

Abstract
Epigenetic enzymes are often dysregulated in human tumors through mutation, altered expression, or inappropriate recruitment to certain loci. The identification of these enzymes and their partner proteins has driven the rapid development of small-molecule inhibitors that target the cancer epigenome. Herein, we discuss the influence of aberrantly regulated histone deacetylases (HDACs) in tumorigenesis. We examine HDAC inhibitors (HDACis) targeting class I, II, and IV HDACs that are currently under development for use as anticancer agents following the FDA approval of two HDACis, vorinostat and romidepsin.
Introduction
Histone acetylation is an important determinant of gene expression. Acetylation is generally associated with elevated transcription, while deacetylated histones are often associated with gene repression. Histone deacetylases (HDACs) are critical regulators of gene expression that enzymatically remove the acetyl group from histones. A broader role for HDACs as acyl-lysine “erasers” is also emerging with the discovery of additional acyl-lysine histone modifications such as crotonylation, succinylation, and malonylation (1, 2). The activity of HDACs on nonhistone proteins is also a key aspect of HDAC function (3). However, while a large number of nonhistone HDAC substrates have been identified, the molecular and biological consequences of nonhistone protein deacetylation is yet to be elucidated for the majority of these targets. Three classical HDAC classes (I, II, and IV) containing 11 HDACs have been identified thus far and are classified according to their homology to yeast proteins, subcellular location, and enzymatic activities (these distinctions are further described in Table 1 and Figure 1). The class III HDACs, or sirtuins, possess NAD-dependent catalytic sites and have some overlapping functions with the classical HDACs. However, sirtuins are not hindered by conventional HDAC inhibitors (HDACis), and as such will not be discussed herein. The biological outcome of HDAC inhibition is dependent on the HDAC specificity of the compound and intrinsic operation of cell-signaling pathways. HDACis are best characterized as anticancer agents, and investigation into their mechanisms of action and clinical efficacy remains robust and active, and will be discussed further.
Table 1.
Aberrant regulation of HDACs in cancer
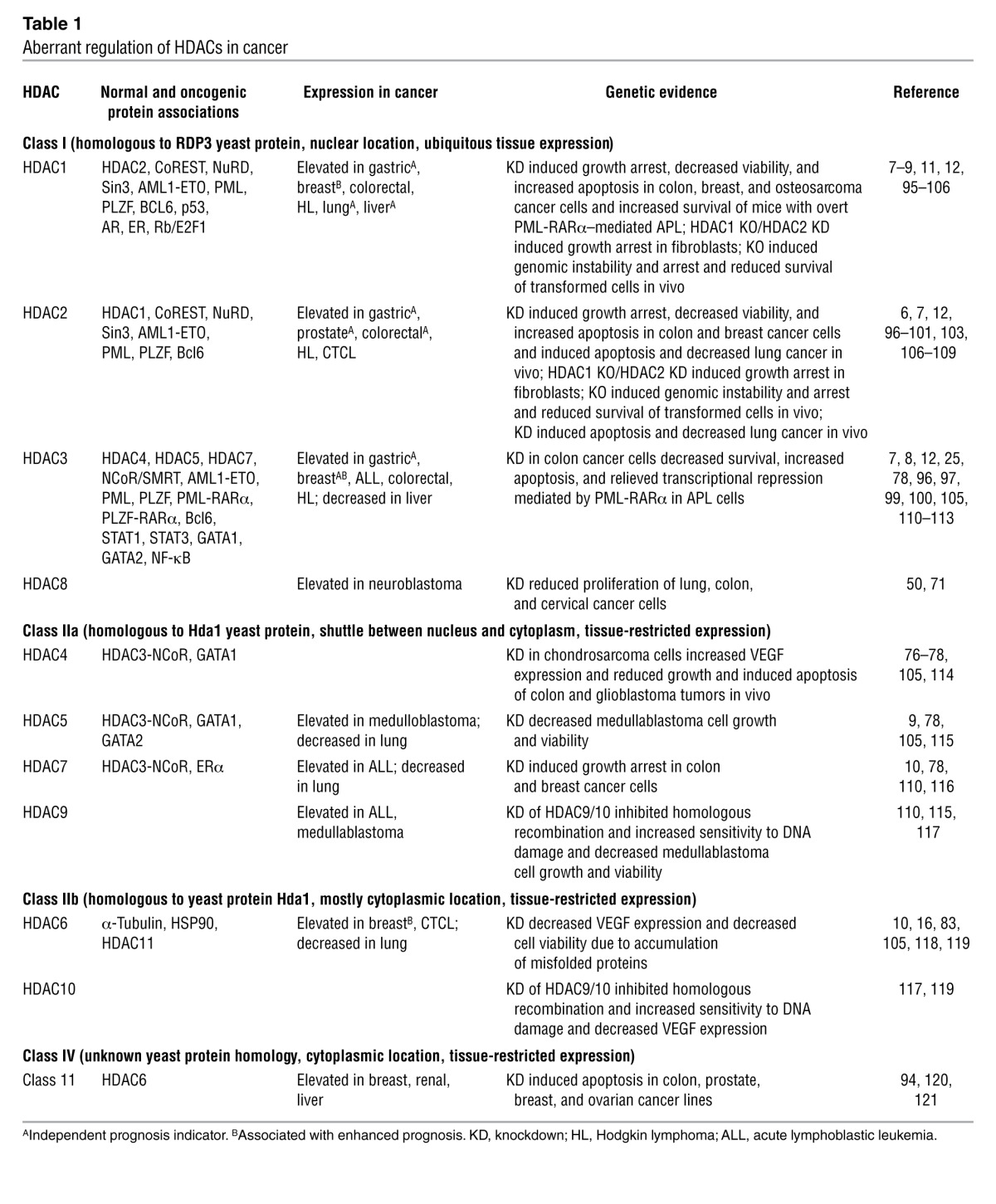
Figure 1. The molecular targets of HDACs, downstream cellular pathways, and anticancer outcomes of HDAC inhibition.

HDAC substrates include histones and nonhistone proteins. Histones are the primary substrates for HDAC1, -2, and -3, while other cellular proteins are targeted by one or more class I and class II HDACs. Some known HDAC targets, downstream molecular changes that occur following HDAC inhibition, and associated biological pathways that mediate antitumor responses are shown. The HDACs, substrates, and molecular responses are color matched to illustrate functional relationships. The best-characterized biological consequences of HDACi treatment of tumor cells are shown on the lowest tier of the diagram. SMC3, structural maintenance of chromosomes 3; GCMa, glial cells missing homolog 1; HAT, histone acetyltransferase; HP1, heterochromatin protein 1.
Rationale for targeting HDACs in cancer
An early indication that genome-wide changes in histone acetylation may underpin cancer onset and progression came from a study demonstrating a global loss of monoacetylation and trimethylation on histone H4 in cancer cells (4). Numerous correlative studies have demonstrated aberrant expression of HDACs in human tumors (Table 1), and expression of HDAC1, -5, and -7 can serve as a molecular biomarker of tumor versus normal tissue (5). Interestingly, in several cancer types, such as prostate (6), colorectal (7), breast (8), lung (9, 10), liver (11), and gastric cancer (12), overexpression of individual HDACs correlated with significant decreases in both disease-free and overall survival and was able to predict poor patient prognosis independent of other variables such as tumor type and disease progression. Indeed, the overexpression of HDACs has been linked to key events of tumorigenesis such as the epigenetic repression of the tumor suppressor gene CDKN1A (encoding the cyclin-dependent kinase inhibitor p21waf1/cip1; ref. 13) and key genes encoding DNA damage repair enzymes such as breast cancer 1, early onset (BRCA1) and ataxia telangiectasia and Rad3 related (ATR) (14). However, it is clear that HDAC overexpression is not always a negative prognostic marker; indeed, elevated HDAC6 levels predicted better prognosis in patients with estrogen receptor–positive (ER-positive) breast cancer (15). Furthermore, despite potent clinical activity of pan-HDACis in cutaneous T cell lymphoma (CTCL), the overexpression of HDAC6 correlated with improved prognosis, and acetylated H4 was associated with more aggressive lesions (16). Thus, the level of HDAC expression and/or histone acetylation may not necessarily indicate clinical sensitivity to oncology drugs, including HDACis.
Aberrant activity of HDACs is, however, linked to key oncogenic events. The genetic knockdown of individual HDACs, most notably HDAC1, -2, -3, and -6, in a variety of tumor types such as colon, breast, and lung and acute promyelocytic leukemia (APL) induced apoptosis and cell cycle arrest (further described in Table 1), implicating HDAC activity as a key mediator of survival and tumorigenic capacity in these settings. Direct deacetylation of the tumor suppressor p53 by HDACs leads to decreased transcriptional activity (17), and the upregulation of oncogenes such as BCL2 is induced by HDAC-mediated deacetylation and activation of the transcription factors SP1 and C/EBPα (18). The aberrant recruitment of HDACs to certain gene loci through binding to oncogenic fusion proteins has also been proposed as an important mechanism of tumorigenesis. For example, AML1-ETO (a fusion of the acute myeloid leukemia 1 [AML1] and eight twenty-one [ETO] proteins resulting from t[8;21]) and PML-RARα (a fusion of promyelocytic leukemia [PML] and retinoic acid receptor α [RARα] fusion arising from t[15;17]) gain corepressor activity upon aberrant recruitment of HDAC complexes (containing nuclear receptor corepressor [NCoR; ref. 19]) and gain subsequent leukemogenic potential upon aberrant recruitment of mSin3a and silencing mediator for retinoid and thyroid receptors (SMRT) (20). Indeed, HDACis are potent inducers of apoptosis and terminal differentiation in leukemias expressing AML1-ETO and PML-RARα (21, 22).
Intriguingly, downregulation and deletion of HDACs may play a role in cancer progression. HDAC4 is routinely decreased in gastric tumors, HDAC1 somatic mutations were detected in approximately 8% of dedifferentiated human liposarcomas (23), and a frame-shift mutation causing dysfunctional HDAC2 expression was observed in human epithelial cancers with microsatellite instability (24). Together these data suggest a putative tumor suppressor role for certain HDACs. Indeed, recent studies by our laboratory and others using HDAC knockout and knockdown technologies provide strong functional evidence supporting tumor suppressor roles for HDAC1, -2, and -3 (25–27). Genetic knockout of Hdac1 and Hdac2 enhanced spontaneous tumorigenesis in a dose-dependent manner by relieving a p53-dependent oncogenic barrier (26), and HDAC1 genetic and pharmacologic inhibition accelerated tumorigenesis of precancerous cells harboring oncogenic lesions (27). Bhaskara et al. observed that downregulation of the HDAC3-containing complex NCoR was apparent in approximately 30% of human hepatocellular carcinomas (HCCs), and the tumor suppressor function of HDAC3 was confirmed when liver-specific HDAC3 knockdown resulted in overt HCC (25). HDACs may also suppress the development of metastasis, as HDACi treatment alleviated repression of metastasis-related genes, subsequently promoting the migration of human tumor cell lines in vitro (28, 29) and enhancing metastasis in vivo (28). It will be important to elucidate the impact of these class I HDACs at each step of tumorigenesis in order to explain their apparent paradoxical roles as tumor suppressors in developing tumors and as therapeutic targets in established neoplasms. HDACis are currently being studied in clinical trials for use against non-cancer diseases such as rheumatoid arthritis (30). Based on the limited experimental data available to date, one might predict that patients chronically treated with HDACis may be at risk of developing therapy-induced tumors. However, there is no clinical data as yet demonstrating the development of malignancies following HDACi treatment of patients with noncancerous diseases.
HDACis as anticancer agents
A vast array of both natural and synthetic chemical compounds function as HDACis. Two HDACis, vorinostat and romidepsin, are FDA approved for use against refractory CTCL, and many others are currently being clinically assessed (see Table 2 for a list of recent clinical trials assessing HDACi efficacy against cancer). HDACis are classified into groups based on their chemical structure, including hydroxamic acids (trichostatin A, vorinostat), carboxylic acids (valproate, butyrate), aminobenzamides (entinostat, mocetinostat), cyclic peptides (apicidin, romidepsin), epoxyketones (trapoxins), and hybrid molecules. There are 11 class I, II, and IV HDACs, and the target specificity of HDACis and the requirement for specific and selective inhibition of HDACs to achieve therapeutic efficacy remain topics of debate. Traditionally, tests examining the bioactivity of HDACis in vivo have included assessment of histone acetylation levels by Western blot or immunohistochemistry, and more recently, fluorescence-based reporter assays examining HDAC activity in the peripheral white blood cells of HDACi-treated cancer patients have been developed (31). These non-HDAC class–selective bioassays might prove useful for determining HDACi pharmacodynamics in clinical trials. However, reliable determination of HDACi target specificity in vitro using standard assays with recombinant HDAC proteins has been hindered due to protein misfolding, lack of enzymatic activity, and most importantly, the inappropriate assessment of isolated HDACs that exist as multiprotein complexes in physiologic conditions. Furthermore, the substrate specificity of HDACs, in particular class IIa HDACs, is yet to be fully elucidated, which means that the precise surrogate biomarkers of individual HDAC inhibition are also not defined. Importantly, the activity of class I HDACs increases upon complex formation (32, 33), and recent pivotal studies using sophisticated chemoproteomics and chemical phylogenetic approaches demonstrated that commonly used HDACis have a higher degree of specificity than originally proposed (34, 35). HDACis can be used as “bait” to explicitly discriminate between multiprotein complexes containing the same HDAC target (34). Interestingly, class IIa HDACs were rarely identified as direct targets of HDACis, possibly due to their low catalytic activity, suggesting a role for this class in the recognition of and binding to acetylated histones (histone “reading”) with subsequent recruitment of additional epigenetic modifiers, as opposed to active deacetylation (34, 35). Corroborating these findings in vivo remains a further challenge, but the development of the HDAC chemical phylogenetic tree (35) and identification of specific substrates of individual HDACs, such as SMC3 for HDAC8 (36), may lead to more focused in vivo investigations into HDAC and HDACi selectivity and specificity.
Table 2.
HDACis in current clinical trials
Studies utilizing genetic knockdown or knockout of HDACs have provided clues as to the most important HDACs to target in cancer. Depletion of HDAC1, -2, -3, -4, or -11 was sufficient to induce apoptosis in a range of tumor cells lines, whereas HDAC1, -2, and -4 were required to maintain cancer cell survival in vivo (specific experiments are listed in Table 1). It will be crucial to determine whether the antitumor effects observed upon knockout or knockdown of HDACs listed in Table 1 was due to lack of HDAC activity specifically or was linked to disruption of HDAC-containing complexes that might have epigenetic or other regulatory activities. This distinction is particularly relevant to HDAC1, -2, and -3, which are key catalytic subunits of various complexes and are recruited by oncogenic fusion proteins (protein associations of individual HDACs are listed in Table 1 and illustrated in Figure 2). Furthermore, the pharmacologic inhibition of HDACs will almost certainly not be equivalent to genetic knockdown/knockout (37), especially in terms of effects on multiprotein complex formation and function, and for less catalytically active HDACs such as those in class IIa. Additional investigations will likely result in the identification of the key HDAC-containing complexes required by tumor cells for survival, the specific functions of HDACs within these complexes, and the molecular and biological consequences of pharmacologic inhibition of these HDACs. These findings will hopefully allow for a more rational approach to stratifying patients for HDACi treatment. Furthermore, it may be possible to investigate the correlation between inhibiting specific HDAC-dependent complexes in particular tumor types with clinical outcomes following HDACi treatment, allowing for the identification of novel HDACi response biomarkers.
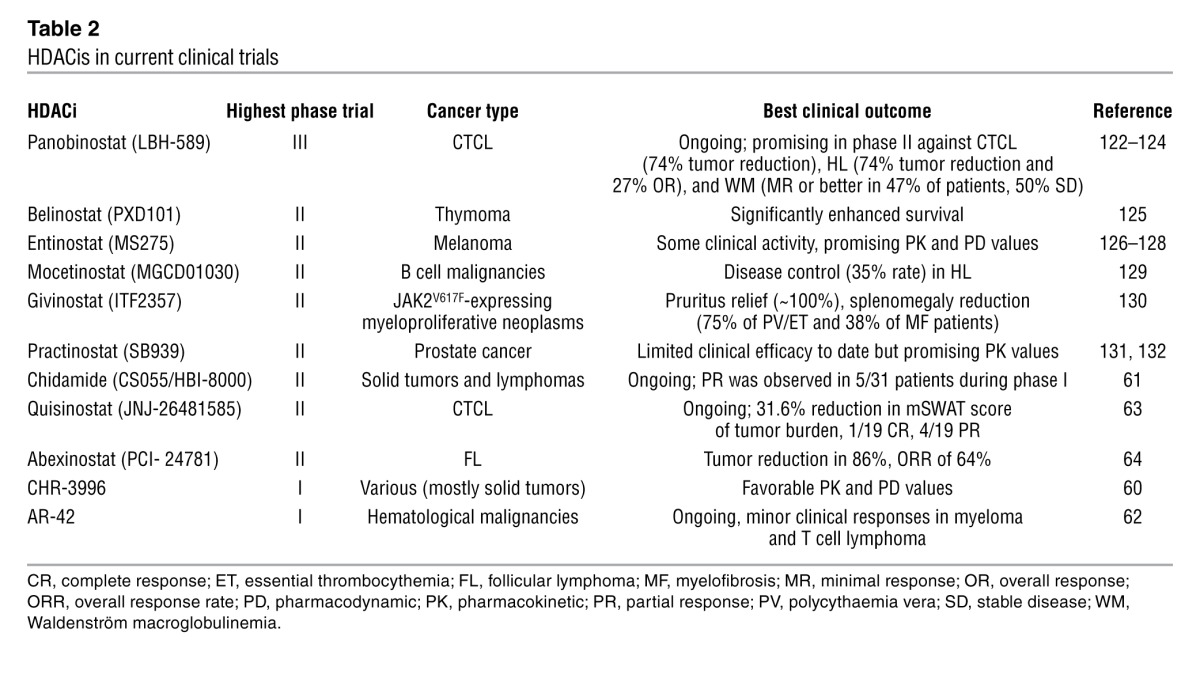
Figure 2. The specificity of HDACis for HDACs and associated protein complexes.

The specificity of HDACis for HDAC isoforms has been recently reclassified using sophisticated chemoproteomics and chemical phylogenetic approaches, revealing that HDACis have surprising affinity for both HDACs and individual HDAC-dependent multiprotein complexes (34, 35). This new specificity is shown for class I HDAC1, -2, -3, and -8 as well as class IIb HDAC6 and -10; specificity is calculated from average KDapp values generated by Bantscheff et al. and Bradner et al. (34, 35). Illustration based on data from Deubzer et al. (94), Bolden et al. (40), Bantscheff et al. (34), and Bradner et al. (35). BHC80, BRAF35-HDAC complex protein; CHD4, chromodomain helicase DNA binding protein 4; CoREST, REST corepressor 1; LSD1, lysine-specific demethylase 1; MTA, metastasis-associated protein; MBD, methyl-CpG binding domain protein; NuRD, nucleosome remodeling and deacetylase; RbAp46, retinoblastoma-binding protein p46; Sap30, Sin3A-associated protein, 30 kDa; SDS, serine dehydratase; TBL1X, transducin β-like 1X-linked; TBL1XR1, transducin β-like 1 X-linked receptor 1; GPS2, G protein pathway suppressor 2.
HDACi mechanisms of action
HDACis can induce tumor cell apoptosis, growth arrest, senescence, differentiation, and immunogenicity, and inhibit angiogenesis, as depicted in Figure 1. It is almost impossible to conceive that a single mechanism of action will be identified that is responsible for antitumor effects across different tumor types mediated by HDACis with diverse specificity profiles. Almost certainly, the biological effects and therapeutic outcomes will depend on the genetic lesions driving the tumor of interest and the HDACi under investigation. While HDACis were historically identified on the basis of their ability to induce tumor cell differentiation (38, 39), induction of tumor cell apoptosis is the biological outcome most often reported (40). Indeed, using preclinical models, we and others have identified a direct link between HDACi-induced tumor cell apoptosis and therapeutic efficacy (22, 41–43). However, even here, divergent views exist regarding the importance of the intrinsic apoptotic pathway mediated by the interplay between proapoptotic and antiapoptotic Bcl-2 family proteins (41, 42, 44), versus the extrinsic pathway mediated by death receptors (e.g., TRAIL receptors, Fas) and their cognate ligands (e.g., TRAIL, FasL) (21, 22). There is a link between altered gene expression and the induction of apoptosis, with histone hyperacetylation observed at promoters of apoptosis-inducing genes such as TNFSF10 (encoding TRAIL; ref. 22) and BMF (encoding the proapoptotic Bcl-2 family member Bmf; ref. 45), and changes in activity of transcription factors due to acetylation, such as inhibition of SP1 and C/EBPα, leading to downregulation of the antiapoptotic protein Bcl-2 (18), following HDACi treatment. It is therefore likely that both the threshold of apoptosis induction and the mechanism by which death can be triggered by HDACis is determined by the interaction between the oncogenic lesions and the intrinsic apoptosis-signaling pathways active within each cell.
Two reports have provided potential insights into biomarkers for response to HDACis. The first demonstrated that enhanced JAK/STAT signaling negatively affected HDACi-induced death of CTCL cells, and constitutive accumulation of STAT1 in the nucleus and high levels of phosphorylated STAT3 correlated with a lack of response to vorinostat in clinical trials (46). The second approach incorporated a sophisticated loss-of-function genetic screen that identified human RAD23 homolog B (HR23B) as important for HDACi-induced apoptosis (47). Subsequent studies showed a correlation between HR23B expression and clinical response to vorinostat (48), and the interaction between Hsp90 and HDAC6 was identified as being the crucial functional effector mechanism delineating relative sensitivity to HDACi-induced apoptosis through regulated HR23B expression (49). Whether there is any functional interplay between JAK/STAT signaling and HR23B remains uncertain. Moreover, as HR23B expression can regulate sensitivity to HDACis that are very weak inhibitors of HDAC6 (e.g., apicidin, romidepsin) (48), it is unlikely that direct effects on the HDAC6/Hsp90 functional interaction account for the mechanistic role of HR23B in regulating HDACi-induced apoptosis.
How or why tumor cells are more sensitive to HDACi-induced apoptosis compared with matched, normal cells remains an intriguing and largely unanswered question. Previous studies indicated that tumor cells treated with HDACis preferentially accumulate ROS compared with treated normal cells, concomitant with enhanced expression of the reducing molecule thioredoxin in normal but not tumor cells (50). Recently, we utilized donor-matched normal and transformed cells treated with vorinostat to identify a tumor cell–selective, proapoptotic gene expression signature containing effectors of the intrinsic apoptotic pathway that conferred tumor cell–selective apoptosis mediated by vorinostat and romidepsin (51). It is not clear why matched tumor and normal cells selectively regulate a subset of genes that confer tumor cell–selective, HDACi-mediated apoptosis. It is tempting to speculate that the cancer epigenome is altered in such a way as to predispose to altered expression of apoptotic genes in response to the transformation process, but definitive mechanistic evidence remains to be obtained.
HDACis in the clinic
The most successful clinical application of HDACis has been the use of vorinostat and romidepsin against refractory cutaneous and peripheral T cell lymphoma (52–55). In addition to these two FDA-approved agents, the butyrates, valproic acid, and newer compounds such as panobinostat (LBH589), givinostat (ITF2357), mocetinostat (MGCD01030), belinostat (PXD101), pracinostat (SB939), and entinostat (MS275) have been extensively studied in the clinic with varying results (56–58). Although some of these agents often demonstrate more potent antitumor effects than vorinostat and romidepsin in preclinical testing, none have thus far demonstrated novel mechanism(s) of action, vastly superior clinical activity, or more favorable toxicity profiles, and accordingly none have yet been registered for clinical use (56–58). While toxicities are largely manageable, in some instances cardiotoxicities as well as constitutional, hematologic, and gastrointestinal effects can be dose limiting (59). The efficacy of more than 20 different HDACis tested in the clinic has been largely restricted to hematological malignancies, with positive effects observed in Hodgkin lymphoma, multiple myeloma, and AML (some examples of these trials are listed in Table 2 and ref. 56). Despite positive responses in patients with hematologic malignancies, the effects of HDACis used as monotherapies against solid tumors has been disappointing, with few durable responses observed (56, 58). Current and future clinical studies using HDACis in combination with other agents, in particular with agents like bortezomib (velcade) in myeloma and other hematological malignancies, are ongoing (56, 57).
A second generation of orally available HDACis have been developed based on the chemical structure of clinically efficacious agents such as the hydroxamic acids (vorinostat) and the benzamides (entinostat, mocetinostat). Of these, several have entered the clinic including the class I–specific agents CHR-3966 (60), chidamide (CS055/HBI-8000) (61), class I– and class II–specific AR-42 (62), and hydroxamides quisinostat (JNJ-26481585) (63) and abexinostat (PCI- 24781) (64), and some preliminary results are described in Table 2. While still ongoing, early preclinical studies indicate these agents are more potent than the parental compounds, with improved pharmacodynamic and pharmacokinetic values, and are potentially less toxic. However, given that these new agents possess the same HDAC specificity profile (based on both chemoproteomic approaches [summarized in Figure 2] and traditional enzyme-based assays) as HDACis already in the clinic, it is unclear whether these newer agents will have improved clinical outcomes. The potency and reasonable toxicity profile of these agents suggests they may instead be useful in combinatorial therapeutic approaches.
Novel and emerging HDACis in preclinical development
Class I HDACis.
HDAC1, -2, and -3 are frequently overexpressed in human tumors, and knockdown of HDAC1 or -2 is sufficient to reduce tumor growth in vivo (see Table 1 for important tumor-specific examples). These studies provide a strong rationale for the development of class I– or isoform-specific inhibitors for the treatment of cancer. The recruitment of HDAC1-, HDAC2-, and/or HDAC3-containing transcription regulatory complexes to leukemogenic fusion proteins indicates class I–specific agents may be particularly suited to the treatment of tumors driven by discrete oncogenic proteins. The pharmacologic development of class I–specific or isoform-specific HDACis is currently underway, aided by clarification of the HDAC2 catalytic site following cocrystallization (65). Chemical modeling based on the HDAC2 crystal structure has allowed the generation of a series HDAC2-specific benzamides, and the inclusion of a sulfhydryl group may allow specific inhibition of HDAC1 (65, 66). Given the close sequence homology and observed functional redundancy mechanisms and compensatory expression relationships that occur between HDAC1 and -2, it will be of interest to determine whether these compounds confer different antitumor effects.
HDAC3 is emerging as a strong candidate for selective pharmacologic target inhibition in the oncology setting. The HDAC3-dependent NCoR complex can be recruited by the oncogenic fusion proteins PML-RARα, PLZF-RARα, or AML1-ETO in AML (19, 67). Furthermore, HDAC3 is required for the expression of almost half of the LPS-induced inflammatory genes in macrophages, and STAT1 levels were reduced in Hdac3–/– macrophages (68), suggesting a possible link. Conceivably, HDAC3 could act as the primary STAT1 and STAT3 deacetylase, which regulates the constitutive expression, deacetylation, and subsequent activation of STAT proteins in certain cancer types, a phenotype associated with resistance to HDACis (46). Other clues that suggest HDAC3 is an attractive target come from studies of Friedreich ataxia, a neurologic disorder caused by aberrant epigenetic silencing of the mutated frataxin (FXN) gene mediated by HDAC3. Treatment of mice bearing the Fxn mutation with an HDAC3-specific inhibitor, RG2833, resulted in decreased disease progression and enhanced survival, findings that led to the instigation of phase I trials (69, 70). It will be of interest to examine the efficacy of RG2833 in patients with cancer, especially in comparison to HDACis with high specificity for HDAC3 and NCoR, such as entinostat and mocetinostat (as represented in Figure 2 and ref. 34). Additionally, it will be important to determine whether patients develop immune-mediated complications, given the role of HDAC3 in inflammation (68). Moreover, the recent identification of putative tumor suppressor roles for HDAC1, -2, and -3 (25–27) raises some concerns about the clinical use of highly selective compounds that target these class I HDACs.
The role of HDAC8 in cancer is not as well established as other class I HDACs. HDAC8 has not been identified in any major corepressor complexes, and very few HDACis developed to date target HDAC8 with any potency (as depicted in Figure 2 and ref. 34). However, HDAC8 may have a particular role in solid tumors, as expression is enhanced in brain tumors, and knockdown revealed a role for maintaining the survival of lung, colon, and cervical cancer cells (Table 1 and refs. 50, 71). Thus, future studies are needed to determine whether inhibition of HDAC8 in solid tumors can enhance the traditionally suboptimal efficacy of HDACis in these malignancies (58). Solving the crystal structure of HDAC8 has assisted with in silico screening for appropriate chemical inhibitors (50, 72), the most promising of which is PCI-34051. Preclinical studies have suggested that PCI-34051 has activity against T cell malignancies, and while the mechanism is yet to be fully elucidated, HDAC8 may be linked to PLCγ1, a signal transducer specifically associated with the T cell receptor; together these proteins may induce apoptosis in T cell lymphomas (73). The development of PCI-34051 may allow for a better understanding of HDAC8 in cancer cells and can be utilized to identify any possible protein complexes containing HDAC8. It will be of interest to determine the potential effects of HDAC8 inhibition on normal T cells during treatment of solid malignancies. Assessment of PCI-34051 in T cell–mediated autoimmune diseases such as systemic lupus erythematosus (74) and colitis (75), in which pan-HDACis are currently undergoing investigation, may also be warranted.
Inhibitors of class II HDACs.
The normal function of class IIa HDACs appears to be highly tissue specific (as indicated in Table 1), and there is a growing understanding that class IIa HDACs have weak deacetylase activity and may therefore more likely function as histone readers. Knockdown of HDAC4, -5, -7, and -9 induced the growth arrest of a variety of human solid tumor cell lines, and the depletion of HDAC4 alone was sufficient to induce apoptosis in colon and neuroblastoma tumors in vivo (Table 1), suggesting an important role for these HDACs in maintaining the survival of tumor cells (76, 77). Key to the non-catalytic activities of class IIa HDACs may be their capacity to act as scaffold proteins, incorporating enzymatically active HDACs into multiprotein complexes. For example, HDAC4, -5, and -7 can recruit NCoR via recognition of the HDAC3 catalytic domain (78). Together these studies suggest there is a rationale for targeting class IIa HDACs as an anticancer approach. Ongoing research investigating the nonhistone protein targets of HDACs combined with the description of both HDAC4 (79) and HDAC7 (80) reader sites will likely allow for more detailed functional assessment of these proteins in cancer development. The recent discovery of trifluoromethyloxadiazole (TFMO), an agent with nanomolar potency against class II HDACs, will also enhance research in this area dramatically (81).
HDAC6 does not deacetylate histones, and accordingly, the anticancer effects of the HDAC6-specific inhibitors tubacin and rocilinostat (ACY-1215) are not associated with disrupted epigenetic control of gene transcription (82). A primary function of HDAC6 appears to be the regulation of protein degradation both via the aggresome (a structure that forms in response to misfolded proteins) and the regulation of Hsp90 chaperone activity. HDAC6 contains a ubiquitin-binding domain, unique among the HDACs, and tethers polyubiquitylated proteins to dynein motor for transport to the aggresome. Knockdown of HDAC6 led to the accumulation of polyubiquitylated, misfolded proteins due to lack of aggresome formation (83). HDAC6 also maintains activation of Hsp90 by deacetylation, and upon knockdown of HDAC6, Hsp90 becomes dissociated from its client proteins, leading to the ubiquitylation and degradation of immature, misfolded proteins (84). Several oncoproteins rely on Hsp90 for stability (e.g., Bcr-Abl, Her2); thus inhibition of HDAC6 can lead to their ubiquitin-mediated degradation, implicating HDAC6 as a promising anticancer target (85). However, inhibiting HDAC6 with tubacin was not sufficient to induce cell death (82), as protein aggregates can be degraded by the proteaseome. Indeed, the rational combination of tubacin or rocilinostat with the proteasome inhibitor bortezomib has been tested in preclinical models of multiple myeloma, with striking, synergistic anticancer effects observed (86, 87). The particular sensitivity of multiple myeloma to this combination therapy is thought to be due to the requirement of the malignant plasma cells to appropriately express and fold large amounts of immunoglobulin proteins; however, additional mechanisms are also likely to contribute (88). The combination of rocilinostat and the proteasome inhibitor bortezomib is currently under clinical investigation in multiple myeloma, and early results are promising (89). HDAC6 is responsible for α-tubulin deacetylation (90), thus providing a sensitive biomarker for rocilinostat activity during clinical trials.
Hybrid molecules targeting HDACs and other oncogenic proteins and pathways
New medicinal chemistry approaches are being utilized to design more potent and specific inhibitors of class I HDACs. Compounds utilizing novel chemical scaffolds such as adamantane and noradamantane have been shown to inhibit HDACs at picomolar concentrations, and to inhibit tumor cell growth with limited toxicities in vivo at low nanomolar ranges (91). Chemical hybrid molecules containing both HDACi activity and an additional anticancer module are under development, and while the HDACi component is relatively broad spectrum, targeting HDACs concurrent with inhibition of PI3K (CUDC-907), tyrosine kinase (CUDC-101), or topoisomerase II (daunorubicin-vorinostat), or with 1α,25-vitamin D (triciferol) for nuclear receptor targeting (92), may be of interest for particular cancer subtypes. Additionally, an ester-HDACi hybrid has been developed consisting of an HDACi and an esterase-sensitive chemical motif, which is specifically hydrolyzed by human monocytes and macrophages, trapping the active drug inside the cell (93). While this technology is currently useful for the inhibition of HDAC-regulated inflammatory cytokines and the treatment of inflammatory diseases, it also provides an impetus to utilize the hybrid technology to develop more cell type–specific HDACis for use in cancer.
Conclusions
The view that genetic lesions resulting in epigenetic deregulation is a major driver of tumor onset and progression is rapidly gaining acceptance by oncologists and cancer researchers. The development of HDACis as anticancer agents and the FDA approval of two agents, vorinostat and romidepsin, have provided the impetus to develop more potent HDACis and to target other epigenetic enzymes for oncology. While existing HDACis with somewhat broad target specificity have proven effective for the treatment of a relatively small population of patients with defined hematologic malignancies, their usefulness as single agents for the treatment of most other tumors has been marginal at best. Correlative data linking HDAC expression with patient prognosis and gene knockout as well as knockdown studies in preclinical mouse models of cancer are providing further clues as to the oncogenic and/or tumor suppressor role of HDACs. Furthermore, these studies should lead to more sophisticated, evidenced-based development of HDACis with greater target specificity. Whether or not these agents will deliver significantly enhanced therapeutic efficacy and less toxicity as hypothesized remains to be determined.
Acknowledgments
R.W. Johnstone was supported by a National Health and Medical Research Council (NHMRC) Principal Research Fellowship and NHMRC program and project grants and by the Cancer Council Victoria (CCV), the Leukaemia Foundation of Australia, the Victorian Breast Cancer Research Consortium, and the Victorian Cancer Agency. A.C. West was supported by a CCV Fellowship.
Footnotes
Conflict of interest: The Johnstone laboratory receives grant support from Novartis.
Citation for this article: J Clin Invest. 2014;124(1):30–39. doi:10.1172/JCI69738.
References
- 1.Olsen CA. Expansion of the lysine acylation landscape. Angew Chem Int Ed Engl. 2012;51(16):3755–3756. doi: 10.1002/anie.201200316. [DOI] [PubMed] [Google Scholar]
- 2.Tan MJ, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1015–1027. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41(1):185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 4.Fraga MF, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 5.Ozdag H, et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 2006;7:90. doi: 10.1186/1471-2164-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weichert W, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98(3):604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weichert W, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14(6):1669–1677. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 8.Krusche CA, et al. Histone deacetylase-1 and-3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat. 2005;90(1):15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 9.Minamiya Y, et al. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer. 2011;74(2):300–304. doi: 10.1016/j.lungcan.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 10.Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004;112(1):26–32. doi: 10.1002/ijc.20395. [DOI] [PubMed] [Google Scholar]
- 11.Rikimaru T, et al. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology. 2007;72(1–2):69–74. doi: 10.1159/000111106. [DOI] [PubMed] [Google Scholar]
- 12.Weichert W, et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 2008;9(2):139–148. doi: 10.1016/S1470-2045(08)70004-4. [DOI] [PubMed] [Google Scholar]
- 13.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 14.Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett. 2009;274(2):169–176. doi: 10.1016/j.canlet.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Saji S, et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005;24(28):4531–4539. doi: 10.1038/sj.onc.1208646. [DOI] [PubMed] [Google Scholar]
- 16.Marquard L, Gjerdrum LM, Christensen IJ, Jensen PB, Sehested M, Ralfkiaer E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology. 2008;53(3):267–277. doi: 10.1111/j.1365-2559.2008.03109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo JY, Su F, Chen DL, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408(6810):377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 18.Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005;25(5):1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelmetti V, Zhang JS, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18(12):7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin RJ, Nagy L, Inoue S, Shao WL, Miller WH, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391(6669):811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 21.Insinga A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2004;11(1):71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 22.Nebbioso A, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11(1):77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 23.Taylor BS, et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov. 2011;1(7):587–597. doi: 10.1158/2159-8290.CD-11-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ropero S, et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet. 2006;38(5):566–569. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- 25.Bhaskara S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18(5):436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heideman MR, et al. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood. 2013;121(11):2038–2050. doi: 10.1182/blood-2012-08-450916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santoro F, et al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121(17):3459–3468. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]
- 28.Lin KT, Wang YW, Chen CT, Ho CM, Su WH, Jou YS. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin Cancer Res. 2012;18(17):4691–4701. doi: 10.1158/1078-0432.CCR-12-0633. [DOI] [PubMed] [Google Scholar]
- 29.Pulukuri SMK, Gorantla B, Rao JS. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J Biol Chem. 2007;282(49):35594–35603. doi: 10.1074/jbc.M705867200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Vojinovic J, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63(5):1452–1458. doi: 10.1002/art.30238. [DOI] [PubMed] [Google Scholar]
- 31.Bonfils C, et al. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin Cancer Res. 2008;14(11):3441–3449. doi: 10.1158/1078-0432.CCR-07-4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21(18):6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13(15):1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bantscheff M, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29(3):255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 35.Bradner JE, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6(3):238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deardorff MA, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489(7415):313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott GK, et al. Destabilization of ERBB2 transcripts by targeting 3′untranslated region messenger RNA associated HuR and histone deacetylase-6. Mol Cancer Res. 2008;6(7):1250–1258. doi: 10.1158/1541-7786.MCR-07-2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leder A, Leder P. Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell. 1975;5(3):319–322. doi: 10.1016/0092-8674(75)90107-5. [DOI] [PubMed] [Google Scholar]
- 39.Riggs MG, Whittaker RG, Neumann JR, Ingram VM. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature. 1977;268(5619):462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 40.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 41.Ellis L, et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. 2009;114(2):380–393. doi: 10.1182/blood-2008-10-182758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindemann RK, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A. 2007;104(19):8071–8076. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newbold A, Lindemann RK, Cluse LA, Whitecross KF, Dear AE, Johnstone RW. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Ther. 2008;7(5):1066–1079. doi: 10.1158/1535-7163.MCT-07-2256. [DOI] [PubMed] [Google Scholar]
- 44.Vrana JA, et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18(50):7016–7025. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Adachi M, Kawamura R, Imai K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2005;13(1):129–140. doi: 10.1038/sj.cdd.4401686. [DOI] [PubMed] [Google Scholar]
- 46.Fantin VR, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785–3794. doi: 10.1158/0008-5472.CAN-07-6091. [DOI] [PubMed] [Google Scholar]
- 47.Fotheringham S, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15(1):57–66. doi: 10.1016/j.ccr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Khan O, et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci U S A. 2010;107(14):6532–6537. doi: 10.1073/pnas.0913912107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.New M, et al. A regulatory circuit that involves HR23B and HDAC6 governs the biological response to HDAC inhibitors. Cell Death Differ. 2013;20(10):1306–1316. doi: 10.1038/cdd.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vannini A, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101(42):15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolden JE, et al. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013;4:e519. doi: 10.1038/cddis.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duvic M, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olsen EA, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 54.Whittaker SJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 55.Piekarz RL, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: An update. Mol Oncol. 2012;6(6):657–682. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6(6):637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu TZ, et al. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013;9(2):255–269. doi: 10.2217/fon.12.173. [DOI] [PubMed] [Google Scholar]
- 59.Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4(4):505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banerji U, et al. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin Cancer Res. 2012;18(9):2687–2694. doi: 10.1158/1078-0432.CCR-11-3165. [DOI] [PubMed] [Google Scholar]
- 61.Dong M, et al. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother Pharmacol. 2012;69(6):1413–1422. doi: 10.1007/s00280-012-1847-5. [DOI] [PubMed] [Google Scholar]
- 62. Hofmeister CC, et al. Phase I study of AR-42 in relapsed multiple myeloma and lymphoma. Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, Georgia, USA. https://ash.confex.com/ash/2012/webprogram/Paper47235.html . Accessed November 15, 2013.
- 63. Child F, et al. Phase 2 Multicenter trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA cutaneous T-cell lymphoma. Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, Georgia, USA. https://ash.confex.com/ash/2012/webprogram/Paper46292.html . Accessed November 15, 2013.
- 64. Evens AM, et al. A phase II multicenter study of the histone deacetylase inhibitor (HDACi) abexinostat (PCI-24781) in relapsed/refractory follicular lymphoma (FL) and mantle cell lymphoma (MCL). Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, Georgia, USA. https://ash.confex.com/ash/2012/webprogram/Paper50038.html . Accessed November 15, 2013
- 65.Bressi JC, et al. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett. 2010;20(10):3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 66.Zhang L, Li MY, Zhang LH, Xu WF. Discovering the binding modes of natural products with histone deacetylase 1. Med Chem. 2013;9(1):126–132. doi: 10.2174/157340613804488314. [DOI] [PubMed] [Google Scholar]
- 67.Grignani F, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391(6669):815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 68.Chen XF, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109(42):E2865–E2874. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sandi C, et al. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol Dis. 2011;42(3):496–505. doi: 10.1016/j.nbd.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soragni E, Xu CP, Plasterer HL, Jacques V, Rusche JR, Gottesfeld JM. Rationale for the development of 2-aminobenzamide histone deacetylase inhibitors as therapeutics for Friedreich ataxia. J Child Neurol. 2012;27(9):1164–1173. doi: 10.1177/0883073812448533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oehme I, et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009;15(1):91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 72.Somoza JR, et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12(7):1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 73.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22(5):1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 74.Reilly CM, et al. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J Immunol. 2004;173(6):4171–4178. doi: 10.4049/jimmunol.173.6.4171. [DOI] [PubMed] [Google Scholar]
- 75.Tao R, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13(11):1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 76.Wilson AJ, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19(10):4062–4075. doi: 10.1091/mbc.E08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mottet D, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28(2):243–256. doi: 10.1038/onc.2008.371. [DOI] [PubMed] [Google Scholar]
- 78.Fischle W, et al. Enzymatic activity associated with class IIHDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9(1):45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 79.Bottomley MJ, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283(39):26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schuetz A, et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283(17):11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lobera M, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9(5):319–325. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- 82.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100(8):4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115(6):727–738. doi: 10.1016/S0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 84.Kovacs JJ, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 85.Bali P, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90 — a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280(29):26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 86.Santo L, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hideshima T, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102(24):8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011;10(11):2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Raje N, et al. Rocilinostat (ACY-1215), a selective HDAC6 inhibitor, alone and in combination with bortezomib in multiple myeloma: Preliminary results from the first-in-humans phase I/II study. Presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, Georgia, USA. https://ash.confex.com/ash/2012/webprogram/Paper52013.html . Accessed November 15, 2013. [Google Scholar]
- 90.Hubbert C, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 91.Gopalan B, et al. Discovery of adamantane based highly potent HDAC inhibitors. Bioorg Med Chem Lett. 2013;23(9):2532–2537. doi: 10.1016/j.bmcl.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 92.Delcuve GP, Khan DH, Davie JR. Targeting class I histone deacetylases in cancer therapy. Expert Opin Ther Targets. 2013;17(1):29–41. doi: 10.1517/14728222.2013.729042. [DOI] [PubMed] [Google Scholar]
- 93.Needham LA, et al. Drug targeting to monocytes and macrophages using esterase-sensitive chemical motifs. J Pharmacol Exp Ther. 2011;339(1):132–142. doi: 10.1124/jpet.111.183640. [DOI] [PubMed] [Google Scholar]
- 94.Deubzer HE, et al. HDAC11 is a novel drug target in carcinomas. Int J Cancer. 2013;132(9):2200–2208. doi: 10.1002/ijc.27876. [DOI] [PubMed] [Google Scholar]
- 95.Zhang ZH, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res Treat. 2005;94(1):11–16. doi: 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- 96.Adams H, Fritzsche FR, Dirnhofer S, Kristiansen G, Tzankov A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin Ther Targets. 2010;14(6):577–584. doi: 10.1517/14728221003796609. [DOI] [PubMed] [Google Scholar]
- 97.Hiebert SW, et al. Mechanisms of transcriptional repression by the t(8;21)-, t(12;21)-, and inv(16)-encoded fusion proteins. Cancer Chemother Pharmacol. 2001;48(suppl 1):S31–S34. doi: 10.1007/s002800100302. [DOI] [PubMed] [Google Scholar]
- 98.David G, Alland L, Hong SH, Wong CW, DePinho RA, Dejean A. Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein. Oncogene. 1998;16(19):2549–2556. doi: 10.1038/sj.onc.1202043. [DOI] [PubMed] [Google Scholar]
- 99.Dhordain P, et al. Corepressor SMRT binds the BTB/POZ repressing domain of the LAZ3/BCL6 oncoprotein. Proc Natl Acad Sci U S A. 1997;94(20):10762–10767. doi: 10.1073/pnas.94.20.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson AJ, et al. Histone deacetylase 3 (HDAC3) and other class IHDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281(19):13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 101.Yamaguchi T, et al. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24(5):455–469. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Santoro F, et al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121(17):3459–3468. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]
- 103.Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN. Genetic dissection of histone deacetylase requirement in tumor cells. Proc Natl Acad Sci U S A. 2009;106(19):7751–7755. doi: 10.1073/pnas.0903139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Senese S, et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27(13):4784–4795. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 106.Wilting RH, et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29(15):2586–2597. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Song J, et al. Increased expression of historic deacetylase 2 is found in human gastric cancer. Apmis. 2005;113(4):264–268. doi: 10.1111/j.1600-0463.2005.apm_04.x. [DOI] [PubMed] [Google Scholar]
- 108.Jung KH, et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J Cell Biochem. 2012;113(6):2167–2177. doi: 10.1002/jcb.24090. [DOI] [PubMed] [Google Scholar]
- 109.Harms KL, Chen XB. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67(7):3145–3152. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 110.Moreno DA, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;150(6):665–673. doi: 10.1111/j.1365-2141.2010.08301.x. [DOI] [PubMed] [Google Scholar]
- 111.Hong SH, David G, Wong CW, Dejean A, Privalsky ML. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor α (RAR α) and PLZF-RAR α oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1997;94(17):9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kramer OH, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009;23(2):223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Atsumi A, Tomita A, Kiyoi H, Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RAR α as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem Biophys Res Commun. 2006;345(4):1471–1480. doi: 10.1016/j.bbrc.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 114.Sun XJ, Wei L, Chen Q, Terek RM. HDAC4 represses vascular endothelial growth factor expression in chondrosarcoma by modulating RUNX2 activity. J Biol Chem. 2009;284(33):21881–21890. doi: 10.1074/jbc.M109.019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Milde T, et al. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 2010;16(12):3240–3252. doi: 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- 116.Malik S, et al. Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol Cell Biol. 2010;30(2):399–412. doi: 10.1128/MCB.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kotian S, Liyanarachchi S, Zelent A, Parvin JD. Histone deacetylases 9 and 10 are required for homologous recombination. J Biol Chem. 2011;286(10):7722–7726. doi: 10.1074/jbc.C110.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang ZH, et al. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10(20):6962–6968. doi: 10.1158/1078-0432.CCR-04-0455. [DOI] [PubMed] [Google Scholar]
- 119.Park JH, et al. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun. 2008;368(2):318–322. doi: 10.1016/j.bbrc.2008.01.056. [DOI] [PubMed] [Google Scholar]
- 120.Rhodes DR, et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002;277(28):25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 122.Duvic M, et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: Results of a phase II trial. Eur J Cancer. 2013;49(2):386–394. doi: 10.1016/j.ejca.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 123. Ghobrial IM, et al. Final results of the phase II Trial of single agent panobinostat (LBH589) in relapsed or relapsed/refractory Waldenstrom macroglobulinemia. Paper presented at: 53rd ASH Annual Meeting and Exposition; December 10–13, 2011; San Diego, California, USA. https://ash.confex.com/ash/2011/webprogram/Paper39751.html . Accessed November 15, 2013. [Google Scholar]
- 124.Younes A, et al. Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30(18):2197–2203. doi: 10.1200/JCO.2011.38.1350. [DOI] [PubMed] [Google Scholar]
- 125.Giaccone G, et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J Clin Oncol. 2011;29(15):2052–2059. doi: 10.1200/JCO.2010.32.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gore L, et al. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14(14):4517–4525. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hauschild A, et al. Multicenter phase II trial of the historic deacetylase inhibitor pyridylmethyl-N-{4- (2-aminophenyl)-carbamoyl -benzyl}-carbamate in pretreated metastatic melanoma. Melanoma Res. 2008;18(4):274–278. doi: 10.1097/CMR.0b013e328307c248. [DOI] [PubMed] [Google Scholar]
- 128.Ryan QC, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23(17):3912–3922. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 129.Younes A, et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12(13):1222–1228. doi: 10.1016/S1470-2045(11)70265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rambaldi A, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150(4):446–455. doi: 10.1111/j.1365-2141.2010.08266.x. [DOI] [PubMed] [Google Scholar]
- 131.Razak ARA, et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br J Cancer. 2011;104(5):756–762. doi: 10.1038/bjc.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yong WP, et al. Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, SB939, in patients with refractory solid malignancies. Ann Oncol. 2011;22(11):2516–2522. doi: 10.1093/annonc/mdq784. [DOI] [PubMed] [Google Scholar]