

Abstract
Epigenetic therapies may play a prominent role in the future management of solid tumors. This possibility is based on the clinical efficacy of existing drugs in treating defined hematopoietic neoplasms, paired with promising new data from preclinical and clinical studies that examined these agents in solid tumors. We suggest that current drugs may represent a targeted therapeutic approach for reprogramming solid tumor cells, a strategy that must be pursued in concert with the explosion in knowledge about the molecular underpinnings of normal and cancer epigenomes. We hypothesize that understanding targeted proteins in the context of their enzymatic and scaffolding functions and in terms of their interactions in complexes with proteins that are targets of new drugs under development defines the future of epigenetic therapies for cancer.
Introduction
The last decade has witnessed an explosion in our understanding of epigenetic modifications in cancer, including a growing appreciation of its complexity and plasticity (1–4). These developments have poised us for an exciting time to translate this knowledge into the concept of “epigenetic therapy” for cancer. To date, such therapies have gained traction largely within the sphere of hematopoietic malignancies; however, an improved understanding of the determinants of sensitivity to existing drugs, both in laboratory and early clinical trial paradigms, suggests true potential for solid tumors (5, 6). Moreover, recent studies may pave the way for newly emerging drugs and novel drug combinations in solid tumor treatment.
Key facets of the epigenome
The basic machinery.
Epigenetics provides a mechanism to mediate heritable changes in patterns of gene expression without intrinsic changes in the DNA sequence (1–4, 7–9). Epigenetic control, including interactions between DNA methylation, histone modifications, and nucleosome remodeling, determines access to DNA, which then maintains cellular transcription patterns and genome structural integrity. A steady-state arrangement of coding and noncoding regions of DNA is established to regulate gene expression and maintain stable cell phenotypes (1–4, 7–9). These interactions play a central role in normal development and adult cell renewal (2, 8, 10).
The extensive alterations in epigenetic interactions that occur in cancer are broadly termed the “cancer epigenome” (1, 2, 4). Abnormalities in DNA methylation and chromatin become evident early in cancer initiation and progression, including in cancer risk states. (2, 4, 11, 12). A recent data-driven view of these abnormalities suggests a widespread breakdown in epigenetically mediated boundaries in linear and three-dimensional arrangements of nuclear DNA that normally maintain separation of more actively transcribed DNA regions from repressed regions (2, 13–15).
The machinery of epigenetic control resides in chromatin, the structure of DNA and histone proteins, which is regulated by DNA modifications, such as methylation, and histone modifications, including acetylation, ubiquitylation, sumoylation, and methylation. The core element of chromatin is the nucleosome, which includes 147 base pairs of DNA coiled around a histone octamer made of pairs of histones H2A, H2B, H3, and H4 (3, 8). These nucleosomes participate in establishment of chromatin boundaries in the genome through regulated modifications on the amino acids on the extending tails and more internal regions of histone proteins (3, 8, 16). Additionally, the spacing of nucleosomes determines three-dimensional aspects of DNA packaging such that dense, closely compacted nucleosomes, often in association with highly methylated DNA, suggests transcriptional repression, and looser nucleosome positioning, or open chromatin, is typical of actively transcribed domains (17, 18). Finally, chromatin boundaries distinguishing active versus inactive transcription domains are separated by binding of insulator proteins that interact with all of the above (2, 14, 19).
The geography of epigenetic control of the genome.
Early epigenetic research typically concentrated on gene promoter regions. For cancer, this emphasis was focused on the initial epigenetic abnormalities discovered, including alterations in DNA methylation and histone lysine acetylation (2, 4, 20). Although such regions remain vital, recent evidence indicates that epigenetic regulation affects key sites distal to promoters, many controlling the transcription of not only coding but also noncoding regions and affecting the three-dimensional organization of DNA (21). For example, many histone modifications and DNA methylation are critical to the activity of enhancer regions distal to gene start sites and participate in regulating whether genes are poised for transcription or are fully repressed or active (22–25). Understanding this landscape will be important in designing epigenetic therapy strategies for cancer.
The interactions of genetics and epigenetics.
A robust interplay between genetic and epigenetic changes drives tumorigenesis. Early examples include the mutagenic potential of DNA-methylated cytosines and the consequences of epigenetically controlled loss of function in DNA repair genes such as MLH1 and O-6-methylguanine methyltransferase (MGMT) (2, 26–28). More recent findings suggest that essentially all cancer types harbor mutations in genes encoding regulatory proteins responsible for establishing and maintaining epigenomes (2, 29). Even mutations in histones at key amino acids for modifications can seemingly cause cancer (30). While it remains to be definitively determined how such mutations cause specific epigenetic abnormalities in cancer, given the difficulties of therapeutically altering mutations, reversing the downstream consequences of oncogenic mutations in epigenetic regulators is an attractive strategy.
Therapeutically targeting the cancer epigenome
The cancer epigenome–regulating “machinery.”
The potential of epigenetic therapy for solid tumors is intimately tied to the ability to specifically target individual proteins that regulate the cancer epigenome. Epigenetic regulators can be broadly broken down into “writers” (enzymes that establish DNA methylation or histone modifications), “erasers” (proteins that remove these marks), and “readers” (proteins that bind modifications and facilitate epigenetic effects) (refs. 2, 3, 31–33, and Figure 1). In addition, “movers” are protein complexes that position the nucleosomes across the genome (refs. 18, 29, 34, and Figure 1).
Figure 1. Various classes of epigenetic modifiers.
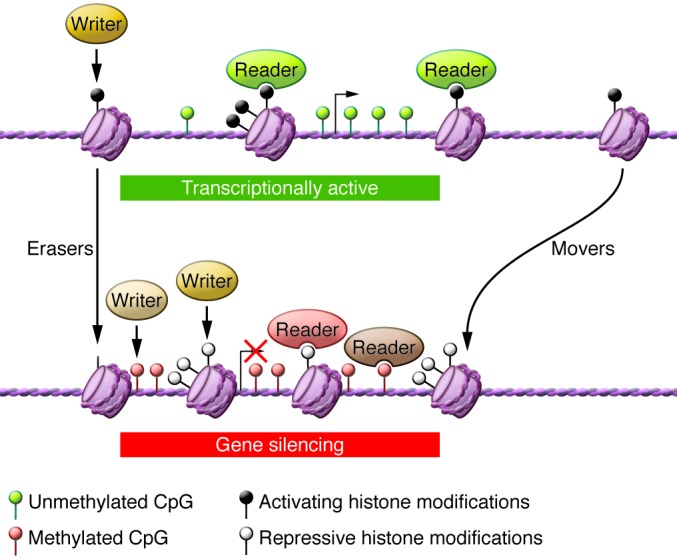
The epigenetic machinery consists of components that catalyze the covalent modifications of histone tails or the DNA (writers). These marks are interpreted by proteins (readers) that bind to the epigenetic marks, resulting in regulation of gene expression. Another class of proteins (erasers) reverses the epigenetic marks by catalyzing their removal. A class of protein complexes (movers) catalyzes the shifting of nucleosomes, resulting in a compact or relaxed chromatin. Here, the broad importance of these proteins in the context of gene silencing is shown. Writers acting separately on histones and DNA result in inactive histone modifications and CpG methylation. Erasers (such as LSD1) remove active histone modifications. Protein readers that bind to active histone (black lollipops) marks (e.g., bromodomain containing 4) are shown in green, those binding to inactive histone (white lollipops) marks (e.g., heterochromatin protein 1, chromobox homolog 7) are shown in pink, and those binding to methylated CpG (green lollipop denotes unmethylated CpG; red lollipop denotes methylated CpG) are shown in brown. The histone movers are shown to play a role in compacting chromatin. Once silenced, a gene will be maintained in the silenced state across cell divisions via recruitment of the writers in every cell cycle.
In cancer, global genome DNA hypomethylation is frequently observed, along with regional hypermethylation involving gene promoters (2, 4, 20). The genome-wide losses often affect chromosome pericentromeric regions, which may elicit structural consequences for chromosomal instability and abnormal transcriptional consequences for repeat sequences (35–38). While cancers can exhibit hypomethylation of normally methylated promoter regions, which may correlate with upregulated transcription, this route of oncogene activation is not frequently observed (39). Cancer-related losses of DNA methylation can also involve gene bodies and regions flanking genes. Unlike transcriptional repression associated with DNA methylation in promoter regions, gene body losses strongly correlate with decreased gene expression (15, 21, 24).
Regional de novo, cancer-specific gains of DNA methylation in cancer have been primarily ascribed to CpG-enriched DNA (CpG islands) in and around gene promoters that are not normally DNA methylated during development (2, 4, 8, 9, 11). This latter point is still often not appreciated and is essential when looking for targetable, cancer-specific events. Such promoter DNA methylation can be associated with decreased gene expression in tumor suppressor genes, acting as an alternative mechanism for transcriptional silencing (2, 4, 8, 9, 11). Nearly all tumors contain hundreds of DNA-hypermethylated genes (40–45) and similar changes in noncoding regions that harbor microRNAs (46, 47). Dissecting the importance of these instead of the gene network alterations in driving tumorigenesis is challenging, given that most of these genes do not harbor known loss-of-function mutations (2, 4, 8, 9, 48). We contend that many of these genes, or groups of genes, are important, especially those that can help prevent loss of self-renewal capacity of tumor stem-like cells and/or prevent cells in the tumor from differentiating (5, 41). A key element of our argument lies in the fact that the majority of genes in cancer with typical de novo, cancer-specific gains in promoter region DNA methylation are developmental genes with key roles in balancing stem cell renewal and lineage commitment during development (41, 49–51).
Targeting DNA methylation abnormalities in cancer involves the protein complexes that regulate and functionally interact with methylated DNA (Figure 2). DNA methylation in promoter regions exerts transcriptional repression in the context of chromatin and nucleosome position, which may target DNA methylation and/or be recruited by it (refs. 2–4, 11, and Figure 2). A key element is the deacetylation of histone tail lysines that is often integral to transcriptional silencing (2, 4, 11, 20, 52–54). Histone deactylases (HDACs), principally HDAC1 and -2, along with the class 3 HDAC, sirtuin 1 (SIRT1), are integrally linked to the initiation and/or maintenance of repression for DNA-hypermethylated genes (55, 56). Growing evidence suggests that these HDACs are recruited to abnormally DNA-methylated promoters in at least four ways (Figure 2). First, the three enzymes that catalyze DNA methylation, DNA methyltransferase 1 (DNMT1), -3A, and -3B, each bind HDAC1 and -2 (57–61). Second, DNA methylation recruits methylcytosine-binding proteins (MBDs; MBD1, MBD2, MBD3, and MeCP2), which also mediate transcriptional repression and bind these HDACs (62, 63). Third, a key component of the nucleosome remodeling and deacetylase (NuRD) repressive remodeling complex, CHD4, which binds HDAC1 and -2, may be integral in maintenance of transcriptional repression of DNA-hypermethylated genes (64). Further, nucleosomes assume a compacted organization over the start sites of DNA-hypermethylated genes (65), and complexes like NuRD are likely critical in controlling this aspect of transcriptional repression. Fourth, SIRT1 and the long-term gene-silencing complex polycomb (PcG) may be integrally linked to the acquisition of DNA-hypermethylated gene promoters during tumorigenesis (56, 66, 67). PcG proteins are key regulators of many developmental genes that are subject to abnormal promoter DNA methylation in cancer. (41, 49, 68). We recently defined one or more complexes linking DNMTs, PcG proteins, and SIRT1 to the vulnerability of genes to DNA hypermethylation in cancer that is recruited to CpG island promoters during inflammatory-like stress, a key cancer risk state (ref. 67 and Figure 2).
Figure 2. Model for initiation of DNA methylation-mediated gene silencing.

(A) Left panel: Methylation of CpGs at the borders of CpG islands in normal cells is mediated by the DNMTs as a complex with the HDACs, PRC4 complex (PRC2 component of PcG including SIRT1) with potential involvement of nucleosome remodelers (NuRD). In normal cells, the DNA methylation machinery is restricted from CpG islands, thus protecting these regions from methylation. Right panel: Movement of the repressive complex into the CpG island with stress and associated gene silencing, marked with a red X. However, most genes revert back to the normal cell state. (B) Left panel: A subset of genes with CpG island promoters in embryonic and adult stem cells have both the active (H3K4me3) and inactive (H3K27me3, PcG mark) marks at their promoters, termed “bivalent” marks, in which the genes have a low but poised expression state. Genes that are hypermethylated in cancers frequently have such bivalent marks in the embryonic and adult stem cells. When repetitive insults to cells, such as inflammation, recruit the DNA methylation machinery into the CpG islands of these vulnerable genes, this can initiate abnormal methylation for some of these genes (far right panel). The NuRD complex, a key platform for HDAC1 and -2, may be a part of the complex that maintains silencing of the methylated CpG islands containing genes. Various components of the epigenetic machinery that mediate gene silencing are being targeted already (red ovals), and others are potential targets (green ovals) for cancer therapy.
Many regulatory proteins involved in abnormal DNA methylation in cancer epigenomes, including DNMTs, HDACs, MBDs, and PcG proteins are targetable and actively being pursued for roles in cancer therapy (Figure 2). Any approach to reversing abnormal DNA methylation changes in cancer needs to consider the complexities of epigenetic regulation, and targeted therapies may best be used in combination.
Consequences of targeting the cancer epigenome: stratifying passenger versus driver events.
Specifically targeting the core processes that regulate epigenetic changes in cancer, predictably, may have complicated molecular and phenotypic consequences. We hypothesize that the therapeutic efficacy will depend on the epigenetic events that drive the tumor phenotype. The overall core characteristic of the cancer epigenome, especially with regard to abnormal DNA methylation changes, appears to reflect a breakdown of boundaries that separate transcriptionally active DNA regions from surrounding regions (refs. 2, 14, 15, 67, and Figure 2). Deep sequencing efforts for DNA methylation in cancer support this hypothesis and show that certain genomic regions, spanning megabase distances on multiple chromosomes, may particularly harbor abnormalities (15, 69). Within these regions, the most prominent change in cancers, which may increase similar mosaic patterns in normal tissues, are large areas of losses of DNA methylation, termed “partially methylated domains” (PMDs) (15, 69). Intriguingly, embedded within these PMDs are very focal, cancer-specific gains of DNA methylation that principally involve the hypermethylated promoter CpG islands (15, 69). Such findings suggest a more variable role for DNA methylation losses and more of a potential driver change for focal DNA methylation gains. A recent study comparing deep sequencing analyses of DNA methylation patterns in metastases versus primary tumor pairs from patients with prostate cancer highly supports this proposal (70). The losses of DNA methylation were not clonally maintained between the two tumor sites and seemed universally spread over all chromosomes. In contrast, the focal gains were clonally maintained, largely involved the promoter CpG islands, and often correlated with decreases in gene expression (70). Although hypomethylation alterations were more numerous than hypermethylation changes, these hypomethylation changes were not enriched for promoter regions.
Differing patterns of DNA methylation abnormalities are emerging from deep sequencing analyses of multiple tumor types (42–45). Tumors can dramatically differ in the numbers of focal, promoter CpG island gains in DNA methylation, as is evident in colorectal tumors (42, 71). These differences likely reflect epigenetic abnormalities imposed on the normal epigenetic control of the cell compartments that give rise to tumor subtypes. This paradigm is evident for breast cancer, in which focal DNA methylation gains are more characteristic of more differentiated, luminal subtypes than tumors with basal or triple-negative characteristics (41, 72, 73); the latter appear to be driven by an epigenome profile dominated by PcG occupancy–mediated gene repression (73). This stratification is important in future consideration of epigenetic therapies because the targeting of PcG activity, and particularly the enzyme enhancer of zeste homolog 2 (EZH2), which catalyzes the prototype PcG repression mark of H3K27 methylation, is an emerging therapy (74–76). Would stressing the latter approach for the triple-negative tumors versus targeting DNA methylation gains in the luminal subtype be the most constructive consideration, or would variable combinations of such therapies be optimal? Indeed, utilizing tumor epigenetic patterns to guide approaches to treatment will be critical for further development of epigenetic therapies in breast cancer and other solid tumors.
Epigenetic therapy as a “reprogramming” approach.
The consequences for even very specific biochemical targeting of each writer, eraser, reader, and mover will likely be diverse. These proteins each contribute and work together to establish and maintain broad patterns of gene expression (2, 3, 77, 78). Furthermore, each protein often has complex biochemical functions. For example, many of the writers and erasers have both enzyme functions to catalyze specific DNA or histone modifications, often even affecting non-nuclear substrates, and each may have scaffolding functions for interacting with companion proteins that determines functions, recruitment, and stability of the complexes (ref. 3 and Figure 1). This complexity is a challenge in defining which protein functions should be targeted. A key example is the use of the oldest drugs, DNA-demethylating agents, which have been used in the clinic for potential epigenetic therapy strategies. These drugs must incorporate into DNA as a modified cytosine where they covalently bind all three biologically active DNMTs to inhibit their catalytic activity for DNA methylation (2). However, the drugs also trigger degradation of DNMTs (79), each of which experimentally can trigger transcriptional repression even with the catalytic sites deleted or mutated (58–61, 80). It is almost always overlooked that these latter properties could well be a significant component of the therapy efficacy for specifically targeting these enzymes for cancer therapy.
These points, while challenging, may actually be the strength of epigenetic therapy if we view it as a means to broadly reprogram the cancer epigenome (Figure 3). The reality of accomplishing this, as has been recently stressed (81), is that a similar comprehensive reprogramming occurs during creation of induced pluripotent stem cells from more mature cells (82). Similarly, for each manifestation of epigenetic abnormalities in cancer, virtually all oncogenic signaling pathways may be affected and subject to reversion for antitumor effects (1, 5, 83). For example, just the abnormality of de novo, focal gains in gene promoter hypermethylation simultaneously involve genes in each of these signaling pathways (1, 5, 11). Furthermore, this concept has implications for other types of targeted therapies, especially those pointed at mutated genes. These often produce robust initial patient responses, but resistance fairly rapidly emerges (84, 85). While in some cases, new mutations in the targeted protein may be the mechanism of drug resistance, compensation by other parts of the pathway or by other pathways potentially mediated by epigenetic mechanisms may account for the drug resistance.
Figure 3. Gene expression changes in multiple pathways associated with epigenetic therapy.

Treatment with well-tolerated, low doses of epigenetic therapy over multiple cycles initiates widespread changes in gene expression in multiple pathways, including cell cycle, cell adhesion, and apoptosis, that may induce tumor regression or sensitize to chemotherapy. These concepts are currently in clinical trials. Activation of the immune responses such as antigen presentation is also a promising strategy for synergy with immunotherapy.
The current status of epigenetic therapy in solid tumors
Lessons from hematopoietic tumors and current studies in solid tumors.
Experience to date with epigenetic therapy in cancer is largely restricted to DNA-demethylating drugs, such as decitabine and 5-azacitidine, and HDAC inhibitors, such as romidepsin and vorinostat, in hematopoietic neoplasms. 5-Azacitidine was approved by the FDA in 2004, and decitabine in 2006, for the leukemia predisposition disorder myelodysplasia (MDS), and these drugs yield good activity in acute myeloid leukemia (AML) (1). Romidepsin has received FDA approval for cutaneous T cell lymphomas and for relapsed peripheral T cell lymphoma (1, 86–88).
Responses to epigenetic therapy in solid tumors in early trials, however, were essentially not seen in older studies. The demethylating drugs were initially developed as chemotherapeutic agents and were used at cytotoxic doses producing significant toxicity (1, 89). Although some responses were seen in isolated cases, it is difficult to reach any conclusions from these data due to the significantly cytotoxic doses that were used.
From the efficacies seen in hematopoietic cancers, especially for the DNA-demethylating drugs, we have proposed two broad principles that underlie targeted epigenetic therapies (5). First, we hypothesized that these agents were initially used in clinical trials at excessive doses, which produced off-target effects and severe toxicities that precluded true efficacy assessment. In the 1990s, doses of 5-azacitidine and decitabine were markedly lowered, permitting prolonged exposure times and exposing patient tumor cells to nM drug levels; this dose reduction was critical to forging the path to FDA approval for MDS (1, 90–94). Indeed, nearly half of patients with high-risk MDS treated with 5-azacitidine therapy showed improved response rates, with increasing responsiveness reported following prolonged treatment (95). Second, patients with MDS/AML generally take months to respond, suggesting, initially, a noncytotoxic mechanism underlying efficacy and raising the possibility that exhaustion of tumor stem-like cell renewal might be involved (5).
In a preclinical model, the use of in vitro, transient exposure of solid tumor cancer cells to nM doses of either 5-azacitidine or decitabine, which cause minimal initial apoptosis, DNA damage, or cell cycle arrest, supported the above hypotheses, including distinct inhibition of tumorigenic stem-like cells (5). Subsequent drug withdrawal produced modest but long-lasting, DNA demethylation and prolonged increases in expression of some genes with promoter DNA hypermethylation. Further, antitumor effects were observed in ex vivo self-renewal assays and for explants in untreated, immunosuppressed mice (5). Genomic analyses suggested an extended cellular reprogramming effect for multiple, key tumorigenic pathways, including decreased cell cycle activity, late increases in apoptosis, increased cellular differentiation, reversal of immune evasion signatures, and changes in cell adhesion properties (ref. 5 and Figure 3).
In the multi-institutional Stand Up To Cancer (SU2C) project, low-dose 5-azacitidne therapy was administered to patients with solid tumors, focusing initially on the biggest killer, advanced, heavily pretreated, non–small-cell lung cancer (NSCLC) (6). We paired this drug with the class I and II HDAC inhibitor entinostat (96), based on lab paradigms that such combinations are additive for facilitating reexpression of DNA-hypermethylated genes by low doses of DNA-demethylating agents (54). This combination is being tested in multiple clinical trials, although the contribution to clinical efficacy for combining the drugs remains to be confirmed. In a phase II trial (6), the initial results were distinctly promising by several parameters. First, the regimen was well tolerated (ref. 6 and Figure 3). Second, early indicators of clinical efficacy have been seen in select patients that are remarkable for their multi-year durability; 3% of patients responded to the above drugs alone, with survival times of 3 to 4 years, while another 25% responded to subsequent chemotherapies for years (6). The possibility of using such therapies to sensitize to subsequent chemotherapies is being tested in other clinical settings, such as use of decitabine to sensitize patients with advanced ovarian cancer to cisplatin (97–100). For all these studies, patient numbers are small, but results have spurred subsequent larger trials, which are now ongoing. It cannot be overemphasized that, based on the above early results in solid tumors, we may be learning a critical concept: while typical clinical criteria such as tumor shrinkage is not unimportant, it may not be the most robust arbiter of efficacy for epigenetic therapies. Long-term consequences from epigenetic therapies, such as significantly improving survival or improving the “tail“ on the survival curve, may be critical to efficacy and/or sensitization to subsequent therapies (Figure 3). Trial designs that do not take these parameters into account may miss critical benefits, such as those observed for lung cancer. The challenge is also to personalize patient responses, to match efficacy with preclinical models and genomics approaches, and to derive biomarker strategies that can predict and/or monitor the long-term consequences of therapy.
The future for solid tumors.
We believe the clinical results to date indicate that targeting DNMTs will play an important role in the epigenetic therapy of solid tumors. Similar trials as those in NSCLC are ongoing in advanced breast and colorectal cancers, and a larger trial to sensitize patients to cisplatin is underway; however, many challenges remain. The current DNA-demethylating agents will likely stay in use, but maximizing their potential may require additional modifications. First, there are pharmacologic considerations. The current DNA-demethylating agents have a short half-life, and the means to extend tumor exposure but maintain low-dose scenarios are needed. In this regard, SGI-110, a novel compound with a pro-drug mechanism that releases decitabine as the final product (101, 102), has shown promising activity in early clinical trials in myeloid malignancies. An additional consideration is the route of delivery of the demethylating drugs, which currently is subcutaneous and intravenous. An oral azacitidine now being tested in solid tumors has shown clinical activity in MDS and chronic myelomonocytic leukemia, and the chronic exposure with this formulation may improve the bioavailability of the drug along with patient compliance (103, 104).
Targeting DNMTs is also an exciting area for future development, given that these proteins interact with regulatory factors controlling the epigenome. Such a strategy may obviate a current problem suggested by our preclinical model. While the low doses of DNMT inhibitors may induce specific targeting of these proteins and cellular reprogramming, the results for demethylation and gene expression consequences are modest (5). Our preclinical model is not a direct therapeutic one, and it is unclear how the transient doses used relate to the precise drug exposure levels seen by tumor cells in the patient therapies currently employed. Moreover, the clinical trials, in general, used combination epigenetic therapy of DNMT inhibitors and HDAC inhibitors. The resulting DNA demethylation and gene expression may be higher in the clinical setting, but preclinical data suggest that current drugs will need additional improvements to maximize efficacy. Raising the doses will likely result in toxicities and off-target effects.
Multiple sound, biochemical hypotheses are relevant to meeting these challenges. Each interacting step that initiates and/or maintains epigenetic regulation is potentially targetable to reverse focal gains in DNA methylation (Figure 2). HDAC inhibitors are currently being added to DNA demethylases to increase lysine deacetylation, although this combination may or may not increase efficacy (54). Perhaps blocking the mechanisms by which HDACs collaborate with DNA methylation in gene reexpression would be better, and targeting MBDs, key scaffolding proteins that recruit and/or maintain HDACs at sites of DNA hypermethylation, can be investigated. Furthermore, the NuRD complex constituents, perhaps again through HDAC involvement, are an attractive target for facilitating gene reexpression activity of low-dose DNA-demethylating agents (64).
Additionally, a simultaneous increase in the active histone modifications histone H3K4 dimethylation (H3K4me2) and trimethylation (H3K4me3) also merits strong consideration. Full repression and absence of proper positioning of these active modifications around proximal gene promoters is a universal finding for de novo, DNA-hypermethylated genes in cancer (41, 105). When DNA-demethylating agents lead to reexpression of the genes, local levels of these active modifications always increase, and their positions surrounding start sites of active genes is assumed (106). Furthermore, high levels of these modifications surrounding gene promoters antagonize DNA methylation (107–109). Several erasers of the H3K4me2 and -3 marks are attractive candidates to target to increase these active marks, especially as these erasers are reported to have oncogenic roles (31, 32, 110, 111). For example, lysine-specific demethylase 1(LSD1), which can erase the H3K4me2 mark and Jumonji, AT-rich interactive domain (JARID proteins) such as lysine demethylase 5a (KDM5a) and KDM5b, which erase the H3K4me3 mark, are implicated in oncogenesis (32, 110–112). The proteins interact with DNMTs, and inhibition of LSD1 has been associated with reexpressing DNA-hypermethylated genes (32). Furthermore, decreasing JARID proteins has been reported to decrease self-renewal of stem-like cells in melanomas and to reverse epigenetically mediated drug resistance associated with tumor cells (110, 111). Drug development for inhibiting these erasers is underway, and consideration of combinatorial approaches with DNA-demethylating agents is warranted. Likewise, increased levels of the repressive mark H3K9 methylation are well associated with recruitment and maintenance of DNA methylation from model organisms to humans (113, 114). Inhibition of the writers of this mark, such as histone methylases like G9a, which associates with DNMTs (114, 115), could be useful. Inhibitor compounds for these proteins are under development.
Targeting EZH2, the PcG component that is the writer for the repressive H3K27me3 mark, also bears consideration for complicated reasons. In cancer, a special relationship exists between the vulnerability of genes to DNA hypermethylation and their regulation by PcG control without promoter DNA methylation in stem and progenitor cells (41, 49, 50, 68). EZH2 is among the PcG proteins that interact with DNMTs in complexes that may attract DNA methylation (56, 67), but once methylation is established, these proteins may actually be reduced or repelled (108). Also, the balance in tumor subtypes between whether abnormal gene suppression may be more dominant for DNA methylation versus PcG control (41, 72, 73) may need to be considered. Nevertheless, potent inhibitors for EZH2 are near clinical trials for hematopoietic neoplasms wherein gain-of-function EZH2 mutations has been recognized (74, 75). How to employ these compounds in solid tumors, and whether combinatorial approaches with DNA-demethylating agents might be efficacious, again merits testing.
Finally, recurrent somatic mutations in epigenetic regulators are increasingly being identified, and how these will drive response to epigenetic therapy is as yet unclear. Mutations in TET2 and isocitrate dehydrogenases IDH1 and IDH2 are seen in gliomas and leukemia (116–122), and IDH1 mutations in gliomas correlate with a DNA CpG island methylator phenotype (116, 123). Preliminary encouraging data suggest that patients with mutations in these epigenetic regulators may particularly benefit from demethylating therapy, with preclinical studies showing decreased tumor growth and induction of tumor differentiation (124, 125). More recently, mutations in the SWI/SNF chromatin remodeling complex, especially in the AT-rich interactive domain 1A (ARID1A), have also been identified in a multitude of solid cancers including pancreas, breast, ovarian, gastric, and colon (42, 126–128), although the relationship between ARID1A mutations and downstream cancer epigenome is still being elucidated.
Moreover, the translation of epigenetic therapy into the clinical arena will be nuanced, requiring personalized approaches along with an understanding of the tumor subtypes and its epigenomic landscape. It is as yet unclear which subsets of patients will benefit from primary epigenetic therapy and whether the mechanisms of response are solely related to reversal of gene silencing or to multiple pathways. Moreover, as epigenetic therapy is being tested to prime for chemotherapy response in clinical trials, one could also conceivably promote drug resistance. As an example, MGMT methylation has been shown to predict response to alkylating agents, and use of demethylating drugs for priming in this subpopulation may in fact promote drug resistance (129).
Conclusions
The challenges are enormous for moving forward in terms of drug development, pharmacokinetic considerations, dosing schedules, and combinatorial concepts. Preclinical models and genomics that take the special effects of epigenetic therapy into account must be used in developing new strategies. How one gauges the phenotypic consequences and readouts for even the most creative approaches to epigenetic therapy may be the most important arbiter of the extent to which epigenetic therapies are pursued or how rapidly such therapies gain traction in the clinic. Failure or misinterpretation of results in these preclinical stages could doom many excellent concepts to premature rejection or encourage vigorous pursuit of concepts that will not work. This caveat is important to clinical trial design; the strength of epigenetic therapies may yield long-term improvements in patient survival and cancer management without producing typical acute clinical responses. We postulate that as these considerations become more refined and judiciously employed, epigenetic therapy may have a bright future in new therapies that will dramatically change the management of cancers with some of the highest morbidity and mortality statistics. As we learn to do this, given that epigenetic abnormalities are apparent early in cancer risk and premalignant states, we may someday derive strategies for the ultimate cancer management goal: its prevention.
Acknowledgments
We wish to thank Kathy Bender and Joann Murphy for their assistance with manuscript preparation. Parts of the authors’ work cited have been supported by various funding agencies. S.B. Baylin was supported by grants from the Stand Up To Cancer (SU2C) Epigenetic Dream Team, the NCI (CA043318, NIEHS ES011858, and NCI CA058184), and the Hodson Foundation. N. Ahuja was supported by grants from the NCI (CA127141), the American College of Surgeons/Society of University Surgeons, the Society of Surgical Oncology, the Stand Up To Cancer (SU2C) Epigenetic Dream Team, and The Jeannik M. Littlefield AACR Grant In Metastatic Colon Cancer.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(1):56–63. doi:10.1172/JCI69736.
References
- 1.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours-lessons from the past. Nat Rev Clin Oncol. 2013;10(5):256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22(1):50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Tsai HC, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epigenetics. Allis C, Jenuwein T, Reinberg D, eds. Cold Spring Harbor, New York, USA: Cold Spring Harbor Laboratory Press; 2006. [Google Scholar]
- 9.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 10.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 11.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7(1):21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 13.Reddy KL, Feinberg AP. Higher order chromatin organization in cancer. Semin Cancer Biol. 2013;23(2):109–115. doi: 10.1016/j.semcancer.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol Cell. 2009;34(3):271–284. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berman BP, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44(1):40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Biran A, Meshorer E. Concise review: chromatin and genome organization in reprogramming. Stem Cells. 2012;30(9):1793–1799. doi: 10.1002/stem.1169. [DOI] [PubMed] [Google Scholar]
- 18.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20(3):267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiwari VK, Baylin SB. Breaching the boundaries that safeguard against repression. Mol Cell. 2009;34(4):395–397. doi: 10.1016/j.molcel.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 21.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 22.Neph S, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489(7414):83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49(5):825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ecker JR, Bickmore WA, Barroso I, Pritchard JK, Gilad Y, Segal E. Genomics: ENCODE explained. Nature. 2012;489(7414):52–55. doi: 10.1038/489052a. [DOI] [PubMed] [Google Scholar]
- 25.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herman JG, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esteller M, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 28.Kolodner RD, et al. Structure of the human MLH1 locus and analysis of a large hereditary nonpolyposis colorectal carcinoma kindred for mlh1 mutations. Cancer Res. 1995;55(2):242–248. [PubMed] [Google Scholar]
- 29.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis PW, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A. 2007;104(19):8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 34.Simon JA, Kingston RE. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell. 2013;49(5):808–824. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ting DT, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331(6017):593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouzinba-Segard H, Guais A, Francastel C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc Natl Acad Sci U S A. 2006;103(23):8709–8714. doi: 10.1073/pnas.0508006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaudet F, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300(5618):489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 39.Smith IM, et al. Coordinated activation of candidate proto-oncogenes and cancer testes antigens via promoter demethylation in head and neck cancer and lung cancer. PLoS One. 2009;4(3):e4961. doi: 10.1371/journal.pone.0004961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuebel KE, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3(9):1709–1723. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Easwaran H, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012;22(5):837–849. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammerman PS, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito Y, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9(6):435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 47.Lujambio A, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67(4):1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 48.De Carvalho DD, et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell. 2012;21(5):655–667. doi: 10.1016/j.ccr.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohm JE, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39(2):237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Widschwendter M, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39(2):157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 51.Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 52.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23(1):62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 53.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11(8):588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21(1):103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 55.Pruitt K, et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2(3):344–352. doi: 10.1371/journal.pgen.0020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4(8):e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling Y, Sankpal UT, Robertson AK, McNally JG, Karpova T, Robertson KD. Modification of de novo DNA methyltransferase 3a (Dnmt3a) by SUMO-1 modulates its interaction with histone deacetylases (HDACs) and its capacity to repress transcription. Nucleic Acids Res. 2004;32(2):598–610. doi: 10.1093/nar/gkh195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25(3):338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 59.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25(3):269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 60.Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem. 2001;276(34):32282–32287. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
- 61.Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24(1):88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- 62.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278(6):4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 63.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cai Y, et al. Oncogene. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes [published online ahead of print May 27, 2013]. doi: 10.1038/onc.2013.178 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.You JS, Kelly TK, De Carvalho DD, Taberlay PC, Liang G, Jones PA. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc Natl Acad Sci U S A. 2011;108(35):14497–14502. doi: 10.1073/pnas.1111309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuzmichev A, et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci U S A. 2005;102(6):1859–1864. doi: 10.1073/pnas.0409875102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Hagan HM, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20(5):606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 69.Hansen KD, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aryee MJ, et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med. 2013;5(169):169ra110. doi: 10.1126/scitranslmed.3005211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weisenberger DJ, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 72.Fang F, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3(75):75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holm K, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12(3):R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCabe MT, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 75.Knutson SK, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 76.Knutson SK, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chi AS, Bernstein BE. Developmental biology. Pluripotent chromatin state. Science. 2009;323(5911):220–221. doi: 10.1126/science.1166261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cui K, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4(1):80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghoshal K, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25(11):4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Clements EG, et al. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012;40(10):4334–4346. doi: 10.1093/nar/gks031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shen H, Laird PW. In epigenetic therapy, less is more. Cell Stem Cell. 2012;10(4):353–354. doi: 10.1016/j.stem.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 82.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nat Biotechnol. 2010;28(10):1033–1038. doi: 10.1038/nbt1010-1033. [DOI] [PubMed] [Google Scholar]
- 84.Poulikakos PI, Rosen N. Mutant BRAF melanomas — dependence and resistance. Cancer Cell. 2011;19(1):11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 85.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 86.Whittaker SJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 87.Piekarz RL, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coiffier B, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631–636. doi: 10.1200/JCO.2011.37.4223. [DOI] [PubMed] [Google Scholar]
- 89.Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. 2010;2(1):71–86. doi: 10.2217/epi.09.44. [DOI] [PubMed] [Google Scholar]
- 90.Silverman LR, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20(10):2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 91.Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10(3):176–182. doi: 10.1634/theoncologist.10-3-176. [DOI] [PubMed] [Google Scholar]
- 92.Kantarjian H, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 93.Kantarjian H, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 94.Issa JP. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: dose, duration, and patient selection. Nat Clin Pract Oncol. 2005;2(suppl 1):S24–S29. doi: 10.1038/ncponc0355. [DOI] [PubMed] [Google Scholar]
- 95.Silverman LR, et al. Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer. 2011;117(12):2697–2702. doi: 10.1002/cncr.25774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saito A, et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A. 1999;96(8):4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fu S, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer. 2011;117(8):1661–1669. doi: 10.1002/cncr.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matei D, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9):2197–2205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matei DE, Nephew KP. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol Oncol. 2010;116(2):195–201. doi: 10.1016/j.ygyno.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Balch C, Fang F, Matei DE, Huang TH, Nephew KP. Minireview: epigenetic changes in ovarian cancer. Endocrinology. 2009;150(9):4003–4011. doi: 10.1210/en.2009-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yoo CB, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67(13):6400–6408. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- 102.Chuang JC, et al. S110, a 5-Aza-2′-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther. 2010;9(5):1443–1450. doi: 10.1158/1535-7163.MCT-09-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Garcia-Manero G, et al. A pilot pharmacokinetic study of oral azacitidine. Leukemia. . 2009;22(9):1680–1684. doi: 10.1038/leu.2008.145. [DOI] [PubMed] [Google Scholar]
- 104.Garcia-Manero G, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29(18):2521–2527. doi: 10.1200/JCO.2010.34.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Van Neste L, et al. Defining a chromatin pattern that characterizes DNA hypermethylated genes in colon cancer cells. Cancer Res. 2008;68(14):5753–5759. doi: 10.1158/0008-5472.CAN-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McGarvey KM, et al. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008;68(14):5753–5759. doi: 10.1158/0008-5472.CAN-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hagarman JA, Motley MP, Kristjansdottir K, Soloway PD. Coordinate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS One. 2013;8(1):e53880. doi: 10.1371/journal.pone.0053880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143(3):470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lindroth AM, et al. Antagonism between DNA and H3K27 methylation at the imprinted Rasgrf1 locus. PLoS Genet. 2008;4(8):e1000145. doi: 10.1371/journal.pgen.1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sharma SV, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roesch A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li Q, et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011;71(21):6899–6908. doi: 10.1158/0008-5472.CAN-11-1523. [DOI] [PubMed] [Google Scholar]
- 113.Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;414(6861):277–283. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- 114.Fahrner JA, Eguchi S, Herman JG, Baylin SB. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002;62(24):7213–7218. [PubMed] [Google Scholar]
- 115.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome — components and functional correlates. Genes Dev. 2006;20(23):3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 116.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013;121(18):3563–3572. doi: 10.1182/blood-2013-01-451781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 119.Fathi AT, Abdel-Wahab O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv Hematol. 2012;2012:469592. doi: 10.1155/2012/469592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Turcan S, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Turcan S, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013;4(10):1729–1736. doi: 10.18632/oncotarget.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Borodovsky A, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4(10):1737–1747. doi: 10.18632/oncotarget.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3(1):35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hegi ME, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]