

Abstract
Drugs targeting the epigenome are new promising cancer treatment modalities; however, not all patients receive the same benefit from these drugs. In contrast to conventional chemotherapy, responses may take several months after the initiation of treatment to occur. Accordingly, identification of good pretreatment predictors of response is of great value. Many clinical parameters and molecular targets have been tested in preclinical and clinical studies with varying results, leaving room for optimization. Here we provide an overview of markers that may predict the efficacy of FDA- and EMA-approved epigenetic drugs.
Introduction
Traditionally, cancer patients have been offered the type of chemotherapy that has shown efficacy in the largest proportion of individuals suffering from that particular type of cancer. However, given the rapidly increasing therapeutic options, we are beginning to envision a paradigm shift in cancer treatment. Today, an increasing number of cancer patients are tested for one or more biomarkers to determine the optimal treatment strategies for the individual patient (1). Still, despite successful incorporation of numerous biomarkers in clinical practice, there is a constant pursuit to identify better markers to predict response to existing and upcoming drugs.
Drugs that target the epigenome are promising novel treatment modalities, but not all patients achieve the same benefit from epigenetic therapy and responses are often not evident until after several months of treatment. Identification of good predictive biomarkers for epigenetic therapy would be of great value because patients with minimal chances of response could be spared long-term treatment with an inefficient drug with unpleasant side effects, and could be offered alternative treatment strategies.
This Review focuses on predictors of response to the two classes of epigenetic drugs currently approved by the European Medicines Agency (EMA) and/or the US FDA for cancer treatment: DNA methyltransferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis). These drugs may be used individually, in combination with each other, or even in combination with conventional chemotherapy.
What characterizes a good biomarker?
A biomarker is generally defined as a substance that can be measured objectively and is an indicator of either a clinically important aspect of a pathogenic process or of a pharmacologic response to a therapeutic intervention. In cancer, most biomarker assays are based on the detection of aberrantly expressed proteins, mRNAs, microRNAs (miRs), or genetic or epigenetic alterations that are specific to the cancer cells. Irrespective of its nature, a biomarker should have high diagnostic sensitivity and specificity as well as a high positive predictive value (PPV) and negative predictive value (NPV) (Table 1). PPV and NPV are highly dependent on the prevalence of the disease. Therefore, PPV and NPV can only be estimated from cross-sectional studies. Conversely, the diagnostic sensitivity and specificity are intrinsic to the test and may therefore also be derived from case-control studies.
Table 1.
Biomarker definitions

It is important to realize that the performance of a biomarker is only as good as the assay employed for its measurement and will be compromised if the assay does not have a sufficiently high analytical sensitivity and specificity. Also, if the substance is found at low levels in unaffected or non-responding individuals, the biomarker assay should preferably be quantitative to define a cut-off that provides optimal diagnostic sensitivity and specificity.
Apart from diagnostic sensitivity and specificity, it is important that the biomarker can be detected in readily accessible tissues or body fluids in order to save the patients from a potentially harmful invasive procedure. Finally, the biomarker assay should be based on a methodology that is user friendly and cost efficient (2).
When conducting and reporting biomarker studies, it is important to realize that several aspects of study design, selection of biomarker assay, and statistical analyses may affect the overall outcome of the study. Specific guidelines have been developed that may be helpful when designing, conducting, and reporting biomarker studies (3). In particular, it is recommended that predictive biomarker studies generally should be conducted within randomized trials and that assays should be used at a more advanced state of development (4).
Predicting response to DNMTis
Recent multicenter studies demonstrated that DNMTis have significant efficacy in the treatment of hematological malignancies (5–9) and have led to the approval of two DNMTis, azacytidine and decitabine, by the FDA and EMA. However, the FDA and EMA have not approved the drugs for similar indications (Table 2). Still, only about 50% of patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) achieve a clinical response to treatment with DNMTis (10, 11). The value of DNMTis in patients that obtain stable disease is still unclear; however, a survival benefit can be observed in patients that obtain hematological improvement. Accordingly, conventional complete remission (CR) and complete remission with incomplete blood count recovery (CRi), as measured by standard parameters (bone marrow blast and peripheral blood cell counts), are not necessarily good markers for predicting outcome (12–14).
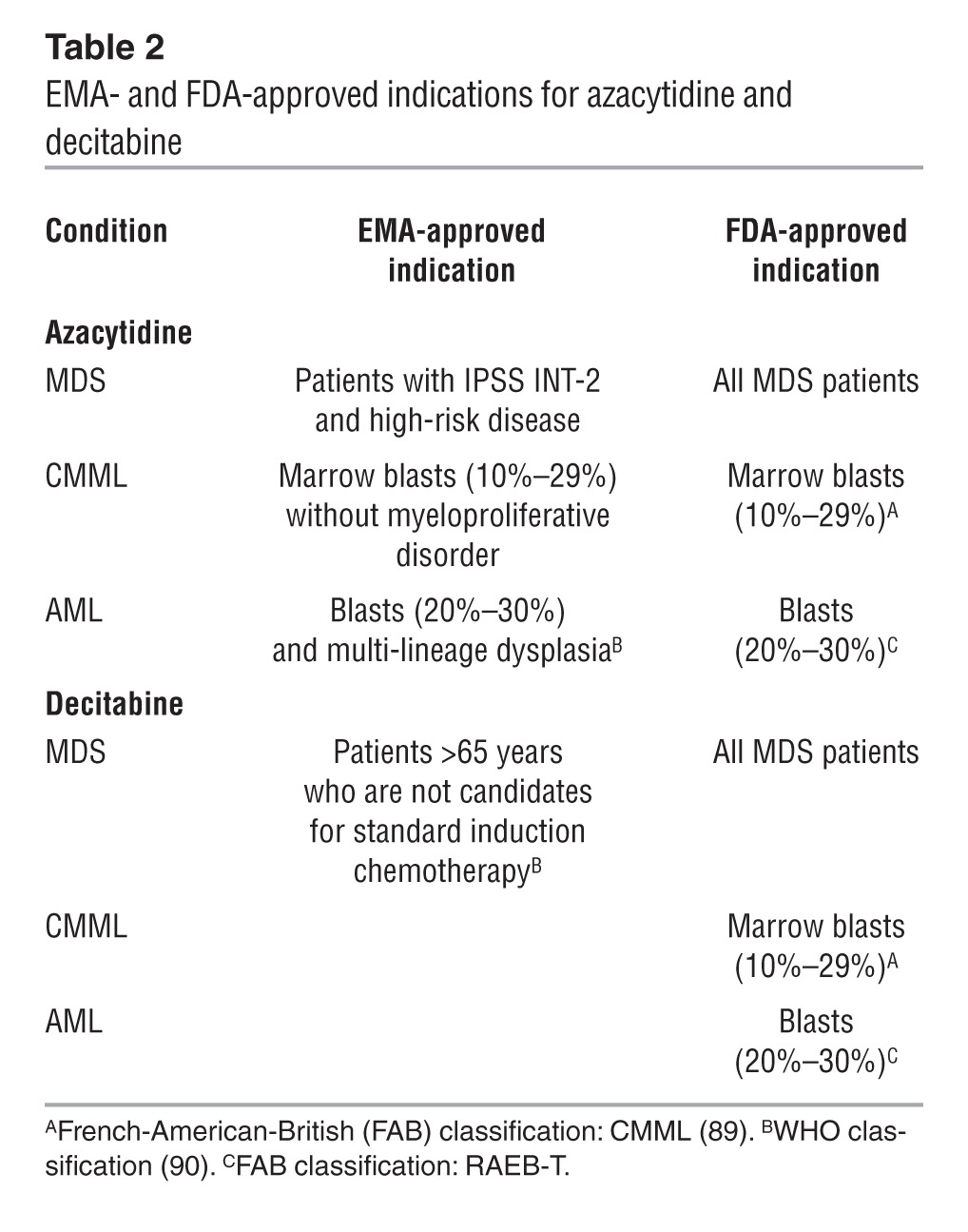
Table 2.
EMA- and FDA-approved indications for azacytidine and decitabine
The varying efficacy of the drugs may relate to different mechanisms of action in individual patients. In vitro studies indicate that DNMTis can reprogram somatic cells by DNA demethylation of aberrantly silenced genes (Figure 1 and ref. 15). However, the exact mechanisms of action of DNMTis in patients are currently unknown; reactivation of epigenetically silenced tumor suppressor genes and genes involved in normal differentiation has been suggested (16), but the data are contradictory (17). We and others have shown that DNMTis can render the malignant cells immunogenic by induction of cancer testis antigens (18–20), suggesting that immune stimulation may be an important contributor to the clinical effect of these agents. Azacytidine, which is mainly incorporated into RNA (80%–90%), may inhibit ribonucleotide reductase, leading to a reduced deoxyribonucleotide pool and impaired DNA synthesis and repair (ref. 21 and Figure 2). Second-generation DNMTis are designed to improve the pharmacological profile. One of these, SGI-110 is currently in phase II clinical trials for the treatment for MDS and AML (22), ovarian cancer (23), and advanced hepatocellular carcinoma (24), while others are mainly being investigated in preclinical settings (25, 26).
Figure 1. Mechanism of action DNMTis.
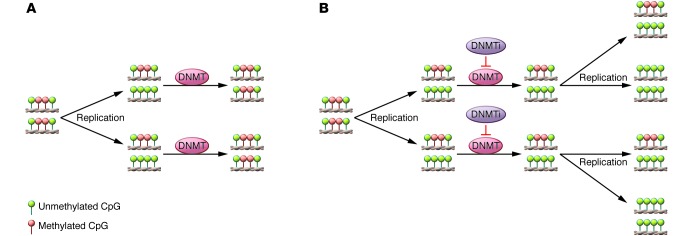
(A) Under normal circumstances, the DNMTs copy the methylation pattern of the parental DNA strand after replication, ensuring that methylation patterns are maintained during cell division. (B) During treatment, DNMTis are incorporated into DNA and RNA, where they covalently bind and thus inactivate DNMTs. After successive cell divisions, the original DNA methylation pattern is lost.
Figure 2. Cellular pathways affected by DNMTis and HDACis.
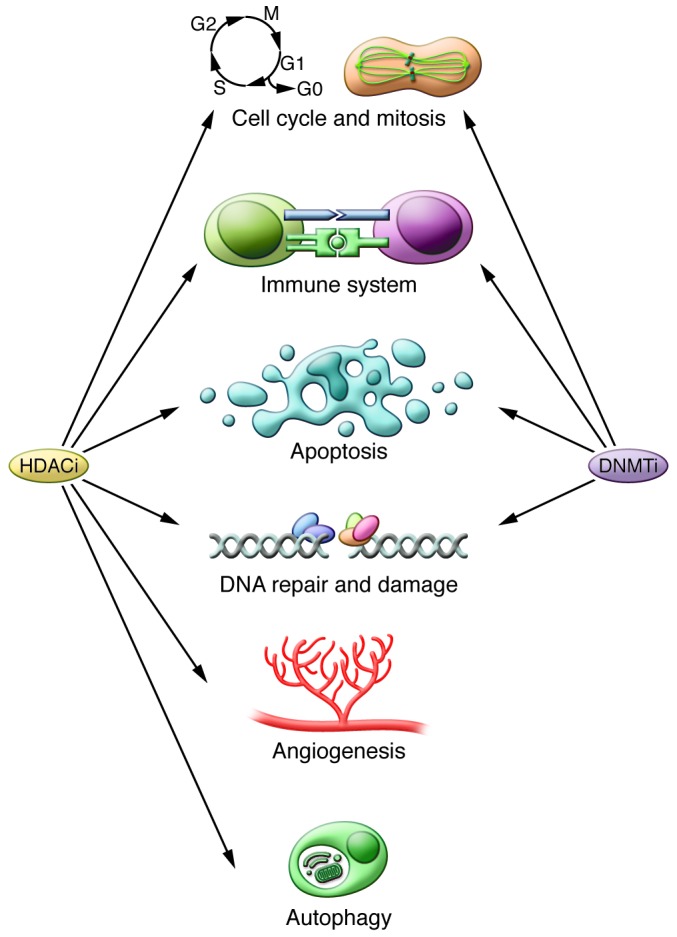
DNA methylation and acetylation of histone and proteins play important roles in multiple cellular pathways, which may be affected by DNMTis and HDACis. Accordingly, the inhibition of HDACs or DNMTs can lead to miscellaneous responses, each of which may require a different biomarker.
Pharmacologic factors with potential impact on DNMTi resistance
Human nucleoside transporters.
Cellular uptake is crucial for the efficacy of azanucleosides. It has been shown in vitro that azacytidine and decitabine use different human nucleoside transporters (hNTs), and that cytotoxicity is dependent on hNT presence (27, 28). These observations suggest that hNTs may be useful biomarkers for the efficacy of DNMTis, but clinical data are still not available.
Cytidine and deoxycytidine kinase.
The next crucial step in DNMTi processing is the initial mono-phosphorylation of azacytidine and decitabine by cytidine kinase and deoxycytidine kinase (DCK), respectively. Accordingly, disruption of DCK may confer decitabine resistance, as demonstrated by a DCK point mutation in the HL60 cell line (29). DCK mutations are rare in patients (30), but a borderline significant lower expression of DCK was observed in non-responders (31).
Cytidine deaminase.
A recent study reported that the expression level and enzymatic activity of cytidine deaminase (CDA) can influence overall survival in patients treated with DNMTis. CDA inactivates both azacytidine and decitabine by irreversible hydrolytic deamination of cytidine/deoxycytidine to uridine/deoxyuridine, and accordingly, high CDA expression/activity decreases the half-life of the drugs. Males have the highest CDA expression/activity, and among 90 MDS patients treated with DNMTis, female patients had significant better overall survival (32). This finding may suggest that CDA is involved in a gender-specific response, although the observations in this study are indirect. However, conflicting results exist, with another study showing gender-specific differences in overall survival (33), while in others a negative impact of male gender was not observed (6, 8, 34, 35). Given that SGI-110 is designed to overcome the effects of CDA (36), it will be interesting to observe whether male patients do relatively better in the SGI-110 trial.
Combined disruption of DNMTi metabolic enzymes.
Few studies of combined disruption of DNMTi metabolic enzymes have been performed, but in one study in 32 patients with MDS, the CDA/DCK ratio was negatively correlated with clinical response to decitabine (31).
Clinical predictors
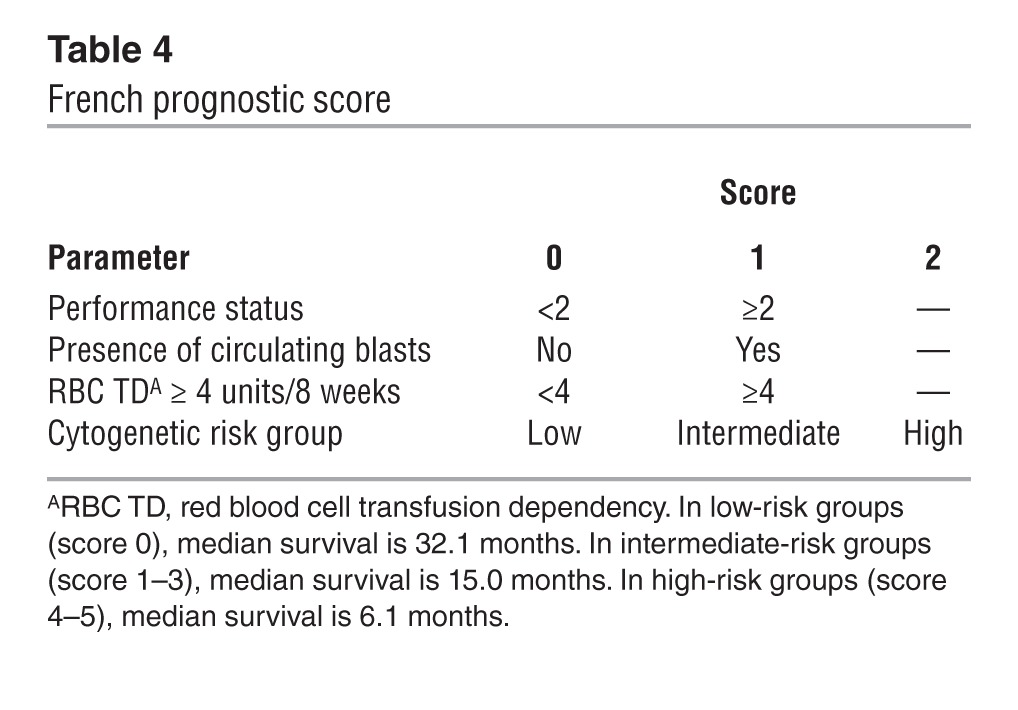
The French prognostic score for MDS patients.
Itzykson et al. evaluated 282 higher-risk MDS patients (International Prognostic Scoring System [IPSS] intermediate-2 [INT-2] and high-risk group; ref. 37) treated with azacytidine, and found that bone marrow blasts >15%, abnormal karyotype, and previous treatment with low-dose cytarabine independently predicted poor response to azacytidine (Table 3). In addition, performance status ≥2, presence of circulating blasts, red blood cell transfusion dependency ≥4 units/8 weeks, and intermediate- or high-risk cytogenetics independently predicted poorer overall survival. Based on these factors, Itzykson et al. developed the French prognostic score for overall survival (Table 4 and refs. 12, 13). This prognostic score was validated in 161 higher-risk MDS patients treated in the AZA001 trial (6), who represented an independent but highly selected patient cohort. The prognostic score has recently been further validated in two independent patient cohorts of 60 (38) and 90 (39) patients, respectively. In addition, this score identified patients who obtained CR; all CRs were observed in the low- or intermediate-risk group (38).
Table 3.
Clinical markers for response to DNMTi
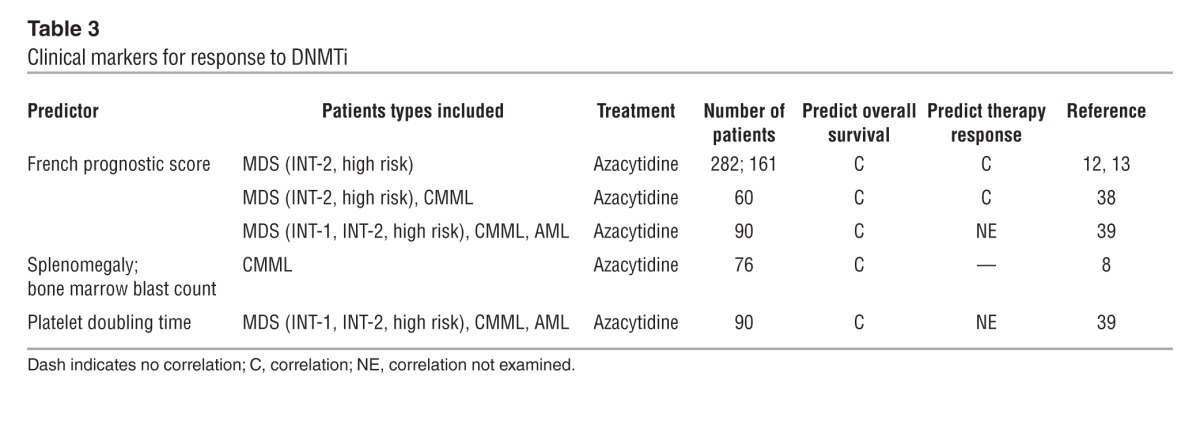
Table 4.
French prognostic score
Clinical predictors in patients with CMML.
The impact of different clinical factors was evaluated in 76 patients with chronic myelomonocytic leukemia (CMML) treated with azacytidine (8). No predictive factors for clinical response were identified, while increased bone marrow blasts, splenomegaly, and high white blood cell counts were associated with significantly shorter survival. However, by multivariate analysis only bone marrow blast count and splenomegaly retained impact on overall survival.
Platelet doubling time.
In a cohort of 90 patients with MDS, CMML, and AML treated with azacytidine, an increase in platelet counts of at least two-fold at the initiation of the second treatment cycle, as compared with the pretreatment values, was associated with significantly better overall survival (39).
Cytogenetic and molecular predictors
Cytogenetic abnormalities.
Poor-risk cytogenetics in MDS and AML has been associated with shorter response duration and shorter overall survival (refs. 12, 38, 39, and Table 5). However, among patients with poor-risk cytogenetics, better clinical response rates and a relatively favorable outcome in patients with deletions or loss of chromosome 7 were observed (6, 7, 12, 35, 40–42). The explanation for this is currently unclear; interestingly, however, chromosome 7 harbors EZH2, which encodes the catalytic component of the polycomb repressive complex 2 histone methyltransferase complex. One study showed that EZH2 may directly recruit DNMTs to promoters (43), which theoretically may lead to global hypomethylation. However, a direct interaction between EZH2 and DNMT has not been consistently substantiated, and at this point no association has been shown between EZH2 mutational status and outcome of azacytidine treatment.
Table 5.
Molecular markers for response to DNMTi
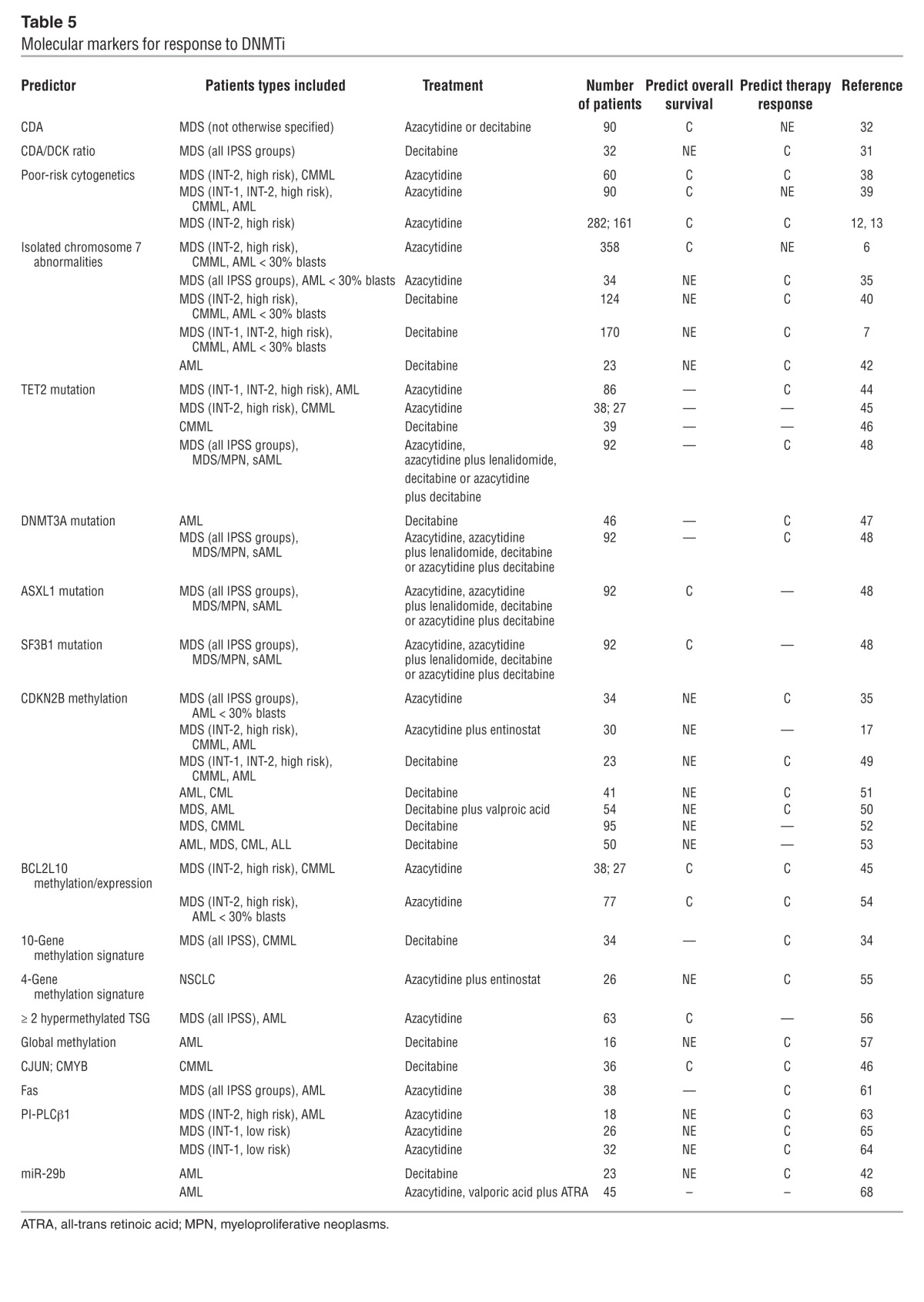
Point mutations.
Mutations in epigenetic regulators are identified in most cancers, and mutations in enzymes that are involved in the regulation of DNA methylation are particularly frequent in hematological malignancies. It seems logical that mutations in these enzymes would influence the response to DNMTis and thus be of prognostic importance, but the results are contradictory. Itzykson et al. observed a correlation between clinical response and mutations in the DNA dioxygenase TET2 in 86 patients with MDS and AML treated with azacytidine (44). Significantly better response rates, but no difference in overall survival, were observed among patients with TET2 mutations. Meanwhile, correlation between TET2 mutational status and clinical response or overall survival was not observed in 38 patients with higher-risk MDS treated with azacytidine and valproic acid (45), or in 39 patients with CMML treated with decitabine (46), respectively.
A positive correlation between mutations in the DNA methyltranferase DNMT3A and clinical response was observed in 46 patients with AML treated with decitabine; this response, however, did not translate into an overall survival benefit (47).
In a recent study, the impact of several point mutations on the response to treatment was examined in 92 MDS, MDS/MPN, and secondary AML (sAML) patients treated with either azacytidine, azacytidine plus lenalidomide, decitabine, or decitabine plus azacytidine (48). TET2 and/or DNMT3A mutations were associated with a better overall response rate and progression-free survival, but not overall survival. Mutations of the putative polycomb associated protein ASXL1 were correlated with poor overall survival, while mutations of the splice factor 3B (SF3B1) were associated with better overall survival. However, these data need confirmation because this patient cohort was heterogeneous with regard to both diagnosis and choice of treatment modalities, and only about 50% of the examined samples were collected before the initiation of DNMTi treatment.
DNA methylation
Several groups investigated whether responses to DNMTis are predicted by pretreatment methylation levels at individual gene promoters, at combinations of genes, or by global screening.
CDKN2B.
The relationship between clinical response to DNMTi and methylation status of the tumor suppressor gene CDKN2B, which encodes the cell cycle inhibitor p15, has been examined in several studies in patients with MDS (17, 35, 49–52). Some reported a positive correlation between low-level pre-treatment CDKN2B methylation and clinical response, while others observed a correlation between CDKN2B demethylation/expression during decitabine treatment and clinical response (16, 49, 52). Yet other groups did not detect any correlation at all (17, 34, 53). The varying results are likely due to variation in patient groups, combinations of epigenetic therapies, and methodologies for monitoring DNA methylation; in particular, not all groups performed quantitative analyses.
BCL2L10.
Methylation of the anti-apoptotic Bcl-2 family member BCL2L10 has been negatively correlated to response to azacytidine and associated with a significantly poorer overall survival in patients with more than 50% BCL2L10 methylation. These results were based on an initial analysis of 38 — and validation in 27 — azacytidine-treated patients with higher-risk MDS (45). By contrast, others showed that patients with azacytidine-resistant MDS/AML have an increased fraction of BCL2L10-positive cells in the bone marrow, and that patients with low BCL2L10 expression had significantly better overall survival (54).
Multiple genes.
In 317 patients with MDS, a methylation signature consisting of 10 hypermethylated genes (CDH1, CDH13, ERα, NOR, NPM2, OLIG2, CDNK2B, PGRA, PDZ, and RIL) was identified among 24 genes previously shown to be methylated in MDS/AML (including several known tumor suppressors). The pretreatment methylation level of these genes was not correlated with clinical response to decitabine, but reduction of methylation after more than four months of treatment (across all 10 genes) was positively correlated to clinical response in a cohort of 34 patients (34).
Promoter methylation of four genes (APC, RASSF1A, CDH13, and CDKN2A) has been shown to correlate negatively to survival in non–small-cell lung cancer (NSCLC). Analysis of methylation status of these genes in plasma samples from 26 patients with NSCLC before treatment with azacytidine and entinostat (HDACi), showed higher clinical response rates in patients with methylation of two or more genes (55).
Among 63 patients with MDS and AML treated with azacytidine, those with methylation of at least two genes from a panel of 24 tumor suppressor genes had a shorter overall survival (56). However, the number of methylated genes did not correlate with the treatment response to azacytidine.
Global methylation.
Another approach has been to examine the methylation status of repetitive elements during treatment, which is independent of the presence of tumor cells after therapy. Several studies have shown significant demethylation of LINE1 and Alu elements during treatment by both azacytidine and decitabine (17, 51, 52). However, prognostic effect of neither pretreatment methylation levels nor methylation changes during treatment has been documented.
A recent study analyzed the global DNA methylation level using MethylCap-seq in 16 patients with AML treated with decitabine. A trend toward a higher baseline methylation level and more pronounced methylation decrease during treatment was observed among responding patients (57).
Gene expression
DNMT3B amplification.
Overexpression of DNMT3B mRNA and protein due to gene amplification is frequently observed in human cancers (58). Interestingly, cell lines harboring the DNMT3B amplification were less sensitive to azacytidine, decitabine, and SGI-110, but clinical data are still not available.
CJUN and CMYB.
The gene expression levels of CJUN and CMYB have been identified as potential biomarkers in a cohort of 36 decitabine-treated patients with CMML (46). cJUN has previously been shown to promote aberrant monocyte transformation (59). CJUN expression was significantly lower in monocytes from responding patients, and higher CJUN expression was correlated to shorter survival (46). Deregulation of CMYB has been implicated in leukemia (60), and higher CMYB expression was also associated with shorter survival (46).
Fas expression.
Expression of the pro-apoptotic protein Fas in CD45lo/CD34+ bone marrow cells from patients with MDS (all IPSS groups) or sAML has been positively correlated with response to azacytidine. A correlation between promoter methylation and Fas expression was also observed. Among 63 patients, low Fas expression at diagnosis (presumably due to hypermethylation) was correlated to clinical response, while no association between Fas expression and overall survival was observed (61). In 38 patients Fas expression was examined before and after at least 3 cycles of azacytidine, and responding patients (23 of 38) had a significant increase in Fas expression.
Phosphoinositide-phospholipase C β1.
Phosphoinositide-phospholipase C β1 (PLCβ1) is a key enzyme in lipid-signaling pathways that acts on cell proliferation and differentiation. PLCβ1 is highly expressed in the early stages of hematopoietic differentiation (62), is hypermethylated in higher-risk MDS patients, and may be a specific target for azacytidine (63). Among 18 patients an increase in PLCβ1 expression and a decrease in PLCβ1 methylation were observed in 9 of 10 patients with hematological response. The same group observed a similar association in two cohorts of 32 and 26 patients with low-risk MDS treated with azacytidine (64, 65). In the latter cohort, the PLCβ1 target cyclin D3 was induced in responding patients, supporting the notion that the PLCβ1 pathway is activated during azacytidine treatment (65). Due to the involvement of PLCβ1 in early hematopoietic differentiation, it is hypothesized that PLCβ1 upregulation by demethylation leads to differentiation.
miR-29b. miR-29b is involved in the regulation of DNA methylation by targeting the DNA methyltransferases DNMT3A/3B and DNMT1 (41, 66).
In a phase II clinical trial in older AML patients treated with decitabine, a positive correlation between the clinical response and high pre-treatment levels of miR-29b was observed (42). In vitro studies from the same group have recently shown that priming of AML cell lines and primary AML blasts with a new HDACi (AR-42) leads to upregulation of miR-29b expression and enhanced anti-leukemic effect of subsequently administered decitabine (67). Yang et al. (68) reported, however, a lack of association between pretreatment miR-29b expression levels and clinical responses to azacytidine in patients with AML. The results obtained by these studies may be explained by the different sources used for miR analysis (peripheral blood vs. bone marrow) and the use of decitabine (42), which may more efficiently downregulate DNMTs.
Predicting response to HDACis
HDACis have considerable antiproliferative and apoptotic activities, making them potential anticancer agents. The HDAC family contains 18 enzymes, grouped into 4 classes that regulate the acetylation level of histones, and several non-histone substrates, including a variety of proteins involved in, for example, cell cycle control, apoptosis, and angiogenesis (69). However, it is still not clear by which key pathways HDACis modify tumor growth in patients (Figure 2). Like the DNMTis, the most promising results are observed in hematological malignancies, with only limited effects in solid tumors. Currently, two HDACis, vorinostat and romidepsin, are FDA approved for treatment of refractory cutaneous T cell lymphoma (CTCL) in patients who have received at least two prior regimens. Romidepsin is also approved for peripheral T cell lymphoma.
Molecular predictors
Acetylation.
At this point, only molecular predictors have been identified as biomarkers for HDACi therapy (Table 6). Thus far, the most extensively studied biomarker for HDACi activity is acetylation levels of the target proteins before and after treatment in peripheral blood or tumor tissue, but no correlation to clinical response has been found (70–74). Indeed, hyperacetylation was generally observed in all patients irrespective of response to HDACi (72–74).
Table 6.
Molecular markers for response to HDACis
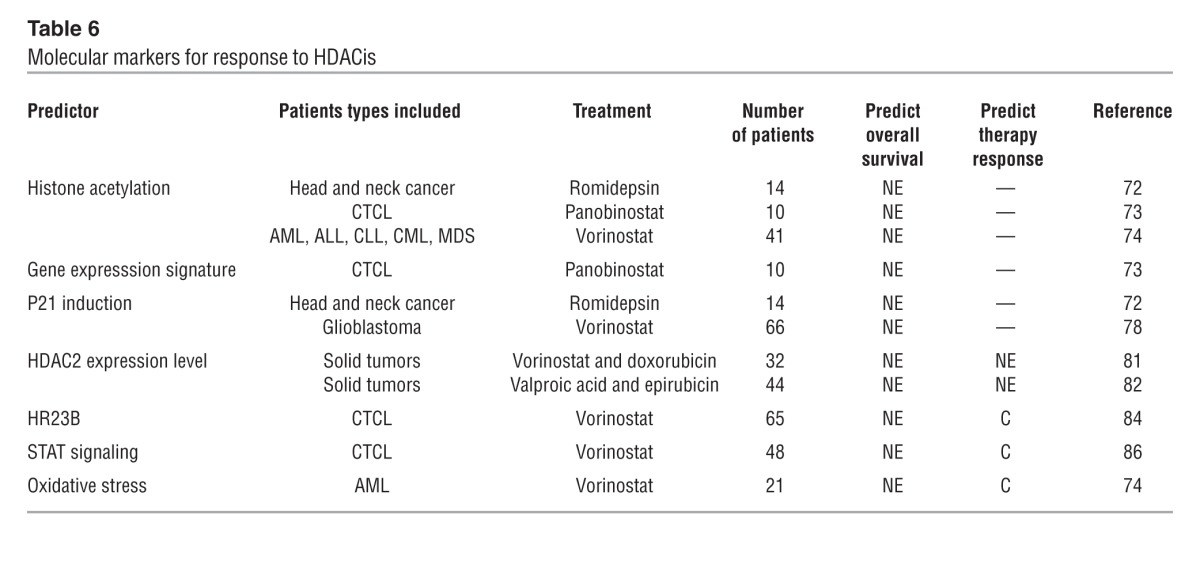
Gene expression signature.
Gene expression profiling of HDACi-treated cell lines indicated that HDACis are only involved in the regulation of 2%–5% of all human genes (75). In 10 patients mRNA expression in CTCL biopsies taken 4, 8, and 24 hours after administration of the pan-HDACi panobinostat showed altered expression (mainly downregulation) in less than 10% of all genes at the four-hour time point, at which peak changes were observed (73). No correlation was observed between gene expression and response, which could obviously be due to the low number of patients. Similar studies have been done in vitro, leading to identification of a nine-gene signature predictive for response in lung cancer cell lines (70), but this signature has not been validated in vivo.
Several studies have demonstrated that many HDACis increase the level of the cell cycle inhibitor p21 both in vitro (76, 77) and in vivo (72, 78), but no correlation between p21 induction and clinical response has been observed. Interestingly, p21 is upregulated independent of p53, and stratification according to disruption of p21 regulatory pathways (e.g., p53 mutation) may identify patients that benefit from HDACi. Due to the variable functions of the HDACs in multiple pathways, gene signatures are likely to vary with the tumor type, the HDACi being applied, and the concentration of the HDACi.
HDAC expression level.
The expression levels of the HDACs themselves have been suggested as a predictive biomarker. Most studies have quantified HDAC expression by immunohistochemistry (IHC), and many HDACs are overexpressed in human cancers (70, 79). Marquard et al. (80) examined the expression levels of HDAC1, -2, and -6 and acetylated histone H4 in 73 CTCL biopsies. Overexpression of HDAC2 and histone H4 acetylation were correlated with more aggressive forms of CTCL. In two clinical trials, a correlation was observed between pretreatment HDAC2 expression and histone acetylation in the tumor tissue (81), and it was suggested that HDAC2 expression potentially can identify patients who will benefit from HDACi treatment (81, 82).
HR23B.
A genome-wide loss-of-function screen indicated that RAD23 homolog B (HR23B) sensitizes tumor cells to HDACis (83). Under normal conditions HDACs inhibit the expression of HR23B. HDACi-mediated HR23B overexpression leads to proteasome overload, aberrant protein degradation, and apoptosis. Accordingly, cells depleted of HR23B are less sensitive to vorinostat-induced apoptosis (84). High HR23B expression by IHC was positively correlated to clinical response (PPV = 71.7%) in a phase II clinical trial with 65 vorinostat-treated CTCL patients (84). Sequential samples from a fraction of these patients showed that HR23B expression remained high throughout the time of response. However, a recent study in malignant pleural mesothelioma cell lines shows that vorinostat induced apoptosis is independent of HR23B (85), indicating that the role of HR23B may be cell type dependent.
STAT signaling.
In a functional screen of 40 human B and T cell lymphoma cell lines, high baseline levels of activated STAT1, STAT3, and STAT5 correlated with resistance to vorinostat (86). STATs are transcription factors that participate in chromatin remodeling and enable transcription of several anti-apoptotic proteins. These factors were evaluated in 48 pretreatment CTCL biopsies from patients enrolled in a vorinostat phase IIb clinical trial, and it was shown that nuclear accumulation of STAT1, and high levels of phosphorylated STAT3, in the malignant T cells correlated with lack of clinical response.
Oxidative stress.
Vorinostat resistance has been linked to increased tolerance of oxidative stress (74). The expression levels of 17 genes involved in antioxidation, selected from preclinical studies, were examined in 21 patients with AML treated with vorinostat in a phase I clinical trial (74). Nonresponders had higher baseline expression levels of these 17 genes compared with patients with hematological improvement, partial response, or CR. The same group showed that a decrease in the cellular glutathione levels increased the sensitivity to vorinostat in cell lines and in primary leukemic cells (87).
Conclusion
Ideally, the identification of good predictive biomarkers allows selection of personalized therapy and thereby maximizes the benefit of treatment. However, despite comprehensive knowledge of the biology and function of epigenetic therapy, the search for specific biomarkers for response and survival is not straightforward. Disappointingly, the novel high-throughput epigenetic screening methodologies have not yet been useful for this purpose.
There may be several reasons why the identification of biomarkers for epigenetic therapy has been less successful. First, most studies have been performed in relatively small and miscellaneous patient cohorts, and the findings need confirmation in larger, independent studies. Second, it is likely that a particular biomarker will only be useful for a specific agent, since each individual epigenetic drug has a different pharmacological profile. Among the DNMTis, decitabine is incorporated into DNA, while azacytidine is mainly incorporated into RNA; the in vitro effects of the two agents differ (18, 88), and it is possible that they have different effects in vivo (10). Similarly, some HDACis inhibit all classes of HDACs, while others target only one or two; e.g., vorinostat is a pan-inhibitor, while romidepsin is a class I inhibitor. Thus, it is likely that each individual drug will require a specific biomarker. Third, epigenetic drugs are being used in combination, which may further complicate the identification of relevant biomarkers. Fourth, each individual patient might respond for different reasons, such as reactivation of tumor suppressor genes, restoration of sensitivity to conventional chemotherapy, induction of immunogenicity, induction of terminal differentiation, or combinations thereof. Finally, great variation in drug sensitivity may exist for each cancer type. The recent next-generation sequencing studies have taught us that each tumor harbors a wealth of mutations, and at this point it is unclear whether some of these will be pertinent biomarkers for the efficacy of epigenetic therapy.
Currently, only the clinical markers have been verified by independent research groups, which is a requirement for implementation in clinical practice. The most promising biomarkers are likely to be measurements of the biological effects during treatment; this, however, may be hampered by the elimination of malignant cells. One solution might be to investigate changes in the constitutive methylation patterns as, for example, LINE1 elements (51), but although significant demethylation is observed during treatment with azanucleosides, there is no evidence of its prognostic value.
In conclusion, there is a lack of proof of a relation between molecular mechanisms of action and biomarkers. Thus, for the time being, there is still much to uncover before the responses to epigenetic therapy can be consistently predicted, but hopefully many large clinical trials in combination with novel high-throughput screening methods will enable us to identify good biomarkers in the near future.
Acknowledgments
This study was supported by The Novo Nordisk Foundation (to K. Grønbæk and M.B. Treppendahl), The Danish Cancer Society (to K. Grønbæk), and The Danish Strategic Research Foundation (to K. Grønbæk and L.S. Kristensen).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(1):47–55. doi:10.1172/JCI69737.
References
- 1.Diamandis M, White NM, Yousef GM. Personalized medicine: marking a new epoch in cancer patient management. Mol Cancer Res. 2010;8(9):1175–1187. doi: 10.1158/1541-7786.MCR-10-0264. [DOI] [PubMed] [Google Scholar]
- 2.Milbury CA, Li J, Makrigiorgos GM. PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55(4):632–640. doi: 10.1373/clinchem.2008.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med. 2012;9(5):e1001216. doi: 10.1371/journal.pmed.1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McShane LM, et al. Reporting recommendations for tumor marker prognostic studies. J Clin Oncol. 2005;23(36):9067–9072. doi: 10.1200/JCO.2004.01.0454. [DOI] [PubMed] [Google Scholar]
- 5.Fenaux P, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–569. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 6.Fenaux P, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantarjian H, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 8.Adès L, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37(6):609–613. doi: 10.1016/j.leukres.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Kantarjian H, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670–2677. doi: 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee Y-G, et al. Comparative analysis between azacitidine and decitabine for the treatment of myelodysplastic syndromes. Br J Haematol. 2013;161(3):339–347. doi: 10.1111/bjh.12256. [DOI] [PubMed] [Google Scholar]
- 11.Schecter J, Galili N, Raza A. MDS: Refining existing therapy through improved biologic insights. Blood Rev. 2012;26(2):73–80. doi: 10.1016/j.blre.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Itzykson R, et al. Prognostic factors for response and overall survival in 282 patients with higher-risk myelodysplastic syndromes treated with azacitidine. Blood. 2011;117(2):403–411. doi: 10.1182/blood-2010-06-289280. [DOI] [PubMed] [Google Scholar]
- 13.Itzykson R, et al. Long-term outcome of higher-risk MDS patients treated with azacitidine: an update of the GFM compassionate program cohort. Blood. 2012;119(25):6172–6173. doi: 10.1182/blood-2012-04-422204. [DOI] [PubMed] [Google Scholar]
- 14.Gore SD, et al. A multivariate analysis of the relationship between response and survival among patients with higher-risk myelodysplastic syndromes treated within azacitidine or conventional care regimens in the randomized AZA-001 trial. Haematologica. 2013;98(7):1067–1072. doi: 10.3324/haematol.2012.074831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 16.Gore SD, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 17.Fandy TE, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu X, et al. Equitoxic doses of 5-azacytidine and 5-aza-2′deoxycytidine induce diverse immediate and overlapping heritable changes in the transcriptome. PLoS One. 2010;5(9):e12994. doi: 10.1371/journal.pone.0012994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almstedt M, et al. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk Res. 2010;34(7):899–905. doi: 10.1016/j.leukres.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Goodyear O, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116(11):1908–1918. doi: 10.1182/blood-2009-11-249474. [DOI] [PubMed] [Google Scholar]
- 21.Aimiuwu J, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012;119(22):5229–5238. doi: 10.1182/blood-2011-11-382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Astex Pharmaceuticals. SGI-110 in Patients With Myelodysplastic Syndromes (MDS) or Acute Myelogenous Leukemia (AML). NIH Web site. http://clinicaltrials.gov/ct2/show/NCT01261312 . Updated August 14, 2013. Accessed November 5, 2013.
- 23. Astex Pharmaceuticals. SGI-110 in Combination With Carboplatin in Ovarian Cancer. NIH Web site. http://clinicaltrials.gov/ct2/show/NCT01696032 . Updated April 14, 2013. Accessed November 5, 2013.
- 24. Astex Pharmaceuticals. SGI-110 in the Treatment of Advanced Hepatocellular Carcinoma (HCC). NIH Web site. http://clinicaltrials.gov/ct2/show/NCT01752933 . Updated April 14, 2013. Accessed November 5, 2013.
- 25.Fahy J, Jeltsch A, Arimondo PB. DNA methyltransferase inhibitors in cancer: a chemical and therapeutic patent overview and selected clinical studies. Expert Opin Ther Pat. 2012;22(12):1427–1442. doi: 10.1517/13543776.2012.729579. [DOI] [PubMed] [Google Scholar]
- 26.Gros C, et al. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie. 2012;94(11):2280–2296. doi: 10.1016/j.biochi.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 27.Rius M, et al. Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol Cancer Ther. 2009;8(1):225–231. doi: 10.1158/1535-7163.MCT-08-0743. [DOI] [PubMed] [Google Scholar]
- 28.Damaraju VL, et al. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids. 2012;31(3):236–255. doi: 10.1080/15257770.2011.652330. [DOI] [PubMed] [Google Scholar]
- 29.Qin T, Jelinek J, Si J, Shu J, Issa J-PJ. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood. 2009;113(3):659–667. doi: 10.1182/blood-2008-02-140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Heuvel-Eibrink MM, Wiemer EA, Kuijpers M, Pieters R, Sonneveld P. Absence of mutations in the deoxycytidine kinase (dCK) gene in patients with relapsed and/or refractory acute myeloid leukemia (AML). Leukemia. 2001;15(5):855–856. doi: 10.1038/sj.leu.2402112. [DOI] [PubMed] [Google Scholar]
- 31.Qin T, et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS One. 2011;6(8):e23372. doi: 10.1371/journal.pone.0023372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahfouz RZ, et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin Cancer Res. 2013;19(4):938–948. doi: 10.1158/1078-0432.CCR-12-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bally C, et al. Azacitidine in the treatment of therapy related myelodysplastic syndrome and acute myeloid leukemia (tMDS/AML): A report on 54 patients by the Groupe Francophone Des Myelodysplasies (GFM). Leuk Res. 2013;37(6):637–640. doi: 10.1016/j.leukres.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Shen L, et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol. 2010;28(4):605–613. doi: 10.1200/JCO.2009.23.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raj K, et al. CDKN2B methylation status and isolated chromosome 7 abnormalities predict responses to treatment with 5-azacytidine. Leukemia. 2007;21(9):1937–1944. doi: 10.1038/sj.leu.2404796. [DOI] [PubMed] [Google Scholar]
- 36.Yoo CB, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67(13):6400–6408. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg P, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–2088. [PubMed] [Google Scholar]
- 38.Breccia M, et al. Application of French prognostic score to patients with International Prognostic Scoring System intermediate-2 or high risk myelodysplastic syndromes treated with 5-azacitidine is able to predict overall survival and rate of response. Leuk Lymphoma. 2012;53(5):985–986. doi: 10.3109/10428194.2011.643408. [DOI] [PubMed] [Google Scholar]
- 39.Van der Helm LH, et al. Platelet doubling after the first azacitidine cycle is a promising predictor for response in myelodysplastic syndromes (MDS), chronic myelomonocytic leukaemia (CMML) and acute myeloid leukaemia (AML) patients in the Dutch azacitidine compassionate named p. Br J Haematol. 2011;155(5):599–606. doi: 10.1111/j.1365-2141.2011.08893.x. [DOI] [PubMed] [Google Scholar]
- 40.Lübbert M, et al. Cytogenetic responses in high-risk myelodysplastic syndrome following low-dose treatment with the DNA methylation inhibitor 5-aza-2′-deoxycytidine. Br J Haematol. 2001;114(2):349–357. doi: 10.1046/j.1365-2141.2001.02933.x. [DOI] [PubMed] [Google Scholar]
- 41.Garzon R, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113(25):6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blum W, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A. 2010;107(16):7473–7478. doi: 10.1073/pnas.1002650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viré E, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 44.Itzykson R, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7):1147–1152. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 45.Voso MT, et al. Role of BCL2L10 methylation and TET2 mutations in higher risk myelodysplastic syndromes treated with 5-azacytidine. Leukemia. 2011;25(12):1910–1913. doi: 10.1038/leu.2011.170. [DOI] [PubMed] [Google Scholar]
- 46.Braun T, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–3831. doi: 10.1182/blood-2011-05-352039. [DOI] [PubMed] [Google Scholar]
- 47.Metzeler KH, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26(5):1106–1107. doi: 10.1038/leu.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Traina F, et al. Leukemia. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms [published online ahead of print September 18, 2013]. doi: 10.1038/leu.2013.269 . [DOI] [PubMed] [Google Scholar]
- 49.Daskalakis M, et al. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood. 2002;100(8):2957–2964. doi: 10.1182/blood.V100.8.2957. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Manero G, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang AS, et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006;66(10):5495–5503. doi: 10.1158/0008-5472.CAN-05-2385. [DOI] [PubMed] [Google Scholar]
- 52.Kantarjian H, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 53.Issa J-PJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103(5):1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 54.Cluzeau T, et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget. 2012;3(4):490–501. doi: 10.18632/oncotarget.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Juergens R, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abáigar M, et al. Prognostic impact of the number of methylated genes in myelodysplastic syndromes and acute myeloid leukemias treated with azacytidine. Ann Hematol. 2013;92(11):1543–1552. doi: 10.1007/s00277-013-1799-9. [DOI] [PubMed] [Google Scholar]
- 57.Yan P, et al. Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood. 2012;120(12):2466–2474. doi: 10.1182/blood-2012-05-429175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simó-Riudalbas L, Melo SA, Esteller M. DNMT3B gene amplification predicts resistance to DNA demethylating drugs. Genes Chromosomes Cancer. 2011;50(7):527–534. doi: 10.1002/gcc.20877. [DOI] [PubMed] [Google Scholar]
- 59.Yang Z, et al. Increased c-Jun expression and reduced GATA2 expression promote aberrant monocytic differentiation induced by activating PTPN11 mutants. Mol Cell Biol. 2009;29(16):4376–4393. doi: 10.1128/MCB.01330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8(7):523–534. doi: 10.1038/nrc2439. [DOI] [PubMed] [Google Scholar]
- 61.Ettou S, et al. Fas expression at diagnosis as a biomarker of azacitidine activity in high-risk MDS and secondary AML. Leukemia. 2012;26(10):2297–2299. doi: 10.1038/leu.2012.152. [DOI] [PubMed] [Google Scholar]
- 62.Follo MY, et al. Synergistic induction of PI-PLCβ1 signaling by azacitidine and valproic acid in high-risk myelodysplastic syndromes. Leukemia. 2011;25(2):271–280. doi: 10.1038/leu.2010.266. [DOI] [PubMed] [Google Scholar]
- 63.Follo MY, et al. Reduction of phosphoinositide-phospholipase C β1 methylation predicts the responsiveness to azacitidine in high-risk MDS. Proc Natl Acad Sci U S A. 2009;106(39):16811–16816. doi: 10.1073/pnas.0907109106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Filì C, et al. Prospective phase II Study on 5-days azacitidine for treatment of symptomatic and/or erythropoietin unresponsive patients with low/INT-1-risk myelodysplastic syndromes. Clin Cancer Res. 2013;19(12):3297–3308. doi: 10.1158/1078-0432.CCR-12-3540. [DOI] [PubMed] [Google Scholar]
- 65.Follo MY, et al. Epigenetic regulation of nuclear PI-PLCβ1 signaling pathway in low-risk MDS patients during azacitidine treatment. Leukemia. 2012;26(5):943–950. doi: 10.1038/leu.2011.300. [DOI] [PubMed] [Google Scholar]
- 66.Fabbri M, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104(40):15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mims A, et al. Increased anti-leukemic activity of decitabine via AR-42-induced upregulation of miR-29b: a novel epigenetic-targeting approach in acute myeloid leukemia. Leukemia. 2013;27(4):871–878. doi: 10.1038/leu.2012.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang H, et al. Levels of miR-29b do not predict for response in patients with acute myelogenous leukemia treated with the combination of 5-azacytidine, valproic acid, and ATRA. Am J Hematol. 2011;86(2):237–238. doi: 10.1002/ajh.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6(6):637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stimson L, La Thangue NB. Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett. 2009;280(2):177–183. doi: 10.1016/j.canlet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 71.Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1(3–4):117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haigentz M, et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012;48(12):1281–1288. doi: 10.1016/j.oraloncology.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ellis L, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14(14):4500–4510. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Manero G, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 75.Glaser KB, et al. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. 2003;2(2):151–163. [PubMed] [Google Scholar]
- 76.Arts J, et al. R306465 is a novel potent inhibitor of class I histone deacetylases with broad-spectrum antitumoral activity against solid and haematological malignancies. Br J Cancer. 2007;97(10):1344–1353. doi: 10.1038/sj.bjc.6604025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noro R, et al. Histone deacetylase inhibitor enhances sensitivity of non-small-cell lung cancer cells to 5-FU/S-1 via down-regulation of thymidylate synthase expression and up-regulation of p21(waf1/cip1) expression. Cancer Sci. 2010;101(6):1424–1430. doi: 10.1111/j.1349-7006.2010.01559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galanis E, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27(12):2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90(1):85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 80.Marquard L, et al. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology. 2008;53(3):267–277. doi: 10.1111/j.1365-2559.2008.03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Munster PN, et al. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer. 2009;101(7):1044–1050. doi: 10.1038/sj.bjc.6605293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Munster P, et al. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res. 2009;15(7):2488–2496. doi: 10.1158/1078-0432.CCR-08-1930. [DOI] [PubMed] [Google Scholar]
- 83.Fotheringham S, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15(1):57–66. doi: 10.1016/j.ccr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Khan O, et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci U S A. 2010;107(14):6532–6537. doi: 10.1073/pnas.0913912107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hurwitz JL, et al. Vorinostat/SAHA-induced apoptosis in malignant mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. Eur J Cancer. 2012;48(7):1096–1107. doi: 10.1016/j.ejca.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 86.Fantin VR, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785–3794. doi: 10.1158/0008-5472.CAN-07-6091. [DOI] [PubMed] [Google Scholar]
- 87.Hu Y, et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood. 2010;116(15):2732–2741. doi: 10.1182/blood-2009-11-256354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Flotho C, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–1028. doi: 10.1038/leu.2008.397. [DOI] [PubMed] [Google Scholar]
- 89.Bennett JM, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51(2):189–199. [PubMed] [Google Scholar]
- 90. Swerdlow SH, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: : IARC Press; ; 2008. [Google Scholar]