

Abstract
Multiple intestinal atresia (MIA) is a rare cause of bowel obstruction that is sometimes associated with a combined immunodeficiency (CID), leading to increased susceptibility to infections. The factors underlying this rare disease are poorly understood. We characterized the immunological and intestinal features of 6 unrelated MIA-CID patients. All patients displayed a profound, generalized lymphocytopenia, with few lymphocytes present in the lymph nodes. The thymus was hypoplastic and exhibited an abnormal distribution of epithelial cells. Patients also had profound disruption of the epithelial barrier along the entire gastrointestinal tract. Using linkage analysis and whole-exome sequencing, we identified 10 mutations in tetratricopeptide repeat domain–7A (TTC7A), all of which potentially abrogate TTC7A expression. Intestinal organoid cultures from patient biopsies displayed an inversion of apicobasal polarity of the epithelial cells that was normalized by pharmacological inhibition of Rho kinase. Our data indicate that TTC7A deficiency results in increased Rho kinase activity, which disrupts polarity, growth, and differentiation of intestinal epithelial cells, and which impairs immune cell homeostasis, thereby promoting MIA-CID development.
Introduction
Multiple intestinal atresia (MIA) is characterized by widespread atresia, extending from the stomach to the rectum, and homogenous calcifications in the abdominal cavity (1). Patients with MIA can develop the first symptoms of the condition in utero. In a subset of patients, MIA is associated with combined immunodeficiency (CID) (2–4). MIA-CID increases a patient’s susceptibility to a broad range of pathogens, mainly bacteria and viruses. These patients have to undergo surgical intestinal resections and derivations within the first days of life, and thus require total parenteral nutrition (TPN) as a consequence of short bowel syndrome. The patients also develop hepatic cholestasis, cirrhosis, and, ultimately, chronic hepatic failure, which can be cured by liver and small-bowel transplantation. Hematopoietic stem cell transplantation can be combined, with a view to curing the CID (4, 5). The disease mechanism for MIA-CID has not been established. Given the rarity of this condition, detailed clinical and pathological descriptions are scarce. Here, we describe clinical, immunological, and pathological data from 6 patients from 6 unrelated pedigrees as well as the inherited etiology of MIA-CID in relation to severe mutations in the gene tetratricopeptide repeat domain–7A (TTC7A). Our genetic findings confirmed 2 recent reports describing 2 TTC7A mutations observed in MIA patients (6, 7). However, no functional data on TTC7A deficiency consequences in the gut were previously reported (6, 7). We therefore performed immunohistochemical staining of patients’ gut biopsies and analysis of gut-organoid cultures grown from intestinal biopsies, which provided insights into the intestinal disease and its relationship with the RhoA signaling pathway.
Results
6 patients from 6 distinct pedigrees were diagnosed with MIA prenatally or at birth after observation of multiple gastrointestinal defects, including a pyloric diaphragm and intestinal atresia (see Methods). These defects required surgical resections in the first months of life and subsequent TPN. 5 of the patients died between 8 months and 4 years of age. At the completion of the present study, patient C3 (aged 8 years) was alive and recently developed mild palmoplantar pachyderma.
Immunological data.
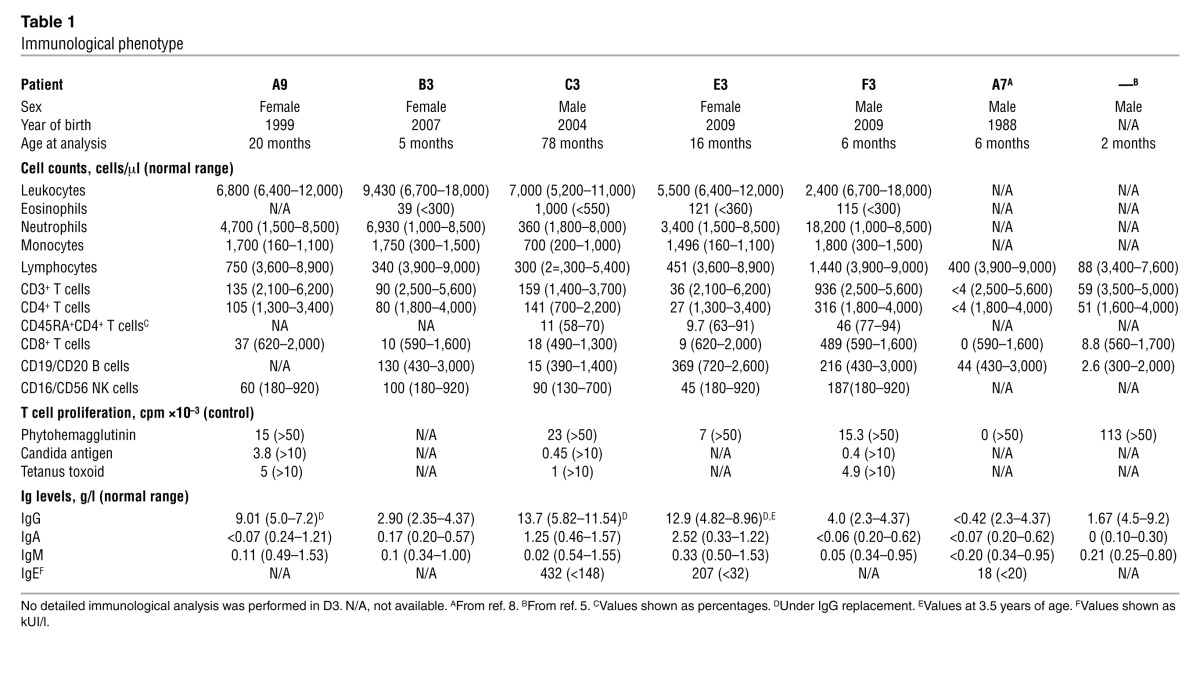
With the exception of F3, all patients displayed early-onset, profound, generalized T cell lymphopenia and milder NK and B cell lymphopenia (Figure 1A and Table 1). Accordingly, the T cell proliferation capacity in response to mitogens, when tested, was poor. Serum IgG, IgA, and IgM levels were very low, whereas IgE levels were elevated in the 2 cases tested. Thus, MIA-CID was a common feature of the 6 patients, as previously reported for other cases (8). A postmortem pathology study revealed hypoplastic thymuses in D3 and E3 (respectively, one-tenth and one-third of the thymus size of age-matched controls). In A4, partial corticomedullary delineation and low CD3+ T lymphocyte cellularity were observed (Figure 1B). Cytokeratin 5 (CK5) and CK8 (markers for medullary and cortical thymic epithelial cells, respectively) were detected in control subjects and A4; however, the CK5+ cells were abnormally distributed throughout the patient’s thymic cortex (Figure 1B). Autopsy data on E3 showed that the mediastinal lymph nodes and mesenteric lymph nodes contained few lymphoid cells; small, dispersed B follicles; and few T cells in the paracortex (Figure 1C and data not shown). There was a polymorphous inflammatory infiltrate rich in eosinophils and macrophages, as revealed by H&E and CD68 staining, respectively (Figure 1C).
Figure 1. Immunological characteristics of MIA-CID patients.

(A) Circulating blood cell counts. Shown are lymphocyte, monocyte, and neutrophil counts as well as total CD3+, CD4+, and CD8+ T cell counts. (B) Thymus pathology. H&E staining (top) revealed poor corticomedullary demarcation and a paucity of lymphocytes and Hassall’s bodies in A4 versus a control subject. Also shown is immunohistochemical staining of CD3, CD8, and CD4 as well as CK5 and CK8 staining for medullary (m) and cortical (c) thymic epithelial cells, respectively. (C) Mediastinal lymph node. H&E staining and immunohistochemical staining for CD20, CD3, and CD68 in a control subject and in E3 after autopsy. Original magnification, ×50 (B), ×400 (B, enlarged H&E views), ×200 (C, H&E), ×100 (C, immunostaining).
Table 1.
Immunological phenotype
TTC7A mutations are associated with MIA-CID.
Based on the hypothesis that the MIA-CID has autosomal-recessive inheritance, we performed genome-wide linkage analysis by homozygosity mapping on families A, B, and C as well as whole-exome sequencing on A9, B3, C3, E3, and F3 (Figure 2A). Linkage analysis revealed a common 4-Mb region located on chromosome 2 (p21) (bp position, g.43,347,681–g.47,320,846, hg19) and a common 7-Mb region on chromosome 10 (bp position, g.17,038,509–g.24,566,092). Both regions had a logarithm of odds score of 1.95. No candidate genes were identified in the common region on chromosome 10. In contrast, whole-exome sequencing of the common region on chromosome 2 identified a homozygous, nonsynonymous variant in TTC7A in B3 and C3. Sanger sequencing of TTC7A’s coding exons confirmed the mutations identified in B3 and C3 and identified other mutations or deletions in the 4 remaining patients, the parents, and the siblings (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI71471DS1). Since exon 2 could not be amplified in samples from A9, sequencing of the cDNA from each of the patient’s parents was performed, which identified a total deletion of exons 2 and 3 in the mother (A1) and exon 2 in the father (A2) (Supplemental Figure 1). The genetic anomalies identified in TTC7A in the 6 patients are summarized in Figure 2B. D3 inherited a missense mutation in exon 14 from her mother, which led to a serine-to-leucine substitution at position 539 (Supplemental Figure 1). The serine at position 539 in the human sequence is highly conserved, and the serine-to-leucine substitution is predicted to be “probably damaging” by PolyPhen2, “deleterious” by SIFT, and “disease causing” by MutationTaster. D3’s paternal mutation was not detectable by exon sequencing of the patient’s DNA. Sequencing of cDNA obtained from her father (D2) highlighted a heterozygous 4-bp deletion in exon 11 associated with exon 12 deletion, predicting premature stop codon (Supplemental Figure 1). One of the heterozygous mutations identified in F3, inherited from his father (F2), is located within intron 12 (g.47102727 T>A) at some distance from a splice site. Sequencing of the father’s cDNA showed a heterozygous splicing defect that causes deletion of exon 12 (Supplemental Figure 1). In the other patients, the identified mutations either create a nonsense codon directly or create a subsequent nonsense codon indirectly via a frameshift in the TTC7A sequence. The identified point mutations and deletions were neither found in in-house exome sequencing data nor in any of the publically available databases (including the dbSNP129 and 1,000 Genomes datasets). Heterozygous individuals were asymptomatic.
Figure 2. Pedigree and TTC7A mutations in MIA-CID families.

(A) MIA-CID family pedigree. Square, male; circle, female; completely filled symbols, affected individuals; half-filled symbols, heterozygous carriers. Affected individuals who did not undergo genetic testing are indicated in gray. Slashes indicate deceased persons; double horizontal lines indicate consanguinity. A7 has been described previously (8). (B) TTC7A gene mutations and their predicted effect on the TTC7A protein. TPRs are indicated on the protein sequence, in accordance with NCBI human sequence annotation (accession no. Q9ULTO-1).
Immunopathology of the gut.
Immunochemistry revealed disruption of the architecture of the mucosa alone, from the stomach to the colon (Figure 3A). The epithelium was pseudostratified, with hyperchromatic nuclei, signs of apoptosis, and partial or total destruction of the glands. These abnormalities were detectable from birth onward. In the small intestine, villi were scarce or absent. The cellular infiltrate within the lamina propria consisted mainly of eosinophils and macrophages. The lymphocyte population was similar to that seen in controls, as shown by staining for CD3 and CD20 (Figure 3B and data not shown). A few lymphoid follicles were present. In the proximal small intestine, the number of mucus-secreting cells and lysozyme-containing cells was normal (data not shown). In patient biopsies, Ki67+ proliferating epithelial cells were detected only in a few crypts (Figure 3B). CK20 staining was detected at the surface of epithelium and partially shed in the gut lumen. It was additionally observed in the glands (Figure 3B), highlighting the epithelium’s abnormal, pseudostratified structure. Villin expression was low, with a very weak signal visible on the luminal side of the epithelium and within a few epithelial vacuoles, instead of at the apex (Figure 3B). Alkaline phosphatase (AP) activity was totally disrupted in patients relative to controls (Figure 3B), indicative of defective apical pole function in the epithelium. Taken together, these data highlight the profound disruption of the epithelial architecture in MIA-CID, with loss of apical pole function associated with infiltration of eosinophils and macrophages in the mucosa.
Figure 3. Immunohistologic studies of the digestive tract of MIA-CID patients.

(A) Pathology studies of the stomach, intestine, and colon. H&E staining of the stomach (antrum) in a control subject (12 years old) and A9 (6 months old). Asterisks denote pseudostratified epithelium; arrow denotes apoptosis in glands. Compared with the proximal small intestine in a control subject (7 days old), that of E3 (2 days old) showed cysts and protrusions of multilayered epithelial cells as well as infiltration with eosinophils. Colon was stained with H&E in a control subject (12 years old) and A9 (6 months old). (B) Immunochemical staining was performed to detect markers for epithelial differentiation as well as AP — as a marker for intestinal epithelium function — in the proximal small intestine in a control subject (1 day old) and patient (2 days old). Staining showed lamina propria infiltration by T cells (CD3) and macrophages (CD68) and Ki67-positive, proliferating epithelial cells as well as expression of CK20, villin, and AP. Original magnification, ×200 (A, stomach and colon, and B), ×50 (A, small intestine), ×400 (A, small intestine, enlarged view).
Intestinal organoids from MIA-CID patients show disrupted apicobasal polarity.
The clinical and pathological findings in MIA-CID patients suggested that TTC7A had a prime role in intestinal epithelial cells. We thus established 3-dimensional organoid cultures as a model for self-renewal and differentiation (9, 10). Small intestine biopsies from C3 and E3 showed efficient initial outgrowth (Supplemental Figure 2), suggestive of the presence of Lgr5 stem cells in atretic regions. However, although control organoids could be expanded over many months, patient organoids deteriorated after several passages; only those of C3 could be grown long enough to be further analyzed. These patient organoids displayed a disturbed epithelial architecture, lacked a central lumen (villus domain) and budding processes (crypts), and formed dense cellular aggregates (Figure 4A). Coimmunofluorescence analysis of Ki67 and CK20 expression revealed loss of proliferation and gain of cytodifferentiation, respectively (Figure 4B), with a mild excess of cell death (as shown by caspase-3 immunostaining; data not shown), which demonstrated TTC7A’s prime role in gut epithelial homeostasis.
Figure 4. MIA-CID patient–derived intestinal organoids show defects in proliferation, differentiation, and epithelial polarity associated with abnormally active ROCK pathway.

(A) Morphologic images of human ileum–derived organoids. C3-derived cultures formed condensed cell aggregates instead of the budding processes (arrows) and central lumen (asterisk) seen in controls, depending on the presence of Y-27632 (Y). (B) Differentiation and proliferation. Dual immunofluorescence of CK20 (red; showing disrupted apicobasal polarity) and Ki67 (green; showing proliferating epithelial cells). Mutations in TTC7A were associated with strong increased differentiation and reduced proliferation. Addition of Y-27632 restored proliferation and suppressed differentiation in patient cultures, but could not rescue abnormal nuclear positioning (blue; DAPI). (C) Growth profile for control- and patient-derived organoids in the presence or absence of Y-27632. Mean organoid number after passaging of 3 independent wells each. Error bars denote SEM. **P < 0.01, ***P < 0.0001, t test. (D) Western blot analysis of patient-derived organoids showed increased ROCK activity. Membranes were probed with anti-ROCK, anti-P-ERM, anti-Rho, and anti-P-MLC. Actin was used as a loading control. (E) Analysis of cell polarization by immunostaining of α6 integrin (green) and F-actin (red) in the apical brush border and of the tight junction marker ZO-1 (green). Control- and patient-derived organoids were cultured in the absence of Y-27632. Nuclei were stained with DAPI (blue). Scale bars: 100 μm.
ROCK inhibition reverses epithelial abnormalities in TTC7A-deficient intestinal epithelial cells.
We previously reported that pharmacologic inhibition of Rho kinase (ROCK) with Y-27632 was required to avoid cell death in isolated intestinal stem cells after the loss of cell-cell contacts (9). We hypothesized that addition of Y-27632 might also support survival of TTC7A-deficient organoids after passaging, a process that involves mechanical shearing and reseeding. Indeed, continuous addition of Y-27632 facilitated the expansion of patient cultures (Figure 4, A–C), but was not essential for control cultures. In patient-derived organoids, ROCK inhibition caused formation of large, cystic structures with renewed Ki67 expression and reduced CK20 expression (Figure 4, A and B, and Supplemental Videos 1 and 2). Western blot analysis showed that untreated patient-derived organoids contained abnormally high levels of phospho–ezrin/radixin/moesin (P-ERM) and phospho–myosin light chain (P-MLC), both of which are downstream targets of ROCK (11), whereas expression of both ROCK and Rho were similar to control values. After treatment with Y-27632, P-ERM and P-MLC expression returned to control levels (Figure 4D).
The MIA-CID patients displayed pseudostratified gut epithelium and disturbed nuclear positioning both in vivo and in vitro. These features are suggestive of defects in apicobasal polarity. It is known that elevated ROCK activity and MLC phosphorylation disrupt epithelial polarity in cell-based models (12). We therefore decided to study polarity markers in organoids after withdrawal of Y-27632. Basolateral expression of α6 integrin and β6 integrin was partially lost; instead, these proteins accumulated at cell-cell junctions inside cell aggregates (Figure 4E and data not shown). The apical brush border actin, which faced the lumen in controls, was deposited at the periphery in patient-derived organoids. The tight junction protein ZO-1, which is normally found below the apical brush border, was detected ectopically (i.e., on both sides of the epithelium; Figure 4E). Culture with Y-27632 rescued the polarity (Figure 5, A–F).
Figure 5. Rescue of normal phenotype of MIA-CID patient–derived organoids and fibroblasts by ROCK inhibition or WT-TTC7A expression.

(A–E) Immunochemical staining of α6 integrin (green) and F-actin (pink) of intestinal organoids. Control- (A and C) and C3-derived ileum organoids (B, D, and E) were cultured for 5 days with or without Y-27632. Nuclei were stained with DAPI. The inset in E shows abnormal F-actin deposition. Scale bars: 50 μm. (F) Quantification of apicobasal polarity after 5 days of growth with or without Y-27632. Normal polarity, epithelial monolayer with α6 integrin outside and F-actin on the luminal side; aberrant polarity, multilayered epithelium with disturbed α6 integrin and/or F-actin deposition; inverted polarity, F-actin facing outward, α6 integrin clustered inside. 50 organoids total were counted per condition. (G) Western blot analysis of ROCK and downstream effectors P-ERM and P-MLC in C3- and control-derived fibroblasts with or without Y-27632 treatment. Lanes were run on the same gel but were noncontiguous. Data are representative of 2 independent experiments. (H) Rescue of the phenotype of MIA-CID patient fibroblasts by WT-TTC7A expression. E3- or control-derived fibroblasts were transiently transfected with WT-TTC7A or empty vector prior to P-ERM and P-MLC expression analysis by Western blot 24 hours later. Actin was used as a loading control. Data are representative of 3 independent experiments. (I) Recovery of normal adhesion and proliferation of MIA-CID patient fibroblasts by WT-TTC7A expression. Measurement of adhesion followed by proliferation by XCELLigence technology of 2 patient- and 2 control-derived fibroblasts transfected with WT-TTC7A or empty vector. Data are representative of 2 different experiments.
Together, our present findings are indicative of an association between increased ROCK activity and spontaneous differentiation and defective apicobasal polarity in patients with TTC7A mutations.
TTC7A-dependent ROCK activation in nonepithelial cells and complementation of the defect.
We next looked at whether the functional link between TTC7A and the RhoA pathway in the intestinal epithelium could be detected in other differentiated cell types, as TTC7A is expressed in multiple tissues (13). Given the profound lymphopenia displayed by MIA-CID patients, lymphocytes could not be evaluated. We analyzed fibroblast cell lines derived from C3 and E3 skin biopsy. Western blotting revealed substantially elevated phosphorylation of P-ERM and P-MLC in cell lysates of patient- versus control-derived fibroblasts (Figure 5G). As observed in intestinal gut epithelial cells, phosphorylation of these proteins was normalized when the patient-derived fibroblasts were incubated in the presence of Y-27632. In addition, expression of a wild-type TTC7A construct (referred to herein as WT-TTC7A) in patient-derived fibroblasts was sufficient to rescue both P-ERM and P-MLC expression to near-control levels (Figure 5H), demonstrating a direct role of TTC7A in regulation of the ROCK pathway. Finally, the patient-derived fibroblasts displayed an increased adhesion and proliferation capacity compared with controls that was reduced by WT-TTC7A expression (Figure 5I).
Discussion
We found that biallelic mutations in TTC7A were responsible for an autosomal-recessive inherited MIA-CID syndrome in 6 patients from unrelated families. All but 1 of the identified genetic anomalies lead to nonsense codons or are predicted to cause a frameshift mutation. The only identified heterozygous missense mutation leads to a serine-to-leucine transition at residue 539; this mutation is predicted to be deleterious by 3 different software tools. The mutation is located in the tetratricopeptide repeat 5 (TPR5) domain, 1 of 2 containing a triple TPR, which is thought to be the minimal functional unit for this motif (14). The TPR-containing protein TTC7A does not have a known function. TPR motifs are protein-protein interaction modules present in numerous functionally different proteins (15). The motifs facilitate specific interactions with one or more partner proteins. Most TPR-containing proteins are involved in complex multiprotein functions in chaperoning, cell cycling, transcription, and protein transport (16, 17). In concert with 2 recent reports on different mutations in TTC7A in patients with MIA (6, 7), our present findings establish a critical role for TTC7A in both the gut and the immune system.
We showed that TTC7A deficiency in the patients described herein affected the immune system and rapidly led to lymphocytopenia, particularly for peripheral T lymphocytes. T cell lymphopenia in MIA-CID might be related to a role for TTC7A in lymphocyte differentiation, proliferation, and survival. The low number of peripheral lymphocytes in these patients precluded further analysis. However, the characteristics of the immunodeficiency associated with a hypomorphic mutation in TTC7A (R. Lemoine, unpublished observations) suggest that TTC7A acts to maintain lymphocyte homeostasis by regulating functions such as cell adhesion, proliferation, and migration. Furthermore, we showed that patients with MIA-CID present a hypoplastic thymus with low lymphocyte cellularity and ectopic distribution of CK5 in the cortex. Severe lymphoid depletion in the thymus of an MIA-CID patient was also recently reported (7). Given that TTC7A expression occurs in many tissues, including the thymic epithelium, we cannot rule out the possibility that defective TTC7A function in the thymic epithelium may also contribute to T cell lymphopenia.
A lack of TTC7A led to dramatic disruption of the gut mucosal architecture in the patients, extending from the stomach to the colon. There was no evidence of a blockade in epithelial cell differentiation. The main anomalies were the presence of many disorganized, pseudostratified cell structures; a low number of villi; and high levels of cell apoptosis. Our study of gut organoids established from 2 MIA-CID patients highlighted TTC7A’s critical role in regulating the balance between growth and differentiation of intestinal epithelial cells. Furthermore, this gut organoid analysis established that TTC7A has a decisive role in the apicobasal polarization of epithelial cells by regulating the RhoA signaling pathway. Apicobasal polarity is regulated through the cytoskeletal proteins (including the integrins) that mediate cell-matrix and cell-cell adhesion (18). The establishment and maintenance of multicellular epithelial tissues require each cell to be polarized with the correct orientation. This is also true for formation of the central lumen. It was previously shown that blockade of β1 integrin in Madin-Darby canine kidney cells inverts the cells’ apicobasal polarity by activating the RhoA signaling pathway and that the latter phenotype can be reversed by treatment with the ROCK inhibitor Y-27632 (12). Similarly, gut tube formation during embryogenesis was shown to depend on ROCK activity (19). Here, we demonstrated that TTC7A is a key player in this signaling pathway and is necessary for coordinated epithelial cell polarity, growth, and differentiation. TTC7A downregulates the activity of ROCK and its effector targets, ERM and myosin II (20, 21). Like other ROCK targets, ERM and myosin II are cytoskeleton regulators that modulate both F-actin and stress fiber contractility as well as integrin activity (Figure 6). These changes influence cell shape, polarization, and motility (22). The pleiotropic role of the RhoA signaling pathway in immune cells is also well established. Myosin II activity was previously shown to reduce T cell motility and favor their arrest upon TCR activation, a necessary step in synapse formation (23). Thus, by interacting with several cytoskeleton regulators, TTC7A-dependent ROCK activity interferes with key cellular functions in epithelial and lymphocyte homeostasis. We suggest that these cellular changes account for the phenotype displayed by MIA-CID patients. How TTC7A regulates the ROCK pathway remains to be determined. The TPR proteins’ ability to participate in several simultaneous interactions suggests that TTC7A could interact with one or more of the effectors or regulators in the ROCK pathway.
Figure 6. ROCK targets responsible for cytoskeleton assembly.

Modulation of cytoskeleton assembly is required for the coordination of cell adhesion, polarization and migration. The active form of RhoA (RhoA-GTP) activates ROCK and interacts with the integrin intracellular signaling pathway. RhoA also modulates cytoskeleton reorganization via focal adhesion kinase (FAK) activation and front-rear polarity via phosphorylation of phosphatase and tensin homolog (PTEN). Phosphorylation of ROCK’s downstream effector MLC is required for the assembly of actomyosin complexes. ROCK downregulates the MLC phosphatase (MLCPase), resulting in an increase in phosphorylated MLC, cell contraction, actin organization, stress fiber formation, and focal adhesions, which confer contractility and migration properties to the cell. Phosphorylation of ROCK’s downstream effector ERM has a role in microvilli formation. Phosphorylated LIM domain kinase (LIM-K) phosphorylates and thus deactivates the actin-depolymerizing protein cofilin. Y-27632 blocks Rho-induced actomyosin activation. These data support a role for TTC7A in the downregulation of ROCK activity. Activation and inactivation are shown as arrowheads and blunted lines, respectively. MLCK, myosin light chain kinase; NHE1, Na+/H+ exchange protein.
In conclusion, our present findings pave the way to better understanding of MIA-CID and highlight a previously unexpected, common mechanism regulating the lymphocyte and epithelial cell differentiation in the gastrointestinal tract and, potentially, the thymus. Since MIA has not always been associated with a CID phenotype, it remains to be seen whether CID was not recognized, given the rapid lethal nature of the disorder, or whether isolated MIA results from specific TTC7A mutations, or even other genetic defects affecting the ROCK pathway.
Methods
MIA-CID patients.
A9 (female) was born to nonconsanguineous parents of mixed European descent. Her 3 older brothers all had the same phenotype and had died in the first months of life (due to infections and post-transfusion graft-versus-host disease, as previously reported; ref. 8). At birth, A9 was diagnosed with several gastrointestinal defects: prepyloric diaphragm, micro–small intestine, microcolon, and colic atresia. At day 3 after birth, she underwent diaphragm resection, hemicolectomy, and a double ileostomy. At 18 months of age, she underwent further surgery (cholecystectomy, pyloroplasty, and gastrostomy). TPN was initiated at 7 months of age. A9 had a chronic subocclusion and developed progressive hepatic cholestasis and cirrhosis. She displayed progressive, generalized, profound lymphopenia and panhypogammaglobulinemia, which required Ig replacement therapy and anti-infective prophylaxis. A thymus could not be visualized on a chest X-ray at 20 months of age. A9 died at 2.5 years of age due to sepsis.
B3 (female) was born in Saudi Arabia to consanguineous parents who had then emigrated to the United Kingdom. A prenatal ultrasound diagnosis of MIA was confirmed at birth and prompted initiation of TPN. During intestinal surgery at 5 weeks of age, a duodenal membrane was found, and more than 50 cm of small intestine (including the ileocecal region) was resected. B3 underwent further intestinal resection at 2 months of age. At 7 weeks of age, she developed sepsis and transient hyperbilirubinemia. An episode of Klebsiella line sepsis occurred at 3 months of age. B3 was transferred to the United Kingdom for medical management at 5 months of age. She was found to have profound, generalized lymphopenia. She received a hematopoietic stem cell transplant (from an HLA 5/6–matched unrelated cord blood donor) at 8 months of age, but developed Pseudomonas pneumonia and severe sepsis, which led to death from a pulmonary hemorrhage at 9 months of age.
C3 (male) was born in France to parents from 2 villages a few miles apart in Sri Lanka. A prenatal diagnosis of intestinal atresia was confirmed at birth. TPN was initiated, but the patient developed progressive hepatic cholestasis and cirrhosis. He displayed rapidly progressive, generalized, profound lymphopenia and severe panhypogammaglobulinemia, which required Ig replacement therapy and anti-infective prophylaxis. C3 did not undergo organ transplantation, because surgery was contraindicated by severe bronchiectasis. From the age of 4 years onward, he developed progressive skin abnormalities with mild pachyderma on both hands and feet.
D3 (female) was born in Norway to nonconsanguineous parents from the same city. She had an enlarged abdomen at birth and was transferred for surgery the next day. D3 presented with multiple atresias — particularly colonic atresia and stenosis — that were removed, and she received TPN for the rest of her life. Postoperatively, she developed a severe infection that was treated with broad-spectrum antibiotics for several weeks. At that point, lymphocytopenia and panhypogammaglobulinemia was detected. In biopsies from both the small intestine and sigmoid, cytomegalovirus was found by immunohistochemistry and PCR. D3 received antibiotic prophylaxis and Ig substitution; however, her clinical condition gradually deteriorated, and she died at 8 months of age from pneumonia with sepsis. Klebsiella pneumonia was found in blood culture and Pneumocystis jiroveci in the lungs at autopsy.
E3 (female) was born in Belgium to nonconsanguineous parents of mixed European descent. She underwent multiple gastrointestinal tract operations, starting in the first few days of life (intestinal resection, duodenostomy, and ileostomy). She suffered from multiple atresia of the proximal digestive tract, with pre- and postduodenal atretic segments. The duodenal loop was massively dilated (extending from the antropyloric region to the duodenojejunal junction [the angle of Treitz]), and a second atresia was found within the duodenojejunal segment. At 10 months of age, E3 developed rapidly progressive, profound, generalized lymphopenia and severe panhypogammaglobulinemia, requiring Ig replacement therapy and anti-infectious prophylaxis. E3 died at 3.9 years of age from acute viral pneumonitis before she could be put on the waiting list for liver/intestinal transplantation.
F3 (male) was the first child born in Germany to nonconsanguineous parents of mixed European descent. A prenatal diagnosis of MIA was confirmed at birth. Intestinal surgery included ileocecal resection and ileostomy at birth and excision of a pylorus membrane 3 days after birth. F3 developed hepatic cholestasis of unclear etiology, rapidly progressive severe generalized lymphopenia, and panhypogammaglobulinemia. Ultrasound assessment showed a normal-sized thymus. F3 also suffered from heart failure due to mitral valve stenosis. A cytomegalovirus pulmonary infection led to death at 7 months of age. See Figure 1 and Table 1 for immunological data for F3.
Whole-exome sequencing and linkage analysis.
We performed a linkage analysis of 3 families (A, B, C) using Affymetrix GeneChip Mapping 250K NspI, as described previously (24). Homozygous regions were detected with a parametric, SNP-based linkage genome browser (MERLIN software, version 1.1.1). Genomic DNA and cDNA were amplified, sequenced (BigDyeTerminator sequencing kit; Applied Biosystems), and analyzed on an ABI Prism 3700 system (Applied Biosystems). Exome analysis (A9, B3, C3, E3, and F3) was performed as described previously (25). Briefly, blood samples were collected, DNA was extracted from leukocytes, and the sequences were aligned with a reference genome (hg19). The pathogenicity of missense mutation was predicted using PolyPhen2, MutationTaster, and Sorting Intolerant from Tolerant (SIFT). The reference sequences in this study are available from GenBank (accession nos. NM_020458.2 [TTC7A transcript] and NP_065191.2 [TTC7A protein]).
3-dimensional intestinal organoid cultures.
Biopsies were manually dissected, and the mucosal layer was cut into small pieces that were washed, embedded in Matrigel, and cultured in conditions described previously (10). The ROCK inhibitor Y-27632 (10 μM; Sigma-Aldrich) was added during establishment and maintenance of the patient cultures as indicated. Organoids were derived from ileum for C3 and duodenum for E3. 2 independent healthy control cultures each were analyzed in parallel. Organoid stainings were performed as described previously (10). Whole-mount staining of PFA-fixed cells was performed for F-actin (phalloidin–Alexa Fluor 647; Life Technologies), and immunostaining was performed using rabbit polyclonal anti-ZO-1 (1:200; Abcam) and monoclonal anti-α6 integrin (GoH3, 1:500; BD). Paraffin sections were boiled in citrate buffer and sequentially incubated with mouse anti-Ki67 (MM1, 1:1,000; Monosan), Alexa Fluor–coupled secondary antibody, and anti-mouse Fab fragments for blocking before incubation in mouse monoclonal anti-CK20 (Ks20.8, 1:25; Dako). Nuclei were counterstained with DAPI, and specimens were analyzed using a Leica SP5 confocal microscope.
Immunohistochemistry.
For morphological studies, antrum, duodenum, and colon paraffin-embedded sections were stained with H&E. Intestine, thymus, and lymph node sections were deparaffinized and rehydrated with successive baths of xylene and ethanol. Antigen retrieval was performed in pH 6.0 citrate buffer for 20 minutes at 100°C (except for CK20 staining, for which a 10-minute incubation in 0.1% Trypsin at 37°C was used). Endogenous peroxidase activity was inhibited with 0.3% hydrogen peroxide (v/v in methanol). Sections were blocked for 30 minutes at room temperature in 1% BSA PBS, then incubated with the primary antibody overnight at 4°C. Primary antibodies used for thymus, lymph nodes, and intestine were mouse anti-CD3 (F7.2.38, 1:200), anti-CD8 (C8/144B, 1:100), anti-CD20 (L26, 1:400), and anti-CD68 (KP1, 1:3,000), all from Dako. Primary antibodies for staining thymic epithelial cells included rabbit anti-CK5 and mouse anti-CK8 (1:1,000; Covance). Markers for intestinal epithelium included mouse anti-CK20 (Ks20.8, 1:500; Dako), anti-villin (1D2C3, 1:100; Santa Cruz Biotechnology Inc.), and anti-AP (8A9, 1:50; Dako). Sections were developed with the Dako REAL System (Dako) containing biotinylated goat anti-mouse and anti-rabbit secondary antibody, streptavidin-labeled HRP, and DAB.
Protein blotting.
Intestinal organoids and SV40-immortalized fibroblasts were lysed in radioimmunoprecipitation/glycerol buffer (50 mM HEPES, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, and 1% sodium deoxycholate) supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma-Aldrich). Cell extracts were separated by SDS-PAGE, blotted, and then stained with the specific antibodies anti-ROCK, anti-P-ERM, and anti-P-MLC (Cell Signaling). After staining with a HRP-conjugated secondary antibody, the immunoblot was developed with an enhanced chemiluminescence detection kit (GE Healthcare Life Sciences).
Rescue experiments.
Full-length TTC7A cDNA was subcloned into the EcoR1 and BamH1 restriction sites of the pEGFP-C1 plasmid (Clontech) using the following TTC7A primers: forward, 5′-AGGAATTCCATGGCTGCGAAGGGCGCGCACGGC-3′; reverse, 5′-GGGGATCCCTCAGAGCTCTCTGGGGATGATGG-3′. Empty vector was used as a control. SV40-immortalized fibroblasts cultured in 10% FCS DMEM were transiently transfected with Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer’s instructions. Western blotting was performed 24 hours after transfection.
Cell adhesion assays using xCELLigence technology.
For adhesion and proliferation assays, 3,000 fibroblasts derived from C3, E3, and control skin biopsy were transiently transfected with WT-TTC7A. After 24 hours, cells were added to E-plate wells containing 50 μl medium, incubated for 20 minutes at room temperature, and then cultured at 37°C in 5% CO2. Cell adhesion was monitored every 5 minutes for up to 20 hours. The electrical impedance was measured as a cell index by the xCELLigence system’s integrated software.
Statistics.
Analyses were performed with PRISM software (version 4 for Macintosh, GraphPad Inc.). Statistical hypotheses were tested using 2-tailed t test. A P value less than 0.01 was considered significant.
Study approval.
Clinical information and blood samples were collected from patients, relatives, and controls, all of whom had given prior informed consent to participate in the study. Genetic studies and data collection procedures were approved by the local investigational review board and the French Advisory Committee on Data Processing in Medical Research.
Supplementary Material
Acknowledgments
We thank Capucine Picard for the immunological study of C3 and Naziha Khen-Dunlop and Joseph Amirandoum for performing intestinal biopsies for organoids. We thank Anne Servais, Vincent Delorme, and C. Mulder for providing biological samples; J. Viala and M. Lecompte-Houcke for providing useful information on patients; and Cécile Masson for participation in exome analysis. We thank all technicians and secretarial staff from the Pathology Department of Necker Hospital, in particular Martine Hennebo, Gisèle Legall, Claire Gandon, Stéphanie Petit, Annie Postel, Nicole Mariage, and Agathe Helan, for their technical support. This research was supported by INSERM, by ANR-08-GENO-020-01, and by an advanced grant from the European Research Council (ERC; PIDImmun reference no. 249816) and the “Imagine Foundation.”
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(1):328–337. doi:10.1172/JCI71471.
References
- 1.Lambrecht W, Kluth D. Hereditary multiple atresias of the gastrointestinal tract: report of a case and review of the literature. J Pediatr Surg. 1998;33(5):794–797. doi: 10.1016/S0022-3468(98)90225-1. [DOI] [PubMed] [Google Scholar]
- 2.Moore SW, de Jongh G, Bouic P, Brown RA, Kirsten G. Immune deficiency in familial duodenal atresia. J Pediatr Surg. 1996;31(12):1733–1735. doi: 10.1016/S0022-3468(96)90066-4. [DOI] [PubMed] [Google Scholar]
- 3.Rothenberg ME, White FV, Chilmonczyk B, Chatila T. A syndrome involving immunodeficiency and multiple intestinal atresias. Immunodeficiency. 1995;5(3):171–178. [PubMed] [Google Scholar]
- 4.Walker MW, Lovell MA, Kelly TE, Golden W, Saulsbury FT. Multiple areas of intestinal atresia associated with immunodeficiency and posttransfusion graft-versus-host disease. J Pediatr. 1993;123(1):93–95. doi: 10.1016/S0022-3476(05)81547-1. [DOI] [PubMed] [Google Scholar]
- 5.Gilroy RK, et al. Donor immune reconstitution after liver-small bowel transplantation for multiple intestinal atresia with immunodeficiency. Blood. 2004;103(3):1171–1174. doi: 10.1182/blood-2003-04-1187. [DOI] [PubMed] [Google Scholar]
- 6.Samuels ME, et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J Med Genet. 2013;50(5):324–329. doi: 10.1136/jmedgenet-2012-101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen R, et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol. 2013;132(3):656–664. doi: 10.1016/j.jaci.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno LA, et al. Severe combined immunodeficiency syndrome associated with autosomal recessive familial multiple gastrointestinal atresias: study of a family. Am J Med Genet. 1990;37(1):143–146. doi: 10.1002/ajmg.1320370133. [DOI] [PubMed] [Google Scholar]
- 9.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 10.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 11.Matsui T, et al. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140(3):647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu W, et al. Involvement of RhoA, ROCK I and myosin II in inverted orientation of epithelial polarity. EMBO Rep. 2008;9(9):923–929. doi: 10.1038/embor.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helms C, et al. The Tetratricopeptide repeat domain 7 gene is mutated in flaky skin mice: a model for psoriasis, autoimmunity, and anemia. Exp Biol Med (Maywood). 2005;230(9):659–667. doi: 10.1177/153537020523000908. [DOI] [PubMed] [Google Scholar]
- 14.Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci. 1995;20(7):257–259. doi: 10.1016/S0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- 15.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21(11):932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Scheufler C, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101(2):199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 17.White RA, McNulty SG, Nsumu NN, Boydston LA, Brewer BP, Shimizu K. Positional cloning of the Ttc7 gene required for normal iron homeostasis and mutated in hea and fsn anemia mice. Genomics. 2005;85(3):330–337. doi: 10.1016/j.ygeno.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien LE, Zegers MM, Mostov KE. Opinion: Building epithelial architecture: insights from three-dimensional culture models. Nat Rev Mol Cell Biol. 2002;3(7):531–537. doi: 10.1038/nrm859. [DOI] [PubMed] [Google Scholar]
- 19.Reed RA, et al. Morphogenesis of the primitive gut tube is generated by Rho/ROCK/myosin II-mediated endoderm rearrangements. Dev Dyn. 2009;238(12):3111–3125. doi: 10.1002/dvdy.22157. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz AA, Govek EE, Bottner B, Van Aelst L. Rho GTPases: signaling, migration, and invasion. Exp Cell Res. 2000;261(1):1–12. doi: 10.1006/excr.2000.5049. [DOI] [PubMed] [Google Scholar]
- 21.de Curtis I, Meldolesi J. Cell surface dynamics — how Rho GTPases orchestrate the interplay between the plasma membrane and the cortical cytoskeleton. J Cell Sci. 2012;125(pt 19):4435–4444. doi: 10.1242/jcs.108266. [DOI] [PubMed] [Google Scholar]
- 22.Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11(4):276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobelli J, Chmura SA, Buxton DB, Davis MM, Krummel MF. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat Immunol. 2004;5(5):531–538. doi: 10.1038/ni1065. [DOI] [PubMed] [Google Scholar]
- 24.Cote M, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119(12):3765–3773. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Goff C, et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2012;44(1):85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.