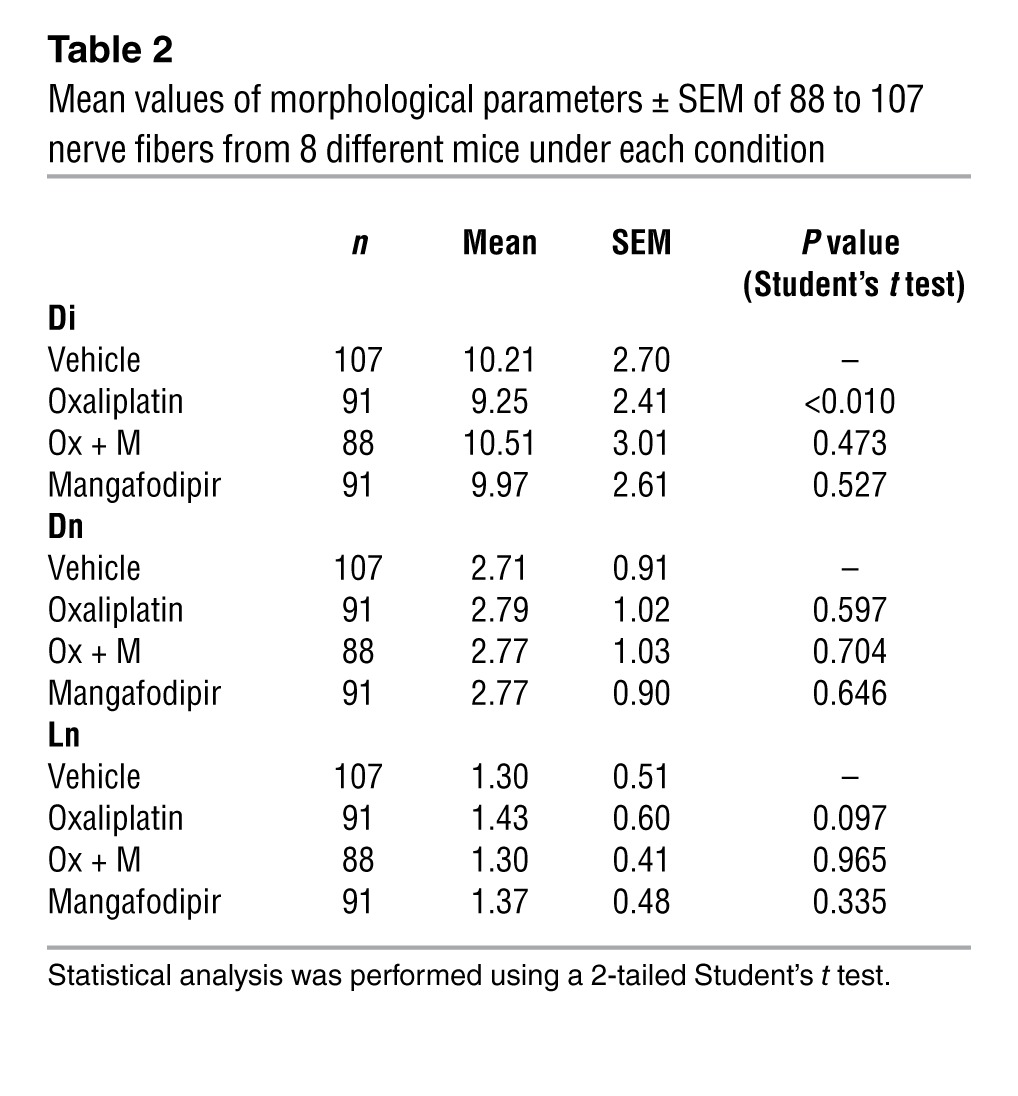
We examined the myelinated axons of sciatic nerves using confocal laser scanning microscopy for optical sectioning followed by 3D reconstruction of myelinated nerve fibers (Figure 2A). The mean values of the three parameters obtained from mice injected for 8 weeks with oxaliplatin revealed a marked decrease in the internodal diameter (P = 0.010), which may reflect a preferential loss of large myelinated nerve fibers. The absence of nodal length modification (P = 0.597) we observed suggests either a preferential loss of large myelinated nerve fibers or an alteration in myelin sheet layers surrounding the axons (Table 2 and Figure 2B). The tendency of nodal length to increase compared with that seen in vehicle-injected animals (P = 0.097, Table 2) is a consequence of membrane hyperexcitability. We did not detect any of these modifications in mice injected for 8 weeks with oxaliplatin plus mangafodipir or with mangafodipir alone.
Figure 2. Morphological characterization of single myelinated nerve fibers isolated from the sciatic nerves of mice injected with vehicle, oxaliplatin, oxaliplatin plus mangafodipir (Ox + M), or mangafodipir (M) for 44 days.

(A) Typical confocal microscopic images under each condition of single nerve fibers acquired by transmitted light (left panels) and of 3D reconstructions by projections of a series of optical sections from single nerve fibers stained with FM1-43 (right panels). The circles indicate the nodes of Ranvier examined. Original magnification, ×3,500. (B) Schematic representation of a single myelinated axon showing the measured morphological parameters: internodal diameter (Di), nodal diameter (Dn), and nodal length (Ln).
Table 2.
Mean values of morphological parameters ± SEM of 88 to 107 nerve fibers from 8 different mice under each condition
Effect of mangafodipir on oxidative stress in mice treated with oxaliplatin.
We determined the advanced oxidized protein products (AOPPs) every 2 weeks in the serum of control and experimental mice. Serum levels remained unchanged in control mice injected with vehicle or mangafodipir (Supplemental Figure 2). In mice injected with oxaliplatin, AOPP levels (μM chloramine T equivalents) increased from week 4 onward (by week 6: 1.83 ± 0.16 μM versus 1.04 ± 0.15 μM in control mice; P = 0.005). We found that the association of mangafodipir with oxaliplatin reduced serum AOPPs (by week 6: 1.50 ± 0.15 μM versus 1.83 ± 0.16 μM in oxaliplatin-treated mice; P = 0.05).
Effect of mangafodipir in patients with oxaliplatin-induced neuropathy: clinical trial.
The phase II clinical trial, which included 23 patients, started in June 2008 and concluded in April 2011. Table 3 details the patients’ baseline characteristics. The oxaliplatin-related neuropathic effects were evaluated in terms of sensitivity to cold objects, discomfort when swallowing cold liquids, throat discomfort, and muscle cramps. The CONSORT flow diagram of patients for whom the treatment was stopped for grade >2 sensory neurotoxicity is shown in Figure 3. The primary endpoint was reached in 77% of patients (17 of 22 patients treated) whose neuropathy improved or stabilized after 4 cycles of mangafodipir and chemotherapy. Mangafodipir reduced the incidence of grade ≥2 neuropathy in 7 of the 22 patients treated (32%) despite the continuation of oxaliplatin for 8 cycles. While at the time of inclusion in the study all patients had grade ≥2 neuropathy, 6 of the 7 patients treated with oxaliplatin and mangafodipir for 8 cycles had grade 0 to 1 neuropathy (Figure 4A). Neuropathy worsened in 6 nonresponder patients, 3 before the fourth cycle and 3 at a later point. Nine patients were excluded because of disease progression that precluded the continuation of oxaliplatin.
Table 3.
Baseline characteristics of cancer patients treated with mangafodipir and oxaliplatin-based chemotherapy
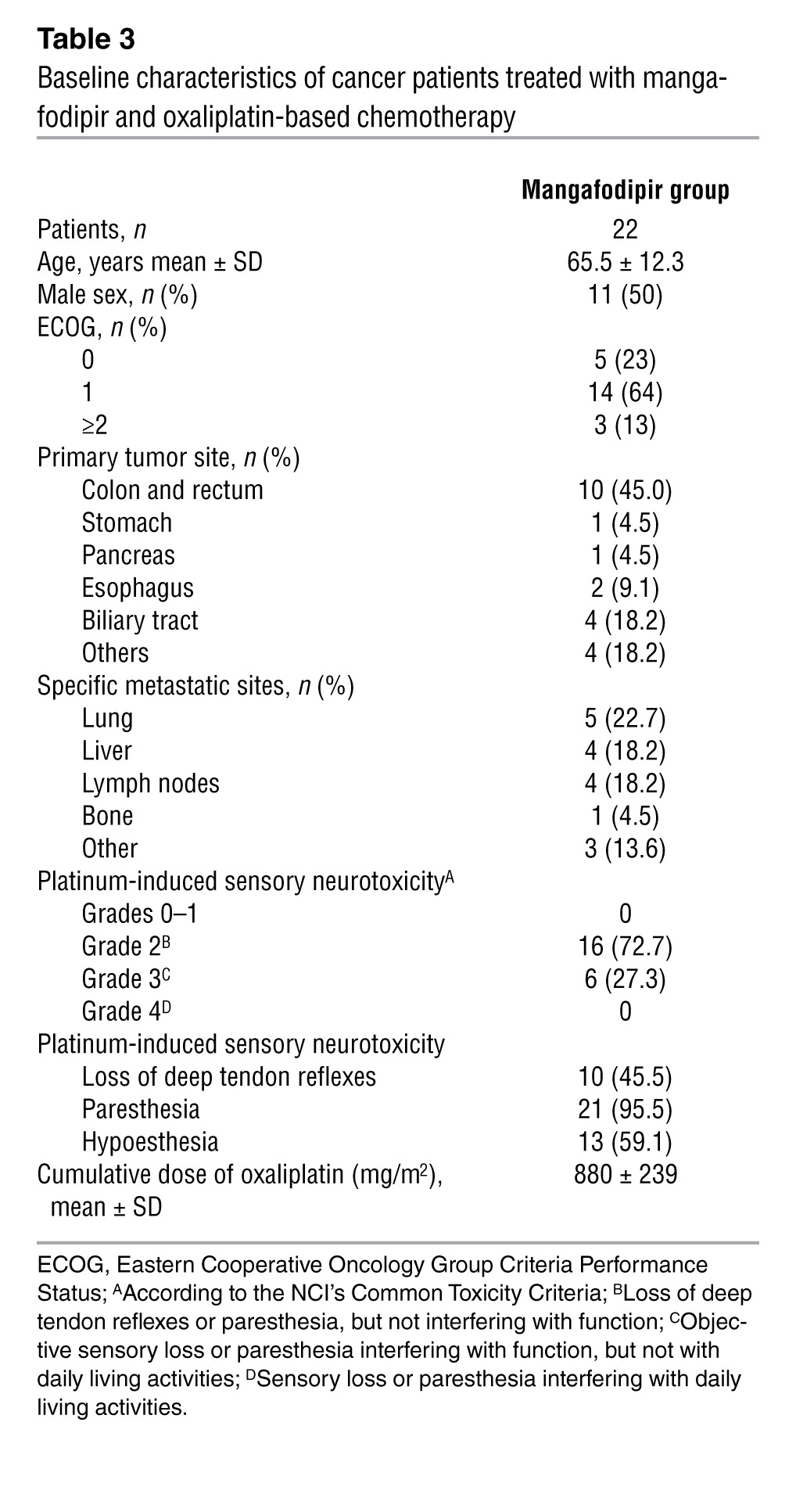
Figure 3. CONSORT statement flow diagram of patients included in the study. n, number of patients.

Figure 4. Sensory neurotoxicity improves with cumulative oxaliplatin plus mangafodipir treatment.

(A) Biological changes in the sera of patients treated with oxaliplatin plus mangafodipir. Plasma manganese changes (B). Oxidative stress markers prior to chemotherapy for serum levels of AOPPs (C), plasma SOD activity (D), GR (E), and thiols (F).
We observed no significant alterations in hematological or biochemical parameters following the introduction of mangafodipir, except for a significant increase in neutrophil cell counts (P = 0.010), a decrease in hemoglobin levels (P < 0.001), and an increase in alkaline phosphatase levels (P < 0.001) (Table 4). Plasma concentrations of manganese remained steady (Figure 4B). In the absence of worsening of neuropathy, oxaliplatin treatment was continued, allowing a median additional progression-free survival of 2.75 months (Figure 5A). Overall survival of patients receiving mangafodipir plus oxaliplatin-based chemotherapy was 33.2 months (Figure 5B).
Table 4.
Biological changes in sera of patients treated with oxaliplatin plus mangafodipir
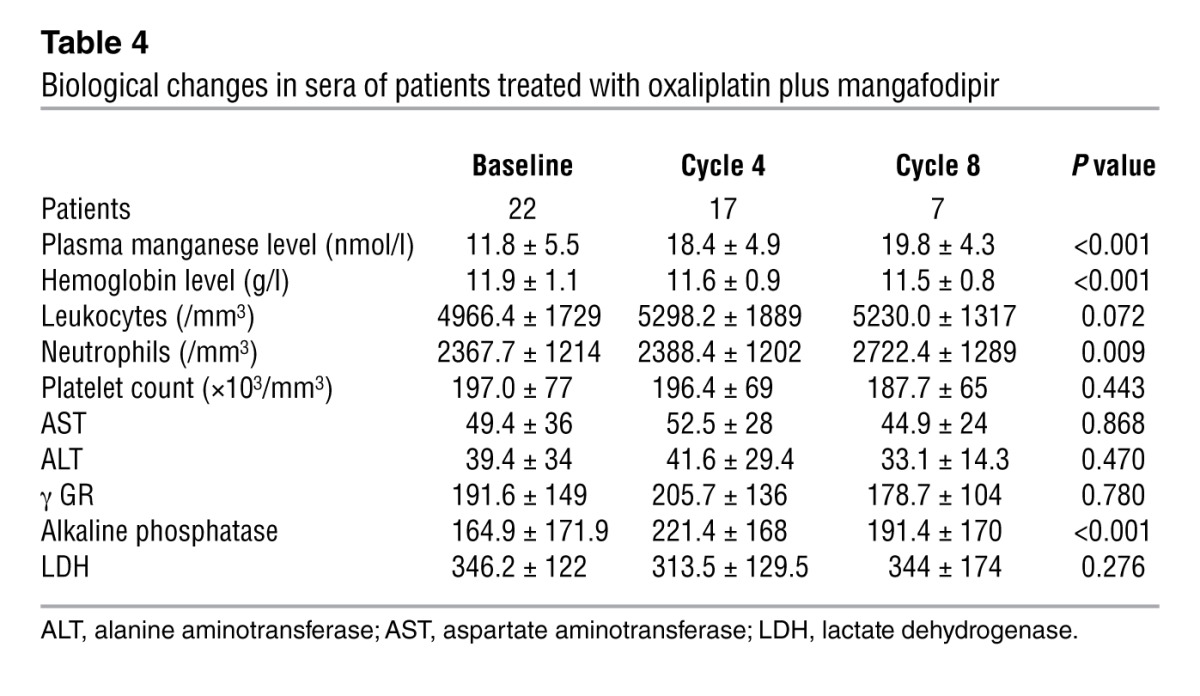
Figure 5. Survival rates of patients treated with oxaliplatin plus mangafodipir.

Progression-free survival (A) and overall survival (B). Cumulative doses of oxaliplatin administered to patients in each group (C).
Effect of mangafodipir on the cumulative dose of oxaliplatin in patients with neuropathy.
The patients enrolled in the study received a mean dose of 880 ± 239 mg/m2 of oxaliplatin prior to inclusion. During the trial, the dosage of oxaliplatin was maintained at 85 mg/m2. Thus, the patients treated with mangafodipir were exposed to a mean additional dose of oxaliplatin of 458 ± 207 mg/m2, allowing a mean cumulative exposure of 1,349 ± 249 mg/m2. We identified no difference in oxaliplatin exposure prior to inclusion in the study between responders and nonresponders (957 ± 408 mg/m2 versus 911 ± 213 mg/m2, P = NS). In contrast, the beneficial effect of mangafodipir in responders allowed a mean additional exposure to oxaliplatin (510 ± 212 mg/m2) that was significantly higher than that in nonresponders (319 ± 113 mg/m2, P = 0.039) (Figure 5C).
Effect of mangafodipir on oxidative stress in patients.
We found that AOPPs determined before each cycle were lower in responders than in nonresponders (P = 0.027) (Figure 4C). Consistently, SOD levels were higher in responders than in nonresponders, suggesting an increased oxidative burst in nonresponders (P = 0.020) (Figure 4D). Further, we observed no differences in GR activities or plasma thiols (Figure 4, E and F).
Discussion
This report shows the beneficial effect of mangafodipir’s association with oxaliplatin in terms of avoiding oxaliplatin-induced neurotoxicity in a mouse model and in patients with grade ≥2 oxaliplatin neuropathy. We believe that this finding may lead to a major breakthrough in the treatment of patients with oxaliplatin, since severe neurotoxicity is the main reason for dose reduction and discontinuation of oxaliplatin.
In order to test our hypothesis that mangafodipir is effective in treating the acute and chronic peripheral neuropathy induced by oxaliplatin, we designed a model of oxaliplatin-induced neurotoxicity in C57Bl/6 mice. All of the clinical and electrophysiological tests we conducted showed that mangafodipir effectively prevented the onset of locomotor and sensitivity disturbances and neuromuscular hyperexcitability in mice treated with oxaliplatin. Furthermore, morphological analyses revealed a decreased internodal (i.e., axonal and myelin sheath layers) diameter of the isolated myelinated nerve fibers in oxaliplatin-treated mice that was abrogated by oxaliplatin’s association with mangafodipir. This involvement of large fibers is a well-known characteristic of platinum-induced neurotoxicity, and our data are in agreement with the morphological changes observed by several groups (19).
The patterns we observed of excitability changes reflecting modulation in axonal membrane potential and ion channel function suggest the involvement of voltage-gated channels in this neuropathy. Previous data have suggested that these changes could result from the dysfunction of voltage-gated ion channels, since either persistent sodium channel activity or decreased potassium conductance can produce axonal hyperexcitability (20). Indeed, in the past decade, it has often been hypothesized that oxaliplatin-induced neuropathy results from defects in voltage-gated sodium channels, resulting in an increase in the amplitude and duration of action potentials that lengthen the refractory period of peripheral nerves (21–23). Nevertheless, Wilson et al. showed no benefit from the potent potassium channel blocker carbamazepine to prevent acute, reversible oxaliplatin-induced neuropathy (20). In vitro studies of various vertebrate nerves showed that dysfunction of voltage-gated potassium and/or sodium channels is involved in oxaliplatin-induced axonal hyperexcitability (7). Thus, the channels involved in the neuropathy secondary to oxaliplatin treatment had been ill defined until recent reports clearly implicated the voltage-gated potassium channels as targets of oxaliplatin, offering new insights into the pharmacological prevention of peripheral neuropathy (24–27). Our mouse model not only confirms the electrophysiological changes in potassium channels, but to the best of our knowledge, also demonstrates for the first time the therapeutic effect of mangafodipir.
Mice treated with oxaliplatin displayed an elevation in serum AOPPs that was probably representative of the pro-oxidative activity of the drug. Since AOPP levels were reduced in mice after 4 weeks of treatment with mangafodipir, we undertook to determine which step of the oxidative cascade was targeted by the molecule. Like mangafodipir, MnTBAP is a chelate of manganese with SOD-like activity, but unlike mangafodipir, it is devoid of vitamin B6 activity (16). Unlike vitamin B6, we found that MnTBAP prevented neuromuscular hyperexcitability, findings that are in accordance with those of Ang et al. following administration of vitamin B complex (28). Therefore, our own data suggest that mangafodipir prevents oxaliplatin-induced neuropathy by scavenging superoxide anions through its SOD-like activity.
Given the data obtained in our animal model, a phase II clinical trial was designed to investigate the curative effect of mangafodipir in patients treated with oxaliplatin. Burakgazi et al. (29) have shown that the two best means of evaluating the severity of oxaliplatin neuropathy are through the use of clinical criteria and the distal leg intraepidermal nerve fiber density assay. The latter was not appropriate for our study, since the patients had grade ≥2 neuropathy at the time of inclusion and very likely had diminished nerve fiber density at the start of the study. Moreover, responder patients in our study received oxaliplatin for 4 months at most, which is not a sufficient duration to expect any improvement in nerve fiber density. Therefore, we used clinical criteria to evaluate the severity of neuropathy.
We found that mangafodipir proved effective in stabilizing or improving grade ≥2 sensory neuropathy in 77% of patients who underwent 4 cycles of oxaliplatin-based chemotherapy. After 8 cycles of chemotherapy, 6 of 7 patients had grade ≤2 neuropathy.
With the addition of mangafodipir to oxaliplatin, the patients already exposed to a mean dose of 880 mg/m2 were able to receive a mean additional dose of 458 mg/m2 of oxaliplatin despite their preexisting neuropathy. The patients were able to continue receiving effective oxaliplatin therapy, resulting in a median progression-free survival of 2.75 months. Moreover, the addition of mangafodipir led to a median overall survival of 33.2 months. Thus, mangafodipir allows a major improvement in the use of oxaliplatin, since severe neurotoxicity is the main reason for dose reduction and discontinuation of oxaliplatin in patients who have a cumulative dose of 750 mg/m2 or more (3, 30, 31).
Further, our data provide evidence that mangafodipir has both curative and preventative properties in oxaliplatin-induced neuropathy, and to the best of our knowledge, this is the first report of successful therapy for this disease.
These results, along with the significant increase observed in neutrophil cell counts with oxaliplatin treatment, are in agreement with previous reports from our team and others that mangafodipir can protect a variety of normal cells from oxidative stress, and in particular from the oxidative stress induced by oxaliplatin, without abrogating the drug’s anticancer effect (2, 16–18, 32, 33). Recently, Karlsson et al. confirmed our preclinical data in patients with regard to hematological protection, but were not able to obtain data regarding the prevention of neuropathy (34).
We found that plasma SOD–like activity was lower in nonresponder patients than in responder patients. Consistently, we observed that the mean serum AOPP concentration in nonresponders was higher, suggesting a more intensive antioxidative effect of mangafodipir in responders than in nonresponders. The nonresponders in our study were either insufficiently exposed to mangafodipir, or had already reached a chronic stage of the disease. Although these results are in line with those obtained in our animal model, they are not truly comparable. Indeed, the doses of oxaliplatin and mangafodipir administered to mice cannot be extrapolated to patients, and the frequency and sites of the mangafodipir injections were not the same. However, altogether, our data suggest that mangafodipir acts upon voltage-gated potassium channels through its antioxidant properties. Moreover, the serum levels of AOPPs could be used as a predictive marker of mangafodipir’s efficacy in preventing oxaliplatin-induced neuropathy.
Chronic exposure to manganese, which appears to be the active component of mangafodipir that prevents neuropathy, can paradoxically lead to neurological side effects, particularly Parkinson-like disease (35). The threshold of manganese toxicity in vivo is unknown, but the maximal increase in plasma concentration (25 nmol/l) reached in our patients was 4,000 times less than the toxic dose in vitro (36). This explains why we observed no clinical or biological side effects of mangafodipir during the trial.
In conclusion, mangafodipir is presently the only available drug that can prevent the worsening of — and that can even ameliorate — oxaliplatin-induced peripheral neuropathy, thus allowing the continuation of platinum-based chemotherapy. Therefore, mangafodipir should be considered a treatment option in patients with grade 2 oxaliplatin–induced neuropathy when oxaliplatin is considered effective against the cancer.
Methods
Animals
C57Bl/6 male mice between 6 and 8 weeks of age were obtained from Janvier (France). Animals received humane care in compliance with institutional guidelines. Control mice received i.p. injections of either 200 μl oxaliplatin (Eloxatine; Sanofi-Aventis) (10 mg/kg) or 200 μl of vehicle alone on Mondays for 8 weeks. Experimental animals received either 200 μl of mangafodipir (10 mg/kg; Teslascan), 200 μl of MnTBAP [manganese(III)tetrakis(4-benzoic acid] (10 mg/kg), or 200 μl of vitamin B6 (10 mg/kg) on Mondays (6 hours after oxaliplatin), Wednesdays, and Fridays for 8 weeks. All molecules were dissolved in the same vehicle (5% glucose solution called “G5”) in which oxaliplatin was reconstituted.
Chemicals
All chemicals were purchased from Sigma-Aldrich except for mangafodipir (GE Healthcare) and oxaliplatin (Sanofi-Aventis).
In vivo study of oxaliplatin-induced neuropathy in mice
Evaluation of motility.
In order to obtain a quantitative measurement of locomotor disturbances induced by oxaliplatin and of the effects of mangafodipir, control and experimental C57Bl/6 mice were placed on a rotarod apparatus (TSE Systems) once a week for 8 weeks. Mice performed three tries per evaluation and were timed while running on the rotating rod (10 rpm) until they stopped or fell.
In vivo study of cold hypoesthesia.
Hypoesthesia was evaluated in mice treated weekly with 10 mg/kg of oxaliplatin for 8 weeks. Cold hypoesthesia was evaluated on a cold plate whose temperature was set at +4°C ± 0.2°C and allowed to stabilize for 20 minutes. A temperature sensor insured accurate temperature readings. Mice were habituated to handling by the investigators and to the testing procedures the week before the experiments. For each session, they were allowed to acclimate for 10 minutes prior to being individually placed onto the cold metal surface enclosed within a clear plexiglass barrier with a top cover. To insure the accuracy of the data, two observers were enlisted. The animal was placed onto the cold plate, and the latency period to escape was measured. The results were expressed as the means of the counts of the observers. A maximal cutoff time of 6 minutes was used to prevent tissue damage.
In vivo study of nociception.
Nociception was evaluated using the von Frey test. The operating principle of this test is standardized (37): once the mouse is calm and motionless, a hind paw is touched by the tip of a flexible fiber of given length and diameter. The fiber is pressed against the plantar surface at a right angle and exerts a vertical force. The force of application increases as long as the investigator is pushing the probe, until the fiber bends. After the fiber bends, continued advancement creates more bend, but not more force of application. This principle makes it possible for the investigator to apply a reproducible force to the skin surface. The scale of force used was from 0.008 to 300 g. Rodents exhibit a paw withdrawal reflex as soon as the paw is touched. The von Frey fiber test was considered positive when the animal indicated a sensation of the fiber by pulling back its paw (37).
In vivo study of cold hyperalgesia.
Cold hyperalgesia was evaluated in mice using the protocol described by Ta et al. (38). Mice were treated daily with 3 mg/kg of oxaliplatin or vehicle for 5 days followed by 5 days of rest, for 2 cycles. Hyperalgesia was evaluated by the cold plate assay at the optimal temperature of –4°C ± 0.2°C. The same equipment was used as for the hypoesthesia evaluation. The total number of brisk lifts of either hind paw was counted as the response to cold hyperalgesia, and two observers were tasked with counting to ensure accuracy. Normal locomotion was distinct, involving coordinated movements of all four limbs that were excluded. A maximal cutoff time of 5 minutes was used to prevent tissue damage. A cold plate test was performed at baseline, during, and after drug treatment at the end of each of the 2 cycles over the 5-day period. Results were expressed as the mean of the observers’ counts.
In vivo study of mouse neuromuscular excitability.
The excitability of the neuromuscular system of control and experimental mice was assessed in vivo 2 and 4 weeks after the initial injections by minimally invasive electrophysiological methods using Qtrac software, as previously described (39). Electrical stimulation was delivered to the sciatic motor nerve by means of surface electrodes, and the CMAP was recorded using needle electrodes inserted into the plantar muscle. To better identify the underlying mechanism(s) involved in the in vivo neuromuscular excitability, five different excitability tests (stimulus-response, strength-duration, and current-threshold relationships, as well as threshold electrotonus and recovery cycle) were performed together. As a whole, more than thirty parameters were determined from the five different excitability tests and were analyzed. It is worth noting that each specific excitability test provides additional and complementary information regarding, on one hand, the functional status of ion channels and electrogenic pumps, and on the other hand, membrane properties (40–42).
In vitro morphological study of mouse single myelinated nerve fibers.
The experiments were carried out for a 6-week period on single myelinated axons isolated from the sciatic nerves of control and experimental mice following the initial injections. Sciatic nerve sections of about 2 cm in length were removed from their sheathes, dissected, and fixed for 1 hour in PBS (1X) with 2% PFA, then rinsed three times with PBS (1X). Sciatic nerves were deposited on microscope slides, myelinated axons were gently teased apart from the main trunk, and preparations were kept at –20°C until use. Just before the experiments, sciatic nerves were rehydrated for about 1 hour with a standard physiological solution containing 154 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 11 mM glucose, and 5 mM Hepes, buffered at pH 7.4 with NaOH. Next, the preparations were exposed for 25 to 30 minutes to the fluorescent dye N-(3-triethylammoniumpropyl)-4-(p-dibutylaminostyryl)pyridinium, followed by 2 μM dibromide (FM1-43; Molecular Probes) dissolved in a standard physiological solution to stain the plasma membranes of the myelinated axons, then the preparations were washed with dye-free solution before imaging. This procedure involved staining nerve plasma membranes for several hours without subsequent destaining. Moreover, the inability of the FM1-43 dye to cross nerve membranes because of its partitioning in the outer membrane leaflet only (43) renders this dye particularly useful for imaging the morphological changes in myelinated axons (43). We used a Zeiss LSM 510 META (Carl Zeiss) multiphoton scanning confocal microscope, mounted on an upright microscope and controlled with the manufacturer’s software and workstation, for optical sectioning of myelinated axons and subsequent 3D high-resolution digital reconstruction of their structure, as previously described (43). Briefly, images were collected using a 63× oil-immersion objective with a 1.40 numerical aperture (Zeiss Plan-Apochromat) following excitation of FM1-43 with the 488-nm wavelength line of an Argon ion laser and then digitized at 12-bit resolution into a 512 × 512 pixel array. Images were then analyzed using ImageJ software (NIH).
In vivo study of the differential effects of MnTBAP and vitamin B6, components of mangafodipir.
Mice treated with 200 μl oxaliplatin (10 mg/kg) weekly for 8 weeks received either 200 μl of MnTBAP (10 mg/kg) or 200 μl of vitamin B6 (10 mg/kg) 3 times a week for 8 weeks. Control mice received only MnTBAP or vitamin B6. The animals were tested once a week using the rotarod, the von Frey test, and cold hypoesthesia, and then at the end of each of the two 5-day cycles as described above.
In vivo study of oxidative stress in the serum of mice with oxaliplatin-induced neuropathy.
The level of oxidative stress was determined in the control mice injected with vehicle and in experimental mice prior to i.p. injections. To measure AOPP levels (44), 200 μl of serum diluted 1:5 in PBS was dispensed into a 96-well microtitration plate, and 20 μl of acetic acid was added to each well. To establish the standard curve, 10 μl 1.16 M potassium iodide (Sigma-Aldrich) was added to 200 μl chloramine-T solution (0–100 μmol/l) (Sigma-Aldrich) followed by 20 μl acetic acid. The absorbance of the reaction mixture was immediately read at 340 nm against a blank consisting of 200 μl PBS, 10 μl 1.16 M potassium iodide, and 20 μl acetic acid. The AOPP concentrations were expressed as micromoles per liter of chloramine-T equivalents.
Clinical trial
The goal of the trial was to test the curative effects of mangafodipir on oxaliplatin-induced peripheral neuropathy in patients treated for cancer with oxaliplatin. The primary objective was to achieve an improvement or a stabilization of oxaliplatin-induced neuropathy during the administration of mangafodipir in at least 50% of the patients. The trial design described by Gehan was applied to the present study (45). The trial was conducted in two steps. Nine patients were included in step 1 to assess whether the treatment met the required standards of success. The sample size in step 2 was based on the success rate (n = 14) achieved in step 1. A mangafodipir response rate of 30% with a β error of 5% was predetermined as the activity level of interest.
Eligible patients included those who suffered from grade ≥2 sensory neuropathy following oxaliplatin-based chemotherapy. These were adults with stage IV carcinoma who, after undergoing oxaliplatin-based chemotherapy, were scheduled to receive at least 4 cycles (2 months) of oxaliplatin-based FOLFOX chemotherapy plus mangafodipir (0.5 ml/kg), with 85 mg/m2 or 100 mg/m2 oxaliplatin every 2 weeks (30, 46, 47). Patients were allowed to receive bevacizumab or cetuximab in combination with the FOLFOX. Participants needed to have adequate hematologic parameters in order to undergo chemotherapy and were required to have serum total bilirubin, creatinine, and calcium concentrations ≤1.5 times the upper normal limit (UNL). Women of childbearing potential were required to have a negative pregnancy test. Patients were not allowed to participate in the trial if they had one or more of the following contraindications: hypercalcemia, previous vinca-alkaloid treatment, treatment with digoxin, heart block or other medical condition which, in the opinion of the treating physician, would make the protocol unreasonably hazardous, or a history of genetic/familial neuropathy. Patients who were considered to be unable to comply with the protocol were excluded from the trial.
At the start of the study and before each 2-week cycle of chemotherapy, patients underwent a medical history and physical examination and laboratory tests (i.e., serum calcium, magnesium, sodium, potassium, creatinine, aspartate aminotransferase (AST), total bilirubin, alkaline phosphatase, and manganese). Oxaliplatin was infused over a 2-hour period. Mangafodipir, aimed at neutralizing the ROS generated by oxaliplatin, was infused in the 30 minutes immediately following the administration of oxaliplatin because of oxaliplatin’s short half-life (about 30 minutes) and rapid tissue uptake. The National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI CTCAE, version 3.0) were used for primary neuropathy assessment. Standardized questions regarding neurotoxic symptoms and examples of answers (48) were used to allow a more accurate scoring of patient-reported symptoms in terms of grades 1, 2, 3, or 4. Patients who experienced no improvement in grade 3 sensory neurotoxicity within 2 weeks were excluded due to sensory neurotoxicity progression. The guidelines also called for a reduction in oxaliplatin dosage to 65 mg/m2 and a reduction in 5-FU dosage by 20% in patients who recovered from grades 3 to 4 gastrointestinal toxicity, grade 4 neutropenia, or grades 3 to 4 thrombocytopenia.
The primary endpoint was the percentage of patients with grade ≤2 chronic sensory neurotoxicity during or at the end of therapy. A maximum of 8 cycles (4 months) of oxaliplatin-based chemotherapy plus mangafodipir was authorized. The patients were divided into two groups: the nonresponders, consisting of patients who experienced persistent grade 3 sensory neurotoxicity; and responders, consisting of patients with a decreasing or constant grade 2 neurotoxicity despite the continuation of oxaliplatin (responder patients).
The secondary endpoint was the cumulative dose of oxaliplatin received by the responder and nonresponder patients, given that all the patients included in the study had previously received oxaliplatin.
Evaluation of oxidative stress in the venous blood of patients
The level of oxidative stress was determined prior to each chemotherapy cycle in venous samples of serum (AOPPs), plasma (thiols), and erythrocyte lysates (SOD and GR activities and total glutathione). Serum, plasma, and erythrocyte lysates were stored at –80°C until use. Serum AOPPs were assayed as described above. Thiol groups were determined using Ellman’s reagent (49). SOD and GR activities were measured with specific kits from Randox Laboratories. Total glutathione was measured using a kit from Cayman Chemical.
Statistics
The statistical significance of differences between experimental groups and controls for either characterization of neuromuscular excitability or morphology or myelinated nerve fibers were analyzed using a 2-tailed Student’s t test for normally distributed continuous variables (Lilliefors test) and a Wilcoxon signed rank test for continuous variables that were not normally distributed. Patients’ baseline characteristics and biological data are reported as the mean ± 1 SD or as a percentage, as appropriate. To compare oxidative stress levels (AOPPs, SOD, GR, and plasma protein thiol oxidation) between responders and nonresponders at baseline, a Wilcoxon signed rank test was performed. Biological parameter modifications under chemotherapy were modeled using linear mixed-effects models. For each biomarker, three random effects structures were tested: random intercept, random slope, and both random intercept and slope. The model that minimized the Akaike information criterion (AIC) was selected as the most appropriate. Analyses were performed using SAS 9.2 software (SAS Institute Inc.). Tests were two-sided, and a P value less than 0.05 was considered statistically significant. *P < 0.05; **P < 0.01; ***P < 0.001.
Study approval
The present study in mice was reviewed and approved by the Comité d’Ethique en Matière d’Expérimentation Animale of the Université Paris Descartes (permit 75-1302) and by the French Departmental Direction of Animal Protection (authorization A91-453). The experiments were performed in accordance with the guidelines established by the French Council on animal care and the guidelines of the EEC86/609 Council Directive (Decree 2001-131) on the treatment of mice under anesthesia by isoflurane (Aerrane; Baxter S.A.) inhalation. The clinical trial was reviewed and approved by the Comité de Protection des Personnes of the Hôpital Tarnier (Paris, France) and registered at Clinicaltrials.gov (NCT00727922) and was conducted in accordance with the principles of the Declaration of Helsinki. All patients provided written informed consent.
Supplementary Material
Acknowledgments
The authors are grateful to Agnes Colle for her excellent typing of the manuscript.
Footnotes
Conflict of interest: Frédéric Batteux and Bernard Weill are co-founders of Protexel, a research and development company aiming to develop mangafodipir as a commercial drug. Frédéric Batteux and Bernard Weill are co-inventors of patents describing mangafodipir as a liver-protective and anticancer agent. They have received no income from Protexel.
Role of funding sources: The studies in the animal model were funded by the Université Paris Descartes and the Ministère de la Recherche et de l’Enseignement Supérieur. The clinical trial was funded by Assistance Publique-Hôpitaux de Paris.
Citation for this article: J Clin Invest. 2014;124(1):262–272. doi:10.1172/JCI68730.
See the related Commentary beginning on page 72.
References
- 1.Masuda H, Tanaka T, Takahama U. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem Biophys Res Commun. 1994;203(2):1175–1180. doi: 10.1006/bbrc.1994.2306. [DOI] [PubMed] [Google Scholar]
- 2.Laurent A, et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005;65(3):948–956. [PubMed] [Google Scholar]
- 3.Gamelin E, Gamelin L, Bossi L, Quasthoff S. Clinical aspects and molecular basis of oxaliplatin neurotoxicity: current management and development of preventive measures. Semin Oncol. 2002;29(5 suppl 15):21–33. doi: 10.1053/sonc.2002.35525. [DOI] [PubMed] [Google Scholar]
- 4.Hartmann JT, Lipp HP. Toxicity of platinum compounds. Expert Opin Pharmacother. 2003;4(6):889–901. doi: 10.1517/14656566.4.6.889. [DOI] [PubMed] [Google Scholar]
- 5.Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev. 2008;34(4):368–377. doi: 10.1016/j.ctrv.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Extra JM, Marty M, Brienza S, Misset JL. Pharmacokinetics and safety profile of oxaliplatin. Semin Oncol. 1998;25(2 suppl 5):13–22. [PubMed] [Google Scholar]
- 7.Grolleau F, Gamelin L, Boisdron-Celle M, Lapied B, Pelhate M, Gamelin E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J Neurophysiol. 2001;85(5):2293–2297. doi: 10.1152/jn.2001.85.5.2293. [DOI] [PubMed] [Google Scholar]
- 8.Holmes J, et al. Comparative neurotoxicity of oxaliplatin, cisplatin, and ormaplatin in a Wistar rat model. Toxicol Sci. 1998;46(2):342–351. doi: 10.1006/toxs.1998.2558. [DOI] [PubMed] [Google Scholar]
- 9.Carozzi VA, Marmiroli P, Cavaletti G. The role of oxidative stress and anti-oxidant treatment in platinum-induced peripheral neurotoxicity. Curr Cancer Drug Targets. 2010;10(7):670–682. doi: 10.2174/156800910793605820. [DOI] [PubMed] [Google Scholar]
- 10.Gamelin L, et al. Prevention of oxaliplatin-related neurotoxicity by calcium and magnesium infusions: a retrospective study of 161 patients receiving oxaliplatin combined with 5-Fluorouracil and leucovorin for advanced colorectal cancer. Clin Cancer Res. 2004;10(12 pt 1):4055–4061. doi: 10.1158/1078-0432.CCR-03-0666. [DOI] [PubMed] [Google Scholar]
- 11.Lersch C, et al. Prevention of oxaliplatin-induced peripheral sensory neuropathy by carbamazepine in patients with advanced colorectal cancer. Clin Colorectal Cancer. 2002;2(1):54–58. doi: 10.3816/CCC.2002.n.011. [DOI] [PubMed] [Google Scholar]
- 12.Mollman JE. Cisplatin neurotoxicity. N Engl J Med. 1990;322(2):126–127. doi: 10.1056/NEJM199001113220210. [DOI] [PubMed] [Google Scholar]
- 13.Goss-Sampson MA, MacEvilly CJ, Muller DP. Longitudinal studies of the neurobiology of vitamin E and other antioxidant systems, and neurological function in the vitamin E deficient rat. J Neurol Sci. 1988;87(1):25–35. doi: 10.1016/0022-510X(88)90051-2. [DOI] [PubMed] [Google Scholar]
- 14.Cascinu S, et al. Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2002;20(16):3478–3483. doi: 10.1200/JCO.2002.07.061. [DOI] [PubMed] [Google Scholar]
- 15.Yang TT, Wang SJ. Pyridoxine inhibits depolarization-evoked glutamate release in nerve terminals from rat cerebral cortex: a possible neuroprotective mechanism? J Pharmacol Exp Ther. 2009;331(1):244–254. doi: 10.1124/jpet.109.155176. [DOI] [PubMed] [Google Scholar]
- 16.Bedda S, et al. Mangafodipir prevents liver injury induced by acetaminophen in the mouse. J Hepatol. 2003;39(5):765–772. doi: 10.1016/S0168-8278(03)00325-8. [DOI] [PubMed] [Google Scholar]
- 17.Coriat R, et al. Mangafodipir protects against hepatic ischemia-reperfusion injury in mice. PLoS One. 2011;6(11):e27005. doi: 10.1371/journal.pone.0027005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandre J, et al. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006;98(4):236–244. doi: 10.1093/jnci/djj049. [DOI] [PubMed] [Google Scholar]
- 19.Roelofs RI, Hrushesky W, Rogin J, Rosenberg L. Peripheral sensory neuropathy and cisplatin chemotherapy. Neurology. 1984;34(7):934–938. doi: 10.1212/WNL.34.7.934. [DOI] [PubMed] [Google Scholar]
- 20.Wilson RH, Lehky T, Thomas RR, Quinn MG, Floeter MK, Grem JL. Acute oxaliplatin-induced peripheral nerve hyperexcitability. J Clin Oncol. 2002;20(7):1767–1774. doi: 10.1200/JCO.2002.07.056. [DOI] [PubMed] [Google Scholar]
- 21.Adelsberger H, Quasthoff S, Grosskreutz J, Lepier A, Eckel F, Lersch C. The chemotherapeutic oxaliplatin alters voltage-gated Na(+) channel kinetics on rat sensory neurons. Eur J Pharmacol. 2000;406(1):25–32. doi: 10.1016/S0014-2999(00)00667-1. [DOI] [PubMed] [Google Scholar]
- 22.Park SB, Goldstein D, Lin CS, Krishnan AV, Friedlander ML, Kiernan MC. Acute abnormalities of sensory nerve function associated with oxaliplatin-induced neurotoxicity. J Clin Oncol. 2009;27(8):1243–1249. doi: 10.1200/JCO.2008.19.3425. [DOI] [PubMed] [Google Scholar]
- 23.Kiernan MC. The pain with platinum: oxaliplatin and neuropathy. Eur J Cancer. 2007;43(18):2631–2633. doi: 10.1016/j.ejca.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 24.Descoeur J, et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med. 2011;3(5):266–278. doi: 10.1002/emmm.201100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimitrov AG, Dimitrova NA. A possible link of oxaliplatin-induced neuropathy with potassium channel deficit. Muscle Nerve. 2012;45(3):403–411. doi: 10.1002/mus.22311. [DOI] [PubMed] [Google Scholar]
- 26.Thibault K, et al. Cortical effect of oxaliplatin associated with sustained neuropathic pain: exacerbation of cortical activity and down-regulation of potassium channel expression in somatosensory cortex. Pain. 2012;153(8):1636–1647. doi: 10.1016/j.pain.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 27.Kagiava A, Kosmidis EK, Theophilidis G. Oxaliplatin-induced hyperexcitation of rat sciatic nerve fibers: an intra-axonal study. Anticancer Agents Med Chem. 2013;13(2):373–379. doi: 10.2174/1871520611313020023. [DOI] [PubMed] [Google Scholar]
- 28.Ang CD, et al. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst. 2008;(3):CD004573. doi: 10.1002/14651858.CD004573.pub3. [DOI] [PubMed] [Google Scholar]
- 29.Burakgazi AZ, Messersmith W, Vaidya D, Hauer P, Hoke A, Polydefkis M. Longitudinal assessment of oxaliplatin-induced neuropathy. Neurology. 2011;77(10):980–986. doi: 10.1212/WNL.0b013e31822cfc59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andre T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–2351. doi: 10.1056/NEJMoa032709. [DOI] [PubMed] [Google Scholar]
- 31.Broomand A, Jerremalm E, Yachnin J, Ehrsson H, Elinder F. Oxaliplatin neurotoxicity — no general ion channel surface-charge effect. J Negat Results Biomed. 2009;8:2. doi: 10.1186/1477-5751-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlsson JO, et al. Cardioprotective effects of the MR contrast agent MnDPDP and its metabolite MnPLED upon reperfusion of the ischemic porcine myocardium. Acta Radiol. 2001;42(6):540–547. doi: 10.1080/028418501127347340. [DOI] [PubMed] [Google Scholar]
- 33.Hirt D, et al. Pharmacokinetic-pharmacodynamic modeling of manganese after a single intravenous infusion of mangafodipir in patients with acute alcoholic hepatitis. Ther Drug Monit. 2009;31(5):557–565. doi: 10.1097/FTD.0b013e3181affd6d. [DOI] [PubMed] [Google Scholar]
- 34.Karlsson JO, Adolfsson K, Thelin B, Jynge P, Andersson RG, Falkmer UG. First clinical experience with the magnetic resonance imaging contrast agent and superoxide dismutase mimetic mangafodipir as an adjunct in cancer chemotherapy-a translational study. Transl Oncol. 2012;5(1):32–38. doi: 10.1593/tlo.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perl DP, Olanow CW. The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol. 2007;66(8):675–682. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- 36.Rovetta F, et al. Organ-specific manganese toxicity: a comparative in vitro study on five cellular models exposed to MnCl(2). Toxicol In Vitro. 2007;21(2):284–292. doi: 10.1016/j.tiv.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Miraucourt LS, Moisset X, Dallel R, Voisin DL. Glycine inhibitory dysfunction induces a selectively dynamic, morphine-resistant, and neurokinin 1 receptor-independent mechanical allodynia. J Neurosci. 2009;29(8):2519–2527. doi: 10.1523/JNEUROSCI.3923-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ta LE, Low PA, Windebank AJ. Mice with cisplatin and oxaliplatin-induced painful neuropathy develop distinct early responses to thermal stimuli. Mol Pain. 2009;5:9. doi: 10.1186/1744-8069-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boerio D, Greensmith L, Bostock H. Excitability properties of motor axons in the maturing mouse. J Peripher Nerv Syst. 2009;14(1):45–53. doi: 10.1111/j.1529-8027.2009.00205.x. [DOI] [PubMed] [Google Scholar]
- 40.Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve. 2000;23(3):399–409. doi: 10.1002/(SICI)1097-4598(200003)23:3<399::AID-MUS12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 41.Krishnan AV, Lin CS, Park SB, Kiernan MC. Assessment of nerve excitability in toxic and metabolic neuropathies. J Peripher Nerv Syst. 2008;13(1):7–26. doi: 10.1111/j.1529-8027.2008.00155.x. [DOI] [PubMed] [Google Scholar]
- 42.Lefaucheur JP, Boerio D, Hogrel JY, Creange A. [Nerve excitability studies in the assessment of dysimmune neuropathies]. Rev Neurol (Paris). 2006;162(spec no 1):3S17–3S26. [PubMed] [Google Scholar]
- 43.Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12(2):363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Witko-Sarsat V, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996;49(5):1304–1313. doi: 10.1038/ki.1996.186. [DOI] [PubMed] [Google Scholar]
- 45.Gehan EA. Design of controlled clinical trials: use of historical controls. Cancer Treat Rep. 1982;66(5):1089–1093. [PubMed] [Google Scholar]
- 46.de Gramont A, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–2947. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 47.Maindrault-Goebel F, et al. Oxaliplatin added to the simplified bimonthly leucovorin and 5-fluorouracil regimen as second-line therapy for metastatic colorectal cancer (FOLFOX6). GERCOR. Eur J Cancer. 1999;35(9):1338–1342. doi: 10.1016/S0959-8049(99)00149-5. [DOI] [PubMed] [Google Scholar]
- 48.Grothey A, et al. Intravenous calcium and magnesium for oxaliplatin-induced sensory neurotoxicity in adjuvant colon cancer: NCCTG N04C7. J Clin Oncol. 2010;29(4):421–427. doi: 10.1200/JCO.2010.31.5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu ML. Measurement of protein thiol groups and glutathione in plasma. Methods Enzymol. 1994;233:380–385. doi: 10.1016/s0076-6879(94)33044-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.