

Abstract
The term epigenetics refers to the heritable changes in gene expression that are not associated with a change in the actual DNA sequence. Epigenetic dysregulation is linked to the pathogenesis of a number of malignancies and has been studied extensively in myelodysplastic syndromes and acute myeloid leukemia. DNA methylation is frequently altered in cancerous cells and likely results in transcriptional silencing of tumor suppressor genes. Re-expression of these genes by inhibition of the DNA methyltransferases has been successful in the treatment of benign and malignant disease. In this Review, we discuss the clinical development of demethylating agents in hematology, with a focus on azacitidine and decitabine.
Introduction
Post-mitotic modification of DNA methylation is a key epigenetic mechanism. This is catalyzed by the enzyme DNA methyltransferase (DNMT), which adds a methyl group to position 5 of the cytosine ring in the CpG dinucleotide. The parental pattern serves as the template for the addition of methyl groups to newly synthesized DNA. The CpG dinucleotides are not evenly distributed in the genome, and they frequently cluster in the promoter regions of genes (1). The CpG-rich promoter regions of active genes are often demethylated, allowing for the binding of transcription factors and gene transcription. Methylation of the CpG islands in the promoter is associated with epigenetic gene silencing, which is involved in control of cell differentiation, imprinting, and X chromosome inactivation (2).
The mechanisms controlling methylation can become dysregulated in cancer. Studies in human tumors have demonstrated differences in DNA methylation between normal and cancerous tissue (1, 2). Genes involved in important regulatory pathways including cell cycle regulation, DNA repair, and tumor suppression are hypermethylated in cancerous cells and therefore transcriptionally silenced. The mechanism that leads to aberrant DNA methylation is not fully understood, but the recent discovery of mutations involving some of the genes regulating methylation, specifically the DNMTs, isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), and the ten-eleven translocation (TET) family of proteins, may help to elucidate some of these mechanisms.
Reversal of epigenetic changes and thus gene reactivation became a therapeutic target. Azanucleoside drugs, which act as DNMT inhibitors, were developed particularly in the myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). In this Review we discuss the clinical development of hypomethylating agents (HMAs) in hematology, with a focus on azacitidine and decitabine, the clinical data that led to their approval by the FDA, and future developments in the field.
Azanucleosides
The azanucleosides are pyrimidine analogs that inhibit DNMTs. These include 5-aza-2′-deoxycytidine (decitabine) and 5-azacytidine (azacitidine). Decitabine is incorporated into newly synthesized DNA strands, and azacitidine is predominantly incorporated into RNA. Part of the 5-azacytidine molecule is incorporated into DNA after conversion to 5-aza-2′-deoxycytidine. This leads to a marked decrease in DNMT activity, which may be the catalyst that allows differentiation to proceed. This is important in disease states such as MDS and AML, in which immature myeloid cells are often arrested in early forms of development (Figure 1).
Figure 1. Biological mechanism of action of azacitidine (AZA) and decitabine (DAC).

(A) Structures of cytidine, azacitidine, and decitabine. (B) DNMTs methylate cytidine. Methylation of cytidine in gene promoter regions blocks the binding of transcription factors, leading to epigenetic silencing. DNMTs aberrantly methylate tumor suppressor genes in hematological malignancies and other cancers. (C) Azacitidine is converted to decitabine and is then incorporated into DNA in place of cytidine during replication. Both azacitidine and decitabine inactivate DNMTs, preventing gene methylation. The resultant hypomethylation leads to the transcription of previously silenced genes. Notably, azacitidine, but not decitabine, can be incorporated directly into RNA, inhibiting protein synthesis.
The development of azanucleosides in benign hematology.
The hemoglobinopathies are inherited, single-gene disorders that result in an abnormal structure of one of the globin chains of the hemoglobin molecule, often resulting in anemia. Demethylating agents have been used in the treatment of hemoglobinopathies to target the β-globin gene, with a resultant rise in fetal hemoglobin. A simultaneous improvement in erythropoiesis and transient increase in hemoglobin concentration were demonstrated with azacitidine treatment of a patient with homozygous β-thalassemia, and hypomethylation of bone marrow DNA was observed near the γ globin chain region (3). Repeated courses of treatment were necessary to maintain increased levels of fetal hemoglobin, and a change in DNA methylation typically occurred after seven days of treatment, which correlated with the onset of increased transcription of γ globin chains and the rise in fetal hemoglobin.
Decitabine has also been studied in the treatment of hemoglobinopathies. Saunthararajah and colleagues used decitabine in four patients with sickle cell disease in deteriorating condition. Decitabine rapidly increased total hemoglobin in all patients, with maximum hemoglobin increases of 35–50 g/l achieved within eight weeks (4). Olivieri and colleagues also studied decitabine in five patients with β-thalassemia intermedia and found that treatment increased both total and absolute fetal hemoglobin (5)
Azanucleosides in the MDSs
Azacitidine.
Azacitidine has been extensively studied in MDS, a clonal hematopoietic stem cell disorder that affects cell growth and differentiation. In addition to the ability to inhibit DNMTs, azacitidine likely is also involved in tumor cell apoptosis and acts as a biologic response modifier. Research to identify mechanisms mediating these effects is underway (6).
In a trial conducted in the Cancer and Leukemia Group B (CALGB), azacitidine was administered at 75 mg/m2/day using continuous i.v. infusion for seven days every four weeks. Responses were identified in 21 (49%) of 43 evaluable patients (7). Subsequently, azacitidine was administered at the same dose and schedule in an outpatient setting as a s.c. injection and demonstrated similar efficacy and toxicity profiles (8). This led to a phase III study comparing azacitidine to supportive care (SC) by the CALGB (9).
A total of 191 patients were randomized to azacitidine or SC. Patients in the SC arm whose disease progressed were permitted to cross over to the azacitidine arm (Table 1). The results of this phase III study demonstrated significantly higher response rates, reduced risk of leukemic transformation, improved quality of life (QoL), and in a landmark analysis, improved survival for patients treated initially with azacitidine compared with SC. Improvement in QoL was statistically significant in the categories of fatigue, physical functioning, dyspnea, psychosocial distress, and positive affect (10). The data from this study led to azacitidine becoming the first FDA-approved drug for the treatment of MDS in 2004.
Table 1.
Phase III clinical trials using HMAs in MDS
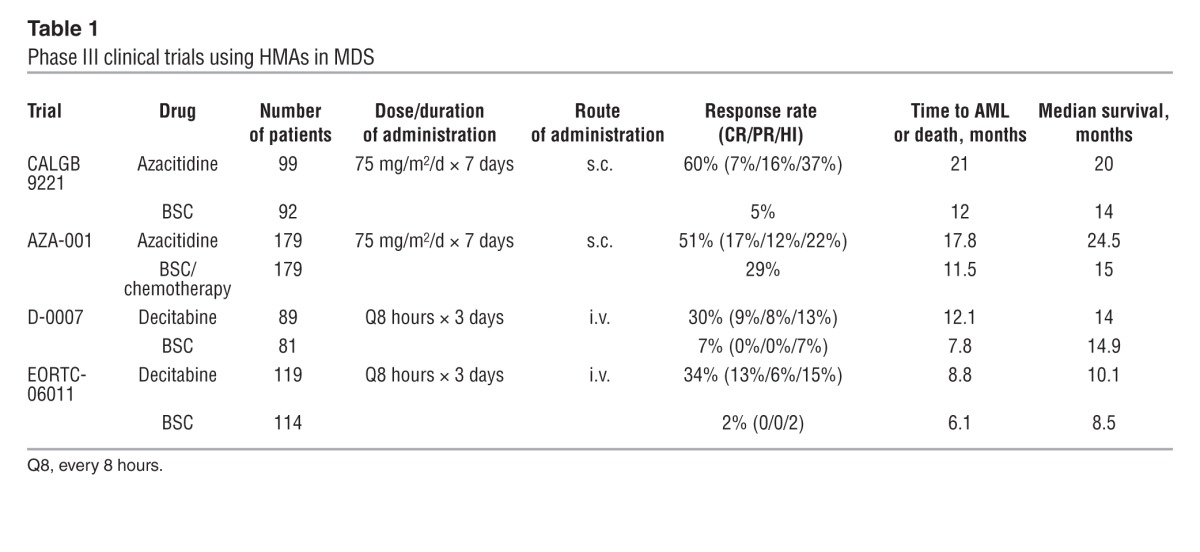
Time to AML or death was significantly delayed for the azacitidine group compared with SC, and transformation to AML occurred as the first event in 15% of patients receiving azacitidine compared with 38% of those randomized to SC. This was the first trial to demonstrate that an agent could reduce or prevent leukemia. Among higher-risk patients, median survival was 19 months in those randomized to azacitidine, compared with 8 months for those receiving SC alone.
A confirmatory phase III, international, multicenter study (AZA-001) was conducted in higher-risk MDS patients to compare azacitidine with three conventional care regimens (CCRs), which included physician’s choice of best SC (BSC), low-dose cytarabine, or induction chemotherapy with anthracycline and cytarabine (11). The median overall survival in the azacitidine arm was 24.5 months compared with 15 months for the CCRs; this survival benefit was demonstrated in all cytogenetic risk groups. Maintenance therapy with prolonged administration of azacitidine is necessary to maximize the therapeutic effect, with 90% of patients who will respond demonstrating response by the end of the sixth treatment cycle (12). Another important observation was that complete remission (CR) is not required in order to achieve a survival advantage. Patients who achieved partial remission (PR) and histologic improvement (HI) demonstrated a survival benefit with azacitidine treatment (13). Patients with stable disease had a survival benefit compared with those with progression, but this was seen in those treated with azacitidine or CCR (14). MDS clones can persist in patients who respond to treatment, suggesting that azacitidine can modulate the function of the cytogenetically abnormal clone and act as a biologic response modifier (15).
The development of an oral formulation of azacitidine is ongoing. In early studies, the oral form demonstrated both biologic and clinical activity in patients with MDS and chronic myelomonocytic leukemia (CMML). Both the oral and subcutaneous forms provided maximal DNA hypomethylation at day 15, with a gradual return of methylation levels near baseline by the end of each cycle (16). Future studies of treatment with azacitidine in combination with other agents in patients with MDS are discussed in further detail below.
Decitabine
In 1981, Rivard and coworkers reported a potent antileukemic effect of high-dose decitabine in the treatment of children with acute leukemias (primarily acute lymphocytic leukemia) that were resistant to conventional therapy (17). Myelosuppression was the observed major toxic effect. Lower-dose decitabine regimens showed promising effects in several early studies. Pinto and colleagues reported an overall response rate (ORR) of 45% (15% CR, 30% PR) in a phase I/II trial of low-dose decitabine in elderly AML/MDS patients (18). In two phase II, multicenter studies of low-dose decitabine in elderly MDS patients, Wijermans and coworkers showed an ORR between 49% and 54% (19, 20). Notably, a significantly higher response rate of 64% was seen in patients with a high-risk International Prognostic Scoring System (IPSS) score (19, 20). Within these phase II trials, 61 patients had chromosomal abnormalities, of which 31% achieved a major cytogenetic response after a median of three treatment courses, which was associated with improved survival (21).
In the first phase III trial of decitabine, 170 patients with MDS (70% of whom had an IPSS score of intermediate-2 [int-2] or high-risk disease; median age 70 years) were randomized to receive either decitabine 15 mg/m2 i.v. for three hours every eight hours for three days, and repeated every six weeks, or BSC. The overall improvement rate in the decitabine arm was 30%, compared with 7% in the BSC arm. In patients with an IPSS score of int-2/high-risk, time to AML or death was significantly delayed. All treatment responders became rbc transfusion independent and platelet transfusion independent. QoL was statistically improved during decitabine treatment compared with the control treatment for global health status, fatigue, and dyspnea scores (22). Following the results of this trial, the FDA approved decitabine for the treatment of all MDS subtypes.
To determine the optimal hypomethylating dose for decitabine, a phase II trial was performed with three schedules of low-dose decitabine in patients with higher-risk MDS and CMML. Patients were randomized to one of three treatment schedules: 20 mg/m2 i.v. daily for five days, 20 mg/m2 s.c. for five days, and 10 mg/m2 i.v. for ten days. One-third (66%) of patients eligible for IPSS classification were int-2 or high risk; of these, 34% achieved CR and 73% had an objective response according to the new, modified International Working Group criteria. The five-day i.v. schedule produced a CR rate of 39% compared with 21% and 24% in the five-day s.c. and ten-day i.v. schedule, respectively, and was selected as optimal (23). In an update of the study, Kantarjian and colleagues reported an overall improvement rate of 70% (24). Steensma and colleagues and investigators in Argentina and South Korea investigated the same dose and schedule as the study above and demonstrated 32% and 35% ORRs, respectively (25, 26).
In a phase III, randomized intergroup trial in Europe (EORTC Leukemia Group and German MDS Study Group), decitabine was compared with BSC in the treatment of patients with high-risk MDS who were ineligible for intensive chemotherapy (27). Patients with a median age of 70 years and almost exclusively with MDS severity of IPSS int-2/high-risk were randomly assigned to receive either decitabine 15 mg/m2 i.v. over four hours, three times a day for three days in six-week cycles, or BSC. The ORR in the treatment arm was 34% versus 2% in the BSC group. The progression-free survival was significantly improved with decitabine, while the median overall survival was not significantly different (10.1 months with decitabine vs. 8.5 months with BSC). Decitabine treatment was also associated with improvement in QoL scores.
In a phase II trial of decitabine in patients with advanced CMML, an ORR of 38% and an overall survival of 48% at two years were reported (28). A multicenter trial of decitabine in 101 Korean patients with MDS and CMML demonstrated an ORR of 55% and a median overall survival of 17.7 months (29).
Azanucleosides in AML
Azacitidine is utilized as a therapeutic option for older patients with AML and has demonstrated efficacy in this subset of patients using the same dose and schedule as for MDS. Trial results are summarized in Table 2. Azacitidine at higher doses was studied in AML patients in the 1970s and 1980s without significant benefit. Reclassification of patients who were treated on the MDS CALGB trials revealed 103 patients with AML according to the WHO criteria (20%–30% myeloblasts). Responses of HI or better occurred in 35%–48% of patients. Median survival was 19.3 months for AML patients treated with azacitidine compared with 12.9 months for those receiving SC (8).
Table 2.
Phase II/III clinical trials using HMAs in AML
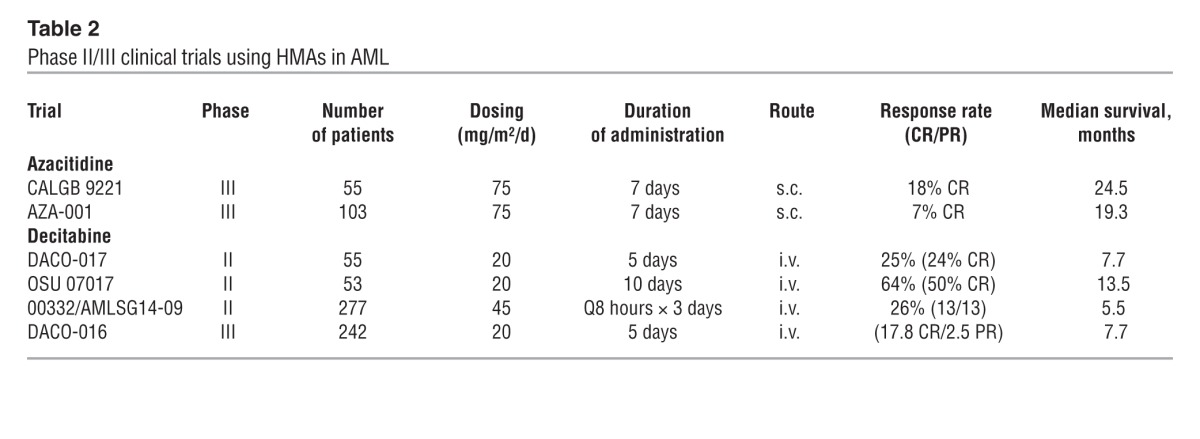
An analysis of 113 patients treated in the AZA-001 study who had 20%–30% blasts in the bone marrow and could also be classified as AML according to the WHO criteria revealed a median overall survival for azacitidine-treated patients of 24.5 months, compared with 16 months for CCRs. The two-year survival rate was 50%, compared with 16%. As in other studies described above, the median age of this patient population was 70 years (30).
In older adult patients with AML with low blast count, azacitidine treatment has resulted in increased overall survival and decreased hospitalizations compared with CCRs. Larger trials are underway to compare hypomethylating approaches to induction chemotherapy.
Ravandi and colleagues compared 5-azacytidine or decitabine treatment with intensive chemotherapy in patients with AML and high-risk MDS with chromosome 5 and 7 abnormalities; their results indicated that HMA may be superior to chemotherapy in patients with chromosomal abnormalities (31). Cashen and colleagues reported an ORR of 25% and a median overall survival of 7.7 months in a phase II, multicenter II trial of decitabine with older, treatment-naive AML patients (32). Another study revealed an ORR of 64% and a CR rate of 50% in AML patients with complex karyotype and 52% in patients with normal karyotype (33). In a phase II, multicenter study of decitabine as first-line therapy for older patients with AML, 26% of patients experienced CR, with a median overall survival of 5.5 months and one-year survival rate of 28% (34). Recently, Kantarjian and colleagues reported the results of a phase III, multicenter trial in older patients with newly diagnosed AML, which compared decitabine treatment to either BSC or low-dose cytarabine treatment (35) The decitabine arm showed a higher response rate compared with the cytarabine/BSC arm, and a subsequent analysis demonstrated an overall survival advantage (35). Although not approved for the treatment of AML in the US, decitabine was approved for this use in Europe based on the results of this study.
Decitabine has also been investigated as a bridge to allogeneic hematopoietic cell transplantation (alloHCT) in patients with MDS and AML and was found to be a feasible and effective strategy to control and downstage the disease in the time of donor search, and to improve the outcome of alloHCT (36, 37). Patients receiving azanucleoside induction therapy achieved rapid and high levels of donor chimerism following transplantation (38). Kim and colleagues compared decitabine to azacitidine in the transplant setting and found that decitabine-treated patients had higher rates of CR (66% vs. 20%) and significantly shorter time to engraftment (11.9 days vs. 16.6 days, P = 0.005) than azacitidine-treated patients (39).
For chronic myeloid leukemia, several studies of decitabine in imatinib-resistant patients have been performed. A phase II study by Issa and colleagues with 35 patients (12 in chronic phase, 17 in accelerated phase, and 6 in blastic phase) showed a complete hematologic response (CHR) in 34% and a partial hematologic response (PHR) in 20% of patients (40). Major cytogenetic responses were observed in 17% and minor cytogenetic responses in 29% (40). Oki and colleagues performed a phase II study of decitabine in combination with imatinib in 28 patients with chronic myeloid leukemia in accelerated phase and myeloid blastic phase (41). In this study, 25 patients were imatinib resistant. The investigators reported a CHR in 32%, PHR in 4%, and HI in 7% of patients (41).
Epigenetic agents in combination
The next logical step in the clinical development of HMAs is to use them in combination with other agents that may provide synergy. One such category of potentially synergistic drugs is the histone deacetylase inhibitors (HDACs). DNA methylation and the hypoacetylation of core nucleosomal histone proteins leads to the tight coiling of chromatin, thereby silencing the expression of a variety of genes, including those implicated in the regulation of cell survival, proliferation, differentiation, and apoptosis. HDAC inhibitors alone or in combination with HMAs restore expression of silenced genes by remodeling the tightly coiled chromatin, leading to the subsequent induction of differentiation, arrest in the progression of the cell cycle, or apoptosis.
A number of clinical trials have tested the combination of HMAs with HDAC inhibitors in both MDS and AML. HDAC inhibitors include valproic acid, vorinostat, etinostat, and mocetinostat. It appears that the synergistic effect is sequence dependent, requiring exposure to the HMA, followed by exposure to the HDAC inhibitor, which is modeled on in vitro systems (Table 3).
Table 3.
Experience with HMAs in combination in MDS/AML
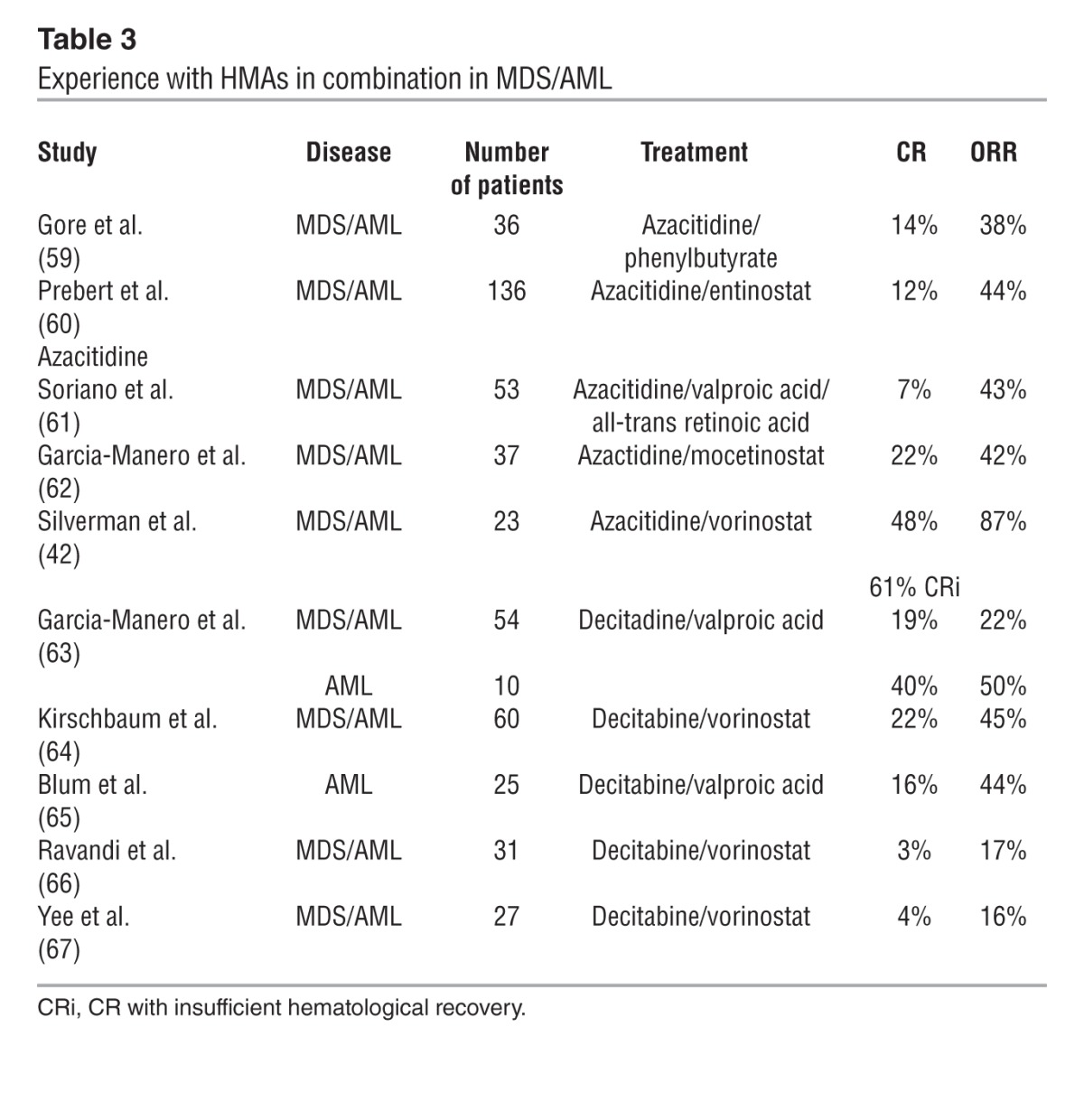
Twenty-eight patients were enrolled in a phase I study that used a combination of azacitidine and vorinostat. Twenty patients had MDS and 8 had AML at the time of entry, with a median age of 68 years. Responses among evaluable patients occurred in 18 of 21 patients, nine with CR, two with CR but insufficient hematological recovery, seven with HI, and two with stable disease. In 57% of patients, the abnormal cytogenetic clone persisted despite response. This study concluded that azacitidine and vorinostat can be safely combined in repetitive cycles, and that the combination is active in both MDS and AML patients with ORRs and complete response rates superior to azacitidine alone (42). Given the mixed results of combinations with decitabine, dosing and treatment schedule is likely important in the synergy of HMAs with HDAC inhibitors.
Azacitidine has also been combined with lenalidomide with promising results in early-phase studies. The rationale stems from the mechanism of action of the two agents: the effects of azacitidine are dependent on cycling cells and lenalidomide inhibits cell cycle progression; therefore, the sequential administration of azacitidine followed by lenalidomide may maximize the synergistic effects (43). In a phase II study conducted by Sekeres and colleagues in patients predominantly with higher-risk MDS, a response rate of 72% was achieved, with 44% of patients obtaining CR (44). However, the median overall survival for the treated population was shorter than the AZA-001 study (Table 1), which may have been due to shorter median follow-up or differences in patient population or study design. In general, the combination of azacitidine and lenalidomide has been feasible and well tolerated using lenalidomide doses of up to 50 mg and standard doses of azacitidine. Initial responses seem to be achieved earlier and ORs appear to be higher than with single-agent azacitidine treatment, although preliminary data suggest that many patients lose their responses early during treatment. A US intergroup study, S-1117, that compares azacitidine to azacitidine combined with either vorinostat or lenalidomide is underway (ClinicalTrials.gov identifier: NCT01522976). A study to combine azacitidine with rigosertib, a benzyl styryl sulfone that has multi-kinase activity and that has shown promising activity in patients with relapsed/refractory MDS, is also underway (ClinicalTrials.gov identifier: NCT01926587).
HMAs have been combined with non-epigenetic agents including gemtuzumab ozogamicin, erythropoietin, filgrastim, romiplostim, arsenic, and sorafinib. These studies have involved small numbers of patients and will require further exploration to determine whether they provide an additive effect to treatment with HMAs alone. The combination of bortezomib with decitabine is noteworthy, as it has demonstrated clinical activity in a limited number of older patients with AML (45). This combination was recently tested in an intergroup study of decitabine with or without bortezomib. However, the trial was terminated early due to toxicity, and publication of the results is pending. Combination therapy with HMAs and other classes of drugs appears to be a promising approach to treat patients with MDS and AML, but requires further investigation.
One common theme throughout clinical development of the HMAs has been the lack of a clear understanding of their mechanism of action. HMAs induce global hypomethylation of the genome as well as changes in methylation in specific genes (46). However, there has been a general lack of clear correlation between clinical response and changes in methylation (47). Most studies measure changes in methylation, DNA damage, and other markers in the first cycle of treatment, while the clinical response is usually not evident until the third treatment cycle or beyond. Concerns about changes in clonal populations later in treatment cycles relative to the initial clone and the impact on the interpretation of changes in cell signaling have been at the heart of this problem. Newer techniques and identification of biomarkers that predict response may alleviate these concerns. As noted above, patients with TET2 mutations have a higher response to azacitidine, and recent data have suggested that microRNA-29b maybe a biomarker for this response (33). Further observations will be required regarding mechanisms of action.
Novel DNMT inhibitors
Ongoing research has led to the development of next-generation DNMT inhibitors. SGI-110 was designed to increase the in vivo efficacy of decitabine. Decitabine is rapidly eliminated in plasma by cytidine deaminase, which limits the amount of time the drug is exposed to cancer cells in vivo. SGI-110 is incorporated into the guanine nucleotide, which improves the stability of decitabine and prevents degradation by cytidine deaminase. This results in improved bioavailability, increased half-life of the drug, and lower dose requirements to achieve a similar effect in vitro. Studies in monkeys have demonstrated sustained hypomethylation when SGI-110 is given as a once-weekly injection for up to four weeks and less myelosuppression compared with decitabine at lower doses (48). Preliminary results from a phase I dose escalation study of SGI-110 in patients with int-2/high-risk MDS or AML (ClinicalTrials.gov identifier: NCT01261312) show that SGI-110 is well tolerated and produces clinical responses in a heavily pretreated population, particularly in patients who demonstrate LINE1 hypomethylation (48).
Zebularine, which inhibits DNA and cytidine deaminase, is another DNMT inhibitor currently being tested in the preclinical setting. In AML cell lines, zebularine inhibits cell proliferation, arrests cells at the G2/M phase, and induces apoptosis at dosages that demethylate the p15INK4B promoter. Zebularine at low doses does not appear to have a hypomethylating effect (49); however, it may potentiate the anti-leukemic activity of decitabine by slowing its intracellular degradation, and therefore may be a promising combination approach (50).
Gene targets
In recent years genomic approaches, including next-generation sequencing and mass spectrometry–based genotyping, have helped to identify recurrent mutations in MDS and AML. Genes involved in the regulation of DNA methylation (DNMT3A, TET2, and IDH1/IDH2) as well as histone function (EZH2, ASXL1, and UTX) are recurrently mutated in MDS and AML, suggesting an important link between genetic and epigenetic mechanisms in these diseases.
DNMT3A mutations are recurrent in patients with AML.
These are largely missense mutations that are associated with poor event-free and overall survival regardless of age, presence of NPM1 or FLT3 mutations, or genetic location. DNMT3A mutations were present in patients with intermediate-risk cytogenetics but absent in those with a favorable-risk cytogenetic profile (51). DNMT3A mutations have also been detected in MDS, and patients with these mutations had worse overall survival and more rapid progression to AML compared with patients with the wild-type form (52). In a mouse model, deletion of Dnmt3a resulted in hematopoietic stem cell expansion and reduced differentiation, along with DNA methylation changes in a subset of genes (53).
Mutations in TET2 have also been described in patients with myeloid malignancies. TET2 catalyzes the production of 5-hydroxymethylcytosine from 5-methyl-cytosine and can ultimately lead to DNA demethylation (54). TET2 is the most frequently mutated gene in MDS identified to date, and the postulated mechanism of action is a loss-of-function mutation (55). The consequence of these mutations on genomic DNA methylation remains controversial, with studies showing inconsistent results. The French MDS group reported a higher response rate among patients with higher-risk MDS and AML with TET2 mutations treated with azacitidine. The ORR was 82% in patients with the mutation versus 45% in patients with the wild-type form. There was no difference in duration of response or overall survival between the two groups (56).
IDH1 and IDH2 catalyze the conversion of isocitrate to α2-glutarate, which participates in the conversion of methylcytosine to hydroxymethylcytosine under the action of TET2. Mutations in IDH1 and IDH2 result in dysregulation of this pathway, thereby inhibiting the function of TET2. These mutations have been identified in MDS, AML, and other myeloid malignancies. The impact of this mutation on overall survival is unclear; however, the largest studies in patients with AML demonstrated a favorable prognosis for patients with IDH2R140Q mutations and no impact of IDH1R132H or IDH2R172K mutations on overall survival (57, 58). There is no clear prognostic importance of IDH1 and IDH2 mutations for patients with MDS. Understanding genetic mutations will provide insight into prognosis, effects on response to therapy, and targets for new therapeutics.
Conclusion
DNA methylation constitutes a major epigenetic regulatory mechanism for inactivating gene expression and has been implicated in the pathogenesis of several hematologic malignancies. Currently, two HMAs have been approved by the FDA for the treatment of MDS. The activity of these agents has demonstrated that epigenetic treatment is effective and changes the natural history and outcome of patients with MDS and possibly AML. Further clinical development of HMAs includes optimization of treatment duration, route of administration, and exploration of rational drug combinations. An improved understanding of the mechanism of genetic mutations will also provide insight into how these agents can be used in a more effective manner.
Acknowledgments
This publication was supported in part by the Henry and Marilyn Taub Foundation (grant to L.R. Silverman) the German Research Foundation, and the DFG (SPP 1463 grant Lu 429/7-1 and CRC 992 Medep grant C04 to M. Lübbert).
Footnotes
Conflict of interest: Shyamala C. Navada receives research support from Onconova Therapeutics. Lewis R. Silverman receives research support from Celgene, Merck, and Onconova Therapeutics.
Citation for this article: J Clin Invest. 2014;124(1):40–46. doi:10.1172/JCI69739.
References
- 1.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley TJ, et al. 5-azacytidine selectively increases gamma-globin synthesis in a patient with beta+ thalassemia. N Engl J Med. 1982;307(24):1469–1475. doi: 10.1056/NEJM198212093072401. [DOI] [PubMed] [Google Scholar]
- 4.Saunthararajah Y, et al. Clinical effectiveness of decitabine in severe sickle cell disease. Br J Haematol. 2008;141(1):126–129. doi: 10.1111/j.1365-2141.2008.07027.x. [DOI] [PubMed] [Google Scholar]
- 5.Olivieri NF, et al. A pilot study of subcutaneous decitabine in beta-thalassemia intermedia. Blood. 2011;118(10):2708–2711. doi: 10.1182/blood-2011-03-341909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- 7.Silverman LR, et al. Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia. 1993;7(suppl 1):21–29. [PubMed] [Google Scholar]
- 8.Silverman LR, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24(24):3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 9.Silverman LR, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20(10):2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 10.Kornblith AB, et al. Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a Cancer and Leukemia Group B study. J Clin Oncol. 2002;20(10):2441–2452. doi: 10.1200/JCO.2002.04.044. [DOI] [PubMed] [Google Scholar]
- 11.Fenaux P, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silverman LR, et al. Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer. 2011;117(12):2697–2702. doi: 10.1002/cncr.25774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.List AF, et al. Effect of azacitidine (AZA) on overall survival in higher-risk myelodysplastic syndromes (MDS) without complete remission [abstract]. J Clin Oncol. 2008;26(15S):Abstract7006. [Google Scholar]
- 14.Gore SD, et al. A multivariate analysis of the relationship between response and survival among patients with higher-risk myelodysplastic syndromes treated within azacitidine or conventional care regimens in the randomized AZA-001 trial. Haematologica. 2013;98(7):1067–1072. doi: 10.3324/haematol.2012.074831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Najfeld V, Silverman LR, Scalise A, Lezon-Geyda K. Modulation of the cytogenetically abnormal myelodysplastic (MDS) clone by azacitidine (Aza C). Blood. 2002;100:97a. [Google Scholar]
- 16.Garcia-Manero G, et al. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29(18):2521–2527. doi: 10.1200/JCO.2010.34.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivard GE, et al. Phase I study on 5-aza-2′-deoxycytidine in children with acute leukemia. Leuk Res. 1981;5(6):453–462. doi: 10.1016/0145-2126(81)90116-8. [DOI] [PubMed] [Google Scholar]
- 18.Pinto A, et al. 5-Aza-2′-deoxycytidine as a differentiation inducer in acute myeloid leukaemias and myelodysplastic syndromes of the elderly. Bone Marrow Transplant. 1989;4(suppl 3):28–32. [PubMed] [Google Scholar]
- 19.Wijermans P, et al. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18(5):956–962. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- 20.Wijermans PW, Krulder JW, Huijgens PC, Neve P. Continuous infusion of low-dose 5-Aza-2′-deoxycytidine in elderly patients with high-risk myelodysplastic syndrome. Leukemia. 1997;11(1):1–5. doi: 10.1038/sj.leu.2400526. [DOI] [PubMed] [Google Scholar]
- 21.Lubbert M, et al. Cytogenetic responses in high-risk myelodysplastic syndrome following low-dose treatment with the DNA methylation inhibitor 5-aza-2′-deoxycytidine. Br J Haematol. 2001;114(2):349–357. doi: 10.1046/j.1365-2141.2001.02933.x. [DOI] [PubMed] [Google Scholar]
- 22.Kantarjian H, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 23.Kantarjian H, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 24.Kantarjian HM, et al. Update of the decitabine experience in higher risk myelodysplastic syndrome and analysis of prognostic factors associated with outcome. Cancer. 2007;109(2):265–273. doi: 10.1002/cncr.22376. [DOI] [PubMed] [Google Scholar]
- 25.Steensma DP, et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27(23):3842–3848. doi: 10.1200/JCO.2008.19.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iastrebner M, et al. Decitabine in myelodysplastic syndromes and chronic myelomonocytic leukemia: Argentinian/South Korean multi-institutional clinical experience. Leuk Lymphoma. 2010;51(12):2250–2257. doi: 10.3109/10428194.2010.524324. [DOI] [PubMed] [Google Scholar]
- 27.Lubbert M, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29(15):1987–1996. doi: 10.1200/JCO.2010.30.9245. [DOI] [PubMed] [Google Scholar]
- 28.Braun T, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–3831. doi: 10.1182/blood-2011-05-352039. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, et al. A prospective multicenter observational study of decitabine treatment in Korean patients with myelodysplastic syndrome. Haematologica. 2011;96(10):1441–1447. doi: 10.3324/haematol.2011.046078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fenaux P, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–569. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 31.Ravandi F, et al. Superior outcome with hypomethylating therapy in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome and chromosome 5 and 7 abnormalities. Cancer. 2009;115(24):5746–5751. doi: 10.1002/cncr.24661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28(4):556–561. doi: 10.1200/JCO.2009.23.9178. [DOI] [PubMed] [Google Scholar]
- 33.Blum W, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A. 2010;107(16):7473–7478. doi: 10.1073/pnas.1002650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lubbert M, et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica. 2012;97(3):393–401. doi: 10.3324/haematol.2011.048231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kantarjian HM, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670–2677. doi: 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Padua Silva L, et al. Feasibility of allo-SCT after hypomethylating therapy with decitabine for myelodysplastic syndrome. Bone Marrow Transplant. 2009;43(11):839–843. doi: 10.1038/bmt.2008.400. [DOI] [PubMed] [Google Scholar]
- 37.Lubbert M, et al. Non-intensive treatment with low-dose 5-aza-2′-deoxycytidine (DAC) prior to allogeneic blood SCT of older MDS/AML patients. Bone Marrow Transplant. 2009;44(9):585–588. doi: 10.1038/bmt.2009.64. [DOI] [PubMed] [Google Scholar]
- 38.Cogle CR, et al. Hypomethylating agent induction therapy followed by hematopoietic cell transplantation is feasible in patients with myelodysplastic syndromes. Clin Adv Hematol Oncol. 2010;8(1):40–46. [PubMed] [Google Scholar]
- 39.Kim DY, et al. Feasibility of hypomethylating agents followed by allogeneic hematopoietic cell transplantation in patients with myelodysplastic syndrome. Bone Marrow Transplant. 2012;47(3):374–379. doi: 10.1038/bmt.2011.86. [DOI] [PubMed] [Google Scholar]
- 40.Issa JP, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23(17):3948–3956. doi: 10.1200/JCO.2005.11.981. [DOI] [PubMed] [Google Scholar]
- 41.Oki Y, et al. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. 2007;109(5):899–906. doi: 10.1002/cncr.22470. [DOI] [PubMed] [Google Scholar]
- 42.Silverman LR, et al. A phase I trial of the epigenetic modulators vorinostat, in combination with azacitidine (azaC) in patients with the myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML): A study of the New York Cancer Consortium. Blood. 2008;112(AML):1252–.abstr3656. [Google Scholar]
- 43.Platzbecker U, Germing U. Combination of azacitidine and lenalidomide in myelodysplastic syndromes or acute myeloid leukemia-a wise liaison? Leukemia. 2013;27(9):1813–1819. doi: 10.1038/leu.2013.140. [DOI] [PubMed] [Google Scholar]
- 44.Sekeres MA, et al. Phase 2 study of the lenalidomide and azacitidine combination in patients with higher-risk myelodysplastic syndromes. Blood. 2012;120(25):4945–4951. doi: 10.1182/blood-2012-06-434639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blum W, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119(25):6025–6031. doi: 10.1182/blood-2012-03-413898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang AS, et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006;66(10):5495–5503. doi: 10.1158/0008-5472.CAN-05-2385. [DOI] [PubMed] [Google Scholar]
- 47.Fandy TE, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kantarjian HM, et al. [November 18, 2013]. Results From the Dose Escalation Phase of a Randomized Phase 1-2 First-in-Human (FIH) Study of SGI-110, a Novel Low Volume Stable Subcutaneous (SQ) Second Generation Hypomethylating Agent (HMA) in Patients with Relapsed/Refractory MDS and AML. Paper presented at: ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, Georiga, USA. https://ash.confex.com/ash/2012/webprogram/Paper49626.html . Accessed .
- 49.Scott SA, Lakshimikuttysamma A, Sheridan DP, Sanche SE, Geyer CR, DeCoteau JF. Zebularine inhibits human acute myeloid leukemia cell growth in vitro in association with p15INK4B demethylation and reexpression. Exp Hematol. 2007;35(2):263–273. doi: 10.1016/j.exphem.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Flotho C, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–1028. doi: 10.1038/leu.2008.397. [DOI] [PubMed] [Google Scholar]
- 51.Ley TJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walter MJ, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25(7):1153–1158. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Challen GA, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44(1):23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langemeijer SM, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 56.Itzykson R, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7):1147–1152. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 57.Green CL, et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011;118(2):409–412. doi: 10.1182/blood-2010-12-322479. [DOI] [PubMed] [Google Scholar]
- 58.Bejar R, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gore SD, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 60. Prebet T, et al. Prolonged Administration of Azacitidine with or without Entinostat Increases Rate of Hematologic Normalization for Myelodysplastic Syndrome and Acute Myeloid Leukemia with Myelodysplasia-Related Changes: Results of the US Leukemia Intergroup Trial E1905. Presented at: 53rd ASH Annual Meeting and Exposition; December 10–13, 2011; San Diego, Califonia, USA; https://ash.confex.com/ash/2010/webprogram/Paper29350.html . Accessed November 18, 2013
- 61.Soriano AO, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110(7):2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Manero G, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112(4):981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia-Manero G, et al. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kirschbaum M, et al. Vorinostat in Combination with Decitabine for the Treatment of Relapsed or Newly Diagnosed Acute Myelogenous Leukemia (AML) or Myelodysplastic Syndrome (MDS): A Phase I, Dose-Escalation Study. Paper presented at: 51st ASH Annual Meeting and Exposition; December 5–8, 2009; New Orleans, Lousiana, USA. https://ash.confex.com/ash/2009/webprogram/Paper23454.html . Accessed November 18, 2013
- 65.Blum W, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25(25):3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 66.Ravandi F, et al. Phase I Study of Suberoylanilide Hydroxamic Acid (SAHA) and Decitabine in Patients with Relapsed, Refractory or Poor Prognosis Leukemia. Blood. 2007;110:897. [Google Scholar]
- 67.Yee KWL, et al. A Phase I Trial of Two Sequence-Specific Schedules of Decitabine and Vorinostat in Patients with Acute Myeloid Leukemia (AML). Blood. 2007;110(3):908. doi: 10.1182/blood-2006-11-057604. [DOI] [PMC free article] [PubMed] [Google Scholar]