

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is often associated with overexpression of TGF-β. Given its tumor suppressor functions, it is unclear whether TGF-β is a valid therapeutic target for PDAC. Here, we found that proliferating pancreatic cancer cells (PCCs) from human PDAC patients and multiple murine models of PDAC (mPDAC) often exhibit abundant levels of phosphorylated retinoblastoma 1 (RB) and Smad2. TGF-β1 treatment enhanced proliferation of PCCs isolated from KrasG12D-driven mPDAC that lacked RB (KRC cells). This mitogenic effect was abrogated by pharmacological inhibition of type I TGF-β receptor kinase, combined inhibition of MEK/Src or MEK/PI3K, and restoration of RB expression. TGF-β1 promoted epithelial-to-mesenchymal transition (EMT), invasion, Smad2/3 phosphorylation, Src activation, Wnt reporter activity, and Smad-dependent upregulation of Wnt7b in KRC cells. Importantly, TGF-β1–induced mitogenesis was markedly attenuated by inhibition of Wnt secretion. In an in vivo syngeneic orthotopic model, inhibition of TGF-β signaling suppressed KRC cell proliferation, tumor growth, stroma formation, EMT, metastasis, ascites formation, and Wnt7b expression, and markedly prolonged survival. Together, these data indicate that RB dysfunction converts TGF-β to a mitogen that activates known oncogenic signaling pathways and upregulates Wnt7b, which synergize to promote PCC invasion, survival, and mitogenesis. Furthermore, this study suggests that concomitantly targeting TGF-β and Wnt7b signaling in PDAC may disrupt these aberrant pathways, which warrants further evaluation in preclinical models.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States, with a dismal overall 5-year survival rate of 6% (1). PDAC most often presents at an advanced stage with metastatic and/or extensive locally invasive disease and is associated with chemoresistance and desmoplasia (2–4). PDACs harbor major driver mutations in the KRAS oncogene (95%) and SMAD4 (55%), TP53 (70%), and CDKN2A (90%) tumor suppressor genes, the latter also being susceptible to epigenetic silencing (3, 5). In addition, there is overexpression of tyrosine kinase receptors and ligands (6), constitutive activation of prosurvival pathways, including AKT and NF-κB (7, 8), reactivation of developmental pathways, such as WNT and Notch (8, 9), and overexpression of transforming growth factor β (TGF-β) isoforms (10). TGF-β overexpression is associated with early recurrence following resection and decreased survival (10), and suppression of TGF-β actions in immune-deficient orthotopic mouse models of PDAC attenuates tumor growth and metastasis (11, 12). However, TGF-β also acts as a tumor suppressor, and in a genetically engineered mouse model (GEM) of PDAC in which oncogenic Kras is combined with p53 haploinsufficiency, disrupting TGF-β signaling enhanced PDAC progression (13). Therefore, the benefit of targeting TGF-β in PDAC is not clearly defined.
Oncogenic KRAS is the initiating molecular alteration in PDAC in humans (hPDAC) and mice (mPDAC) (14–19). KC (which stands for Kras;Pdx1-Cre recombinase) mice carry an oncogenic Kras (KrasG12D) allele that is silenced by an upstream LoxP-Stop-LoxP (LSL) element, but activated following Cre-mediated recombination (14). KC mice develop low-grade pancreatic intraepithelial neoplasia (PanIN) and acinar-to-ductal metaplasia (ADM) by 2 months of age (14). By 10 months, KC mice develop mPDAC at moderate penetrance (14). PanIN are an important feature of PDAC initiation in both humans and GEMs, and PanIN progression to mPDAC is accelerated by deletion of the p53 (16), p16Ink4a/p19Arf (Cdkn2a) (17), Smad4 (15), p16Ink4a (19), and RB (18) tumor suppressors. However, the RB1 gene is rarely mutated in hPDAC (20), and given the high frequency of KRAS and CDKN2A mutations occurring in conjunction with the overexpression of multiple tyrosine kinase receptors and increased cyclin D1 levels (6), the loss of RB function in PDAC presumably does not drive its pathobiology.
We report here that both RB and Smad2 were frequently phosphorylated in Ki67-positive pancreatic cancer cells (PCCs) in hPDAC, indicating that RB was functionally inactivated in proliferating PCCs in the face of robust TGF-β signaling. We also show that in murine PanIN arising in a GEM in which the pancreas only harbors oncogenic Kras (KC mice), there was a paucity of phosphorylated RB (p-RB) and Ki67, but abundant phosphorylated Smad2 (p-Smad2) and p21Waf1, a TGF-β–induced gene that inhibits proliferation (21). By contrast, in mice in which oncogenic Kras was combined with either p53 (KPC mice) or p16Ink4a (KIC mice) loss, we found that many PanIN and PCCs concomitantly exhibited p-RB, Ki67, and p-Smad2, whereas p21Waf1 was not detectable. Moreover, in mice with oncogenic Kras and Rb1 deletion (KRC mice), p-Smad2 was abundant in proliferating PanIN and PCCs, and in all cases of mice with increased p-Smad2 in PanIN and PCCs, stromal p-Smad2 was also abundant.
Using KRC-derived PCCs, which are devoid of RB, we demonstrated that TGF-β1 enhanced proliferation while increasing Smad2/3 phosphorylation and nuclear translocation, as well as activation of Src, PI3K, and ERK. We also show that TGF-β1–induced proliferation was suppressible by RB reexpression or Wnt7b inhibition. Moreover, in a syngeneic orthotopic model of PDAC, we found that SB505124 markedly attenuated PCC proliferation, tumor growth, and metastasis, as well as ascites and stroma formation. Thus, RB dysfunction is common in PDAC, and loss of RB function converts TGF-β from a tumor suppressor to a mitogen that enhances PCC proliferation.
Results
Both RB and Smad2 are phosphorylated in proliferating pancreatic cancer cells.
The ability of RB to inhibit cell cycle progression requires its activation through hypophosphorylation (22), and TGF-β–mediated growth inhibition of human PCCs depends on maintaining RB in a hypophosphorylated state (23). To determine whether RB is inactive in PDAC, we evaluated hPDAC tissue samples for the presence of hyperphosphorylated, inactive RB (Figure 1A). We found that p-RB was present in 44 of 58 PDAC samples in at least 10% of PCCs per high-power field (Figure 1B), suggesting that in 76% of these cancers RB was inactive. Moreover, in 8 of 8 tested PDACs, we found p-RB to be abundant in cancer cell nuclei in 72% of Ki67-positive cells (Figure 1B and Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI71526DS1), pointing to RB inactivation in proliferating PCCs. Phosphorylated Smad2 (p-Smad2) was also abundant in PCCs and stromal cell nuclei in all 8 tested PDACs and colocalized with 84% of Ki67-positive PCCs (Figure 1C and Supplemental Figure 1B). Analysis of thin (3 μm) serial sections revealed that nuclear p-RB and p-Smad2 were abundant in the PCCs, frequently colocalizing with Ki67 (Supplemental Figure 1C). By contrast, we rarely observed p21Waf1 immunoreactivity (Figure 1D).
Figure 1. RB is inactivated in proliferating PCCs exhibiting phospho-Smad2 in human PDAC.
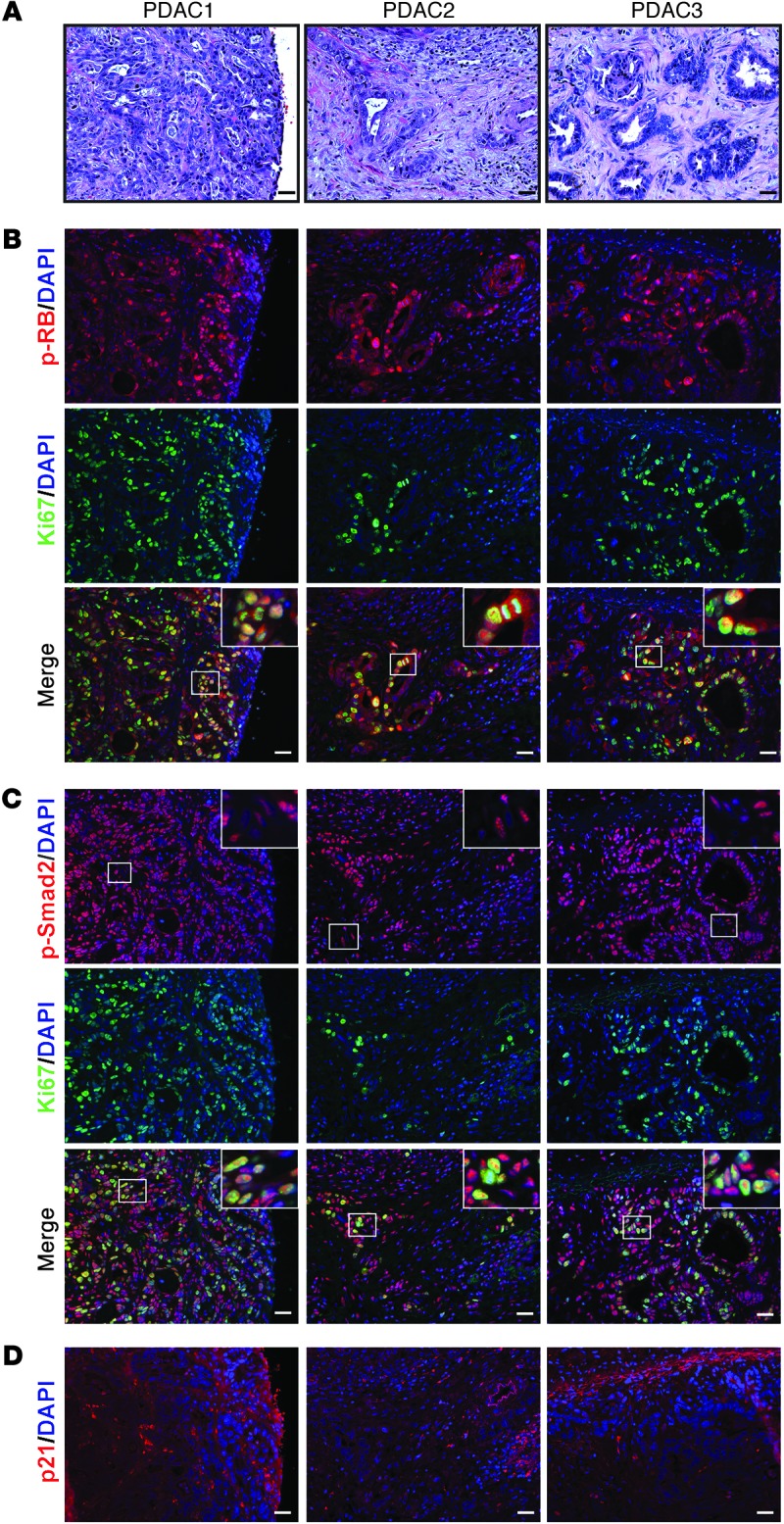
(A) H&E staining. Shown are representative images from 3 of 8 human PDAC tissues (PDAC1–3). (B) p-RB (red) was frequently detected in Ki67-positive cells (green) as evidenced by the overlay (merge). (C) p-Smad2 (red) was abundant in PCCs and adjacent stromal cells (insets show magnified images of boxed areas) and was frequently detected in Ki67-positive PCCs (green) as evidenced by the overlay (merge). (D) p21 (red) was mostly absent in PCC nuclei. Shown in B–D are serial sections of the PDACs in A. Scale bars: 50 μm.
We next examined p-Smad2, p-RB, p21Waf1, and Ki67 expression in murine pancreata to evaluate their status in relation to proliferation in mPDAC precursor lesions. In KC pancreata, p-Smad2 was present in PanIN and stromal nuclei, whereas p-RB was absent, Ki67 was rarely detected, and p21Waf1 was abundant (Supplemental Figure 2). Thus, active RB and upregulated p21Waf1 combined to constrain proliferation in KC PanIN. By contrast, PanIN in KIC and KPC pancreata exhibited Ki67-positive nuclei that frequently expressed p-RB, but not p21Waf1, despite abundant p-Smad2 positivity in PanIN and stromal nuclei and elevated Tgfb1 mRNA levels (Supplemental Figure 2 and Supplemental Figure 3A). In both GEMs, p-RB was detected in 76% of Ki67-positive PCCs that coexpressed nuclear p-Smad2 (Supplemental Figure 3, B–D), whereas in KRC pancreata, which also overexpress TGF-β1 (18), p-Smad2 was detected in stromal cells and all Ki67-positive PanIN and mPDAC cells (Supplemental Figure 4A). Thus, inactivation of RB removes negative growth restraints, including those mediated by p21Waf1, thereby enhancing proliferation in PCCs with active TGF-β signaling.
RB loss impairs TGF-β growth inhibition but not Smad-dependent signaling.
KRAS mutations are very frequent (95%) in hPDAC (24) and are the initiating event in KC, KPC, KIC, and KRC GEMs (14, 16–18). To investigate the effects of loss of RB function on TGF-β signaling in the context of oncogenic Kras, we used three RB-deficient murine PCCs established from KRC tumors (KRC1017, KRC1022-4, and KRC1022-5), two of which (KRC1022-4 and KRC1022-5) were of clonal origin. We found that TGF-β1 failed to inhibit proliferation in KRC1017 and KRC1022-5 cells and enhanced proliferation in KRC1022-4 cells by approximately 32% (Figure 2A). TGF-β1 also failed to induce cell cycle arrest and caused an average increase of 9% in the S phase in the three cell lines (Figure 2B) despite increased p107 expression as compared with p107 levels in murine PCCs established from KC tumors (KC1 and KC2), which express oncogenic Kras, but retain RB (ref. 18 and Supplemental Figure 5A). Moreover, in KC cells, TGF-β1 inhibited growth (Figure 2C) and decreased p-RB levels (Supplemental Figure 5B).
Figure 2. RB loss blocks TGF-β1–mediated growth inhibition.

(A) MTT assay shows that TGF-β1 did not inhibit the growth of KRC1017 (blue line) or KRC1022-5 (green line) cells and stimulated the growth of KRC1022-4 (orange line) cells. (B) TGF-β1 (0.5 nM) enhanced the S phase (open segments) in KRC cells by 9% (KRC1017), 5% (KRC1022-4) and 13% (1022-5). (C) TGF-β1 inhibited the growth of KC cells. *P < 0.009; **P < 0.001. (D) HA expression was detectable in HA-RB–transduced (R) KRC cells, but not parental- (P) or sham-transduced (S) cells. Shown is a representative blot from three independent experiments. ERK2 was used to confirm equivalent lane loading. (E) TGF-β1 (0.5 nM) inhibited proliferation of HA-RB–transduced KRC cells. *P < 0.006. (F) TGF-β1 (0.5 nM) enhanced proliferation of control (CT) or RB-M12–transfected KRC1022-4 cells. **P < 0.001. (A–C, E and F) Data represent the means ± SEM of three independent experiments.
We transduced KRC PCCs with a lentiviral construct encoding HA-tagged murine RB (HA-RB), which expressed Rb1 mRNA at the same level as that observed in KC PCCs (Supplemental Figure 5C) and partially restored TGF-β1–mediated growth inhibition (Figure 2, D and E). By contrast, sham-transduced KRC cells were either refractory to TGF-β1 (KRC1017) or were growth stimulated (KRC1022-4) (Supplemental Figure 4B). Moreover, we found that transfection with a construct encoding RB in which all potential phosphorylated sites were mutated to glutamate (25) did not restore TGF-β1–mediated growth inhibition (Figure 2F). Thus, functional RB is necessary for the cytostatic functions of TGF-β1 in KRC cells, consistent with previous findings in fibroblasts (26). Yet, in serum-starved KRC cells, we found that TGF-β1 induced Smad2 phosphorylation (Figure 3A), enhanced Smad2/3/4 nuclear translocation (Figure 3B), and increased Smad transcriptional activity, as demonstrated using two Smad-dependent luciferase reporter constructs, SBE4-Luc and p3TP-Lux (Supplemental Figure 4C). Moreover, in KRC mPDAC and PanIN, we detected phosphorylated Smad3 (p-Smad3) (Supplemental Figure 4D) in the nuclei, indicating that RB loss did not abrogate canonical TGF-β signaling in vivo.
Figure 3. TGF-β1 enhances Smad2-4 nuclear translocation without inducing p21Waf1 in KRC cells.

(A) TGF-β1 (0.5 nM) enhanced Smad2 phosphorylation in all KRC cells that also expressed Smad3 (lower band, second panel) and Smad4. (B) TGF-β1 (0.5 nM) enhanced Smad2/3 (red) and Smad4 (green) nuclear localization in KRC cells, shown by a merge with DAPI (blue). Scale bars: 50 μm. (B) TGF-β1 (0.5 nM) downregulated cyclin D1 in KRC cells. (C) TGF-β1 (0.5 nM) induced p21Waf1 in KC, but not KRC, cells. TGF-β1 (0.5 nM) did not upregulate p27Kip1 or p15Ink4b in KC or KRC cells. (D) TGF-β1 (0.5 nM) did not increase CDK2, CDK4, or CDK6 in KC or KRC cells, which had increased basal CDK2 levels compared with KC cells. (A and C–E) Representative blots from three independent experiments. ERK2 was used to confirm equivalent lane loading.
RB loss combined with oncogenic Kras leads to aberrant cell cycle regulation by TGF-β1.
TGF-β exerts its antimitogenic functions by inhibiting cyclin-dependent kinases (CDKs) primarily through CDK inhibitor (CKI) upregulation (21), but also through cyclin D1 downregulation (27). These events occur upstream of RB, leading to its activation through dephosphorylation, repressing E2F, and inducing cell cycle arrest. Therefore, we next evaluated the effects of TGF-β1 on cyclin D1, CKI, and CDK expression in KRC cells. We found that cyclin D1 was downregulated by TGF-β1 in KRC cells (Figure 3C), whereas p21Waf1 induction was minimal (Figure 3D). Although TGF-β1 did not induce p27Kip1, p15Ink4b, CDK2, CDK4, or CDK6 in KC or KRC cells (Figure 3, D and E), basal CDK2 levels were increased in KRC cells compared with KC cells (Figure 3E). Moreover, nuclear CDK2 immunoreactivity was present in KRC PanIN and mPDAC, whereas CDK4 and CDK6 exhibited weak immunoreactivity (Supplemental Figure 4E). Thus, in spite of functional pathways that downregulate cyclin D1, RB loss is associated with increased CDK2 expression and impaired TGF-β1–mediated upregulation of p21Waf1.
TGF-β1 promotes the proliferation of KRC cells in 3D culture.
To determine whether TGF-β exerts direct mitogenic effects on KRC cells, we examined TGF-β1 actions on colony formation and growth in 3D culture (28). We found that both KRC1017 and KRC1022-4 cells formed sphere-like colonies that grew between days 7 and 14 (Figure 4A). TGF-β1 colony size was markedly increased within 7 days, with each colony displaying a disorganized morphology associated with bud-like, invasive projections (Figure 4A). Compared with controls, TGF-β1–treated colonies were 76% larger by day 14 (Figure 4B), and nuclei in the control and TGF-β1–treated colonies frequently coexpressed p-Smad2 and Ki67 (Figure 4C). We observed that TGF-β1 also increased proliferating cell nuclear antigen (PCNA) levels in KRC cells grown in 3D culture (Figure 4D), confirming that TGF-β1 enhances PCC proliferation. By contrast, TGF-β1 failed to enhance the growth of RB-restored KRC cells (Figure 4E).
Figure 4. TGF-β1 stimulates KRC cell growth in 3D culture.

(A) Compared with controls, TGF-β1 (0.5 nM) dramatically enhanced growth and altered the morphology of KRC cells by days 7, 10, and 14. Insets (magnified images of boxed areas) show that control colonies are spherical, whereas TGF-β1–treated colonies have budding projections. (B) Compared with controls (black bars), TGF-β1 (0.5 nM) (white bars) significantly enhanced colony growth. *P < 0.0012. (C) Control- and TGF-β1–treated colonies contain nuclear p-Smad2 (red), Ki67 (green), and merge. (D) TGF-β1 (0.5 nM) markedly enhanced PCNA expression in KRC cells grown in 3D culture for 14 days. Shown is a representative blot from three independent experiments. Tubulin was used to confirm equivalent lane loading. (E) Restoring RB in KRC cells (HA-RB) prevented TGF-β1 growth stimulation. (A, C, and E) Representative images from three independent experiments. Scale bars: 50 μm.
SB505124, a TGF-β type I receptor inhibitor (TβRI) (29), decreased the number and size of control KRC colonies, markedly attenuated TGF-β1–induced colony growth (Figure 5, A and B), and prevented TGF-β1–mediated Smad2 phosphorylation (Figure 5C). TGF-β1 also enhanced AKT (S473), p70 S6 kinase (T421/S424 and T389), Src (Y419) (Figure 5D), and ERK1/2 (Supplemental Figure 6A) phosphorylation in KRC cells, indicating that TGF-β1 also activated noncanonical signaling. Moreover, PI3K (LY294002) and Src (dasatinib) inhibition reduced basal and TGF-β1–enhanced growth by 50% and 60%, respectively (Figure 5E), indicating that PI3K and Src activation contributes, in part, to the mitogenic effects of TGF-β1. Although the MEK1/2 inhibitor U0126, which inhibits ERK1/2, and the SAPK inhibitors SB203580 and SP600124, which inhibit p38/SAPK and JNK/SAPK, respectively, failed to block TGF-β1–enhanced cell growth (Figure 6E), the combination of U0126 with PI3K or Src inhibitors suppressed TGF-β1–induced proliferation (Supplemental Figure 6B).
Figure 5. TβRI, PI3K, and Src inhibition attenuates TGF-β1–induced growth of KRC cells.

(A) TGF-β1–stimulated (0.5 nM) KRC growth was attenuated by SB-505124 (2 μM). Shown are representative images from three independent experiments. Scale bars: 50 μm. (B) TGF-β1–enhanced KRC growth (white bars; *P < 0.006) was suppressed by SB-505124 (†P < 0.001). (C) SB-505124 blocked TGF-β1–enhanced Smad2 phosphorylation in KRC cells. Data are presented in separate panels, indicating that lysates were run on the same gel but not in adjacent lanes. (D) Time course (0–60 minutes) shows that TGF-β1 robustly increased Src, AKT, and S6K phosphorylation in KRC cells. Shown are representative blots from three independent experiments. Tubulin was used to confirm equivalent lane loading. (E) Compared with DMSO (D) controls, U0126 (U, 2 μM), SB203580 (SB, 5 μM), and SP600124 (SP, 10 μM) failed to abrogate TGF-β1–enhanced growth (white bars; **P < 0.001), whereas LY294002 (LY, 10 μM) and dasatinib (Das, 0.1 μM) significantly (** vs. *, P < 0.038) attenuated it.
Figure 6. KRC cells display activated TGF-β and Wnt pathways, and blocking Wnt signaling attenuates TGF-β1–enhanced growth.

(A and B) GSEA shows that genes upregulated in KRC cells (red) correlated with genes significantly upregulated by TGF-β, as determined by FWER (A, FWER = 0.001) or TGF-β and Wnt (B, FWER = 0.013). (C) β-catenin and p-GSK3β were present in KRC cells, and compared with controls, TGF-β1 (0.5 nM) increased p-GSK3β levels. (D) TGF-β1 (white bars) increased TOPFlash activity in KRC cells. *P < 0.012. Data represent the means ± SEM from three independent experiments. (E) IWP-2 (2 μM) or sFRP-1 (1 μg/ml) markedly attenuated TGF-β1–enhanced growth of KRC cells. Shown are representative images from day 14 of two independent experiments. Scale bars: 50 μm.
TGF-β1 enhances KRC growth, in part, through Wnt7b.
TGF-β pathways crosstalk with Wnt pathways (30), and aberrant Wnt pathway activation is important in PDAC pathobiology (8, 9, 31, 32). Moreover, KRC cells express all three TGF-βs, including high TGF-β2 levels (18). We therefore performed gene set enrichment analysis (GSEA) of array data, which revealed that compared with KC cells, genes upregulated in KRC cells correlated strongly with activated TGF-β and TGF-β/Wnt signaling (Figure 6, A and B). Wnt pathway activation was evidenced by elevated expression of Wnt7b and Wnt receptors Fzd2, 3, 6, and 9 compared with their expression in KC cells (Supplemental Figure 7). Importantly, KRC cells expressed active β-catenin that was devoid of phosphates at GSK3β-targeted residues and of inactive (phosphorylated) GSK3β (p-GSK3β), and the levels of p-GSK3β were increased by TGF-β1 (Figure 6C). TGF-β1 also increased TOPFlash reporter activity (Figure 6D), indicating that TGF-β1 enhances canonical Wnt signaling in KRC cells. Moreover, we observed that IWP-2, a Wnt secretion inhibitor (33), and secreted Frizzled-related protein 1 (sFRP1), a Wnt antagonist (34), markedly attenuated TGF-β1–stimulated growth of KRC1022-4 cells in 3D culture (Figure 6E).
Given the above, we next focused on Wnt7b, a mitogen (35, 36) whose expression is increased in human PCC lines (36–38). We confirmed by quantitative PCR (qPCR) that KRC cells expressed high Wnt7b mRNA levels as compared with KC cells (Figure 7A). Moreover, Wnt7b mRNA and protein levels were further increased by TGF-β1 (Figure 7, B and C). Conversely, inhibition of TβRI signaling with SB505124 or incubation with an anti–TGF-β2 neutralizing antibody suppressed basal Wnt7b mRNA levels (Figure 7, D and E). In silico analysis revealed that the murine Wnt7b promoter harbored two Smad binding elements (SBEs) at –903 bp (SBE1) and –771 (SBE2). Moreover, in murine PCCs derived from tumors in a Kras-driven GEM that lacks Smad4 (15), we found that basal Wnt7b mRNA levels were low and not inducible by TGF-β1 (Figure 7, A and F), suggesting that PCC-derived TGF-βs may enhance Wnt7b expression through Smad4-dependent pathways. In support of this conclusion, TGF-β1 increased the activity of a wild-type, but not a mutant, Wnt7b promoter-luciferase construct in KRC cells and failed to increase luciferase activity in KSC cells (Figure 7G). Moreover, in ChIP assays, Smad2/3/4 complexes were associated with the Wnt7b promoter in TGF-β1–stimulated KRC cells (Figure 7H). Given that Smad4 is mutated in 55% of PDACs (3), we evaluated PDAC tissues for Wnt7b and Smad4 coexpression. Wnt7b exhibited moderate-to-strong immunoreactivity in PCCs in 28 of 58 PDACs, and only these samples exhibited nuclear Smad4 immunoreactivity (Figure 7I). By contrast, when Smad4 was undetectable, Wnt7b immunoreactivity was negative to weak (Figure 7I).
Figure 7. TGF-β is a driver of Wnt7b expression in KRC cells.

(A) KRC cells (white bars) expressed high Wnt7b mRNA levels compared with KC (control, black bars; *P < 0.04) and KSC (gray bars; *P < 0.02) cells. (B and C) TGF-β1 (0.5 nM) increased Wnt7b mRNA (B, white bars; *P = 0.032) and protein (C) levels in KRC cells. (C) Representative blot from two independent experiments. ERK2 was used to confirm equivalent lane loading. (D and E) SB505124 (D, 2 μM, white bars) and anti–TGF-β2 (E, 5 mg/ml, white bars) reduced Wnt7b mRNA levels in KRC cells (**P < 0.001) compared with their respective controls (black bars). (F) TGF-β1 (0.5 nM, white bars) did not alter Wnt7b mRNA levels in KSC cells. (G) TGF-β1 (0.5 nM) increased wild-type Wnt7b promoter activity (left panel) in KRC cells (*P = 0.002), whereas KSC cells had low basal activity that was not increased by TGF-β1. TGF-β1 failed to increase the activity of an SBE-mutated Wnt7b reporter in KRC cells (right panel). (H) ChIP assays revealed that Smad2-4 (Smad) bound the Wnt7b promoter in TGF-β1–stimulated (0.5 nM) KRC cells. Left panel: quantification; **P < 0.001. Right panel: representative gel image from three independent experiments. (I) hPDAC PCCs with strong cytoplasmic Wnt7b immunoreactivity showed nuclear Smad4 (top panels), whereas hPDACs that lack Wnt7b did not demonstrate Smad4 immunoreactivity (bottom panels). (I) Representative images from two PDACs. Scale bars: 50 μm. (A, B, and D–H) Data represent the means ± SEM from three independent experiments.
We next assessed the role of Wnt7b in TGF-β1–stimulated mitogenesis by silencing Wnt7b in clonal KRC1022-4 cells. siRNA targeting of murine Wnt7b reduced Wnt7b mRNA levels (62%) and abrogated TGF-β1–induced TOPFlash activity (Figure 8, A and B). Moreover, Wnt7b silencing partially restored the growth inhibitory effects of TGF-β1 in KRC cells grown on plastic (Figure 8C) and markedly attenuated TGF-β1–enhanced growth in 3D culture (Figure 8, D and E), indicating that the mitogenic effects of TGF-β1 are mediated in part by Wnt7b.
Figure 8. Wnt7b silencing attenuates TGF-β–activated Wnt signaling and TGF-β–stimulated growth.

(A) Wnt7b siRNA (white bar) reduced Wnt7b mRNA levels in KRC cells by 62% (**P < 0.001) compared with control siRNA (gray bar) or parental (black bar). (B) Compared with control siRNA, Wnt7b siRNA blocked TGF-β1–enhanced (white bars) TOPFlash activity (**P < 0.001). (C) TGF-β1 (0.5 nM, 48 hours) slightly enhanced KRC growth (*P = 0.003) on plastic (2D), but with Wnt7b siRNA, TGF-β1 exerted growth-inhibitory effects. (D) Quantitation of TGF-β1–treated colonies grown in 3D culture for 14 days shows that Wnt7b siRNA abrogated TGF-β1–enhanced KRC growth (*P < 0.05). (E) Wnt7b siRNA (right panels) markedly attenuated TGF-β1–enhanced KRC cell growth compared with control siRNA (left panels). Shown are representative images on day 14 from three independent experiments. Scale bars: 50 mm. (A–D) Data represent the means ± SEM from three independent experiments.
TGF-β1 induces epithelial-to-mesenchymal transition and invasion in KRC cells.
We next sought to determine whether TGF-β1 induces epithelial-to-mesenchymal transition (EMT) and invasion in KRC cells. Within 48 hours, TGF-β1 promoted cell elongation, a spindly morphology, and disruption of tightly packed epithelial-like arrangements (Figure 9A). Although TGF-β1 did not markedly decrease E-cadherin expression (Figure 9B), we found that it caused its redistribution to the cytoplasm with predominant perinuclear localization (Figure 9A). TGF-β1 also upregulated N-cadherin in KRC1017 and KRC1022-5 and the EMT-inducing transcription factors ZEB1 and SNAIL in all three cell lines (Figure 9B). In parallel, we found that KRC cell invasion was enhanced by an average of 758% (Figure 9C). Given the role of Wnts in EMT, invasion, and metastasis (39), we next assessed the role of Wnt7b in TGF-β1–enhanced invasion of KRC cells. In KRC1022-4 cells transfected with nontargeting control siRNA, TGF-β1 enhanced invasion by 768%, and this effect was blocked by siRNA targeting of Wnt7b (Figure 9C). These results indicate that TGF-β1 promotes EMT and enhances invasion in KRC cells in a Wnt7b-dependent manner.
Figure 9. TGF-β1 induces EMT and invasion in KRC cells, whereas TβRI inhibition attenuates tumor growth and prolongs survival.

(A) TGF-β1 (0.5 nM) altered the epithelial morphology of KRC cells (phase contrast). Immunofluorescence for E-cadherin (red) and N-cadherin (green) shows that TGF-β1 altered the localization of E-cadherin (insets) and changed cell morphology. Scale bars: 50 μm. (B) TGF-β1 decreased E-cadherin and upregulated N-cadherin, ZEB1, and SNAIL. Shown are representative blots from three independent experiments. ERK2 was used to confirm equivalent lane loading. (C) TGF-β1 (white bars) enhanced invasion (left panel), and using KRC1022-4 cells, Wnt7b siRNA blocked this effect. *P < 0.032. Data represent the means ± SEM from three independent experiments. (D) High-resolution ultrasound images show that vehicle-treated mice harbored large tumors (T, arrows) and had formed ascites (A), as evidenced by color doppler (blue), whereas SB505124-treated tumors were smaller and ascites were not detectable. Shown are representative images from day 17. (E) Quantitation shows that SB505124 (SB, white bars) significantly attenuated tumor volumes. *P < 0.031. (F) Kaplan-Meier survival curves reveal that SB-505124 significantly prolonged the survival of mice bearing KRC1017- (green versus blue line, P = 0.026) and KRC1022-4–derived (red versus orange line, P = 0.007) tumors. Horizontal line indicates 50% survival. (G) Compared with vehicle (V, black bars), SB505124 (SB, white bars) significantly reduced the percentage of CK19-positive PCCs with Ki67. **P < 0.001. (E and G) Data represent the means ± SEM.
TβRI inhibition attenuates tumor growth in vivo.
We next evaluated the impact of TGF-β signaling blockade on tumor growth and metastasis in the context of an intact immune system using KRC1017 and KRC1022-4 cells in a syngeneic orthotopic model. Treatments began on day 10 after intrapancreatic PCC injection, when the mean volumes of KRC1017 and KRC1022-4 tumors were 57 mm3 and 55 mm3, respectively (Supplemental Figure 8A). By day 17, tumors in vehicle-treated mice had grown by 810% and caused abundant ascites, whereas in SB505124-treated mice, tumors had grown by 195% and were devoid of ascites (Figure 9, D and E). Overall, 100% of vehicle-treated mice succumbed to disease by day 26 (Figure 9F) and frequently (12 of 14) displayed abundant peritoneal seeding (Supplemental Figure 8B). Moreover, some mice (4 of 14) displayed grossly evident liver metastases, and three of these mice harbored multiple pulmonary micro-metastases (Supplemental Figure 8C). By contrast, SB505124-treated mice survived as long as 50 days (Figure 9F) and never exhibited ascites (Figure 9D), peritoneal seeding (Supplemental Figure 8B), or distant metastases. These tumors exhibited decreased PCC proliferation (Figure 9G and Supplemental Figure 9A), stroma formation (Supplemental Figure 8, D and E), and N-cadherin and ZEB1 immunoreactivity (Supplemental Figure 9B). We found WNT7b immunoreactivity to be heterogeneous, with foci of mild, moderate, and strong intensity. Compared with PCCs in vehicle-treated tumors, we observed that Wnt7b immunoreactivity was markedly reduced in the SB505124-treated group (Supplemental Figure 9, C and D), indicating that SB505124 suppressed Wnt7b expression in vivo. Thus, disruption of TGF-β signaling in a syngeneic model with an intact immune system induces multiple beneficial actions.
Discussion
One of the hallmarks of cancer is an insensitivity to growth inhibitory pathways (40). In the case of TGF-βs, loss of growth inhibition has been associated with an enhanced tumor progression attributed to its paracrine effects on the tumor microenvironment (41). Here, we determined that both Smad2 and RB were frequently phosphorylated in proliferating cancer cells and stromal cells in hPDAC. Proliferating cancer cells and PanIN cells in mPDAC in KrasG12D-driven models that lack either p16Ink4a (KIC) or p53 (KPC) also exhibited p-Smad2 and p-RB, and p-Smad2 was also abundant in the stromal cells. Moreover, PanIN, stroma, and PCCs in KRC mPDAC (devoid of RB) were highly proliferative (18) and exhibited many p-Smad2– and p-Smad3–positive nuclei. By contrast, PanIN in a murine model driven by KrasG12D alone were not highly proliferative and exhibited abundant p-Smad2 phosphorylation and low levels of p-RB. Taken together, these observations indicate that RB dysfunction is common in hPDAC and in biologically aggressive GEMs of PDAC (16, 17), that KrasG12D alone does not induce RB dysfunction, and that in addition to exerting paracrine effects on the tumor microenvironment, TGF-βs may exert autocrine effects that activate cell-autonomous pathways in PCCs.
There are multiple mechanisms by which RB can be inactivated or suppressed in PDAC. First, based on loss of the CDKN2A gene, the RB/p16 pathway was reported to be inactive in 98% of PDAC cases (42). Second, the presence of oncogenic KRAS in 95% of PDACs (24), occurring in conjunction with the overexpression of mitogenic tyrosine kinase receptors and their ligands (6), could combine to induce RB phosphorylation and inactivation. Third, 50% of PDAC cases overexpress Smad7, which suppresses TGF-β1–mediated growth inhibition by maintaining RB in a hyperphosphorylated state (23). Fourth, microRNA-132 (miRNA-132) and miRNA-212 are upregulated in PDAC and act to suppress RB expression in PCCs (43). Fifth, PDACs occasionally harbor RB1 mutations (20). In the present study, we confirm our previous finding that RB activation is crucial for TGF-β–mediated growth inhibition (23). Thus, cells devoid of RB (KRC cells) were no longer growth inhibited by TGF-β, and growth inhibition was restored when RB was reexpressed to levels similar to those present in KC cells. Moreover, using a mutant RB construct, we demonstrated that RB genetic ablation is equivalent to phosphorylation-mediated inhibition. Collectively, these findings indicate that in addition to interfering with negative growth constraints, RB loss converted TGF-β to a PCC mitogen.
Previous studies have shown that TGF-β1 stimulates the proliferation of human prostate epithelial cells that harbor mutated HRAS (44), that tyrosine phosphorylation on type II TGF-β receptor (TβRII) leads to the recruitment of GRB2, which allows TGF-β to stimulate proliferation in breast cancer cells (45), and that high levels of the type III TGF-β receptor (TβRIII) enable TGF-β and BMP-2 to enhance proliferation and suppress their ability to upregulate p21 and p27 (46). By contrast, several lines of evidence in the present study demonstrate that in the absence of functional RB, TGF-β1 enhances mitogenesis by activating both noncanonical and canonical TGF-β signaling. First, the mitogenic effect of TGF-β was blocked by TGF-β receptor kinase inhibition with SB505124, indicating that it required TβRI activation. Second, we observed that TGF-β activated noncanonical pathways (41) that have crucial roles in mitogenesis (MEK/ERK), cell survival (PI3K/p-AKT), and invasion/metastases (p-Src[Y419]). Third, we found that TGF-β–induced mitogenesis was partially blocked by targeting either PI3K or Src, MEK and PI3K, or MEK and Src, confirming that these pathways are important mediators of TGF-β–induced mitogenesis. Fourth, TGF-β1–induced proliferation was markedly attenuated by IWP-2–mediated inhibition of Wnt processing and secretion and by sFRP-1–mediated Wnt ligand sequestration, pointing to a Wnt-mediated mechanism for TGF-β–activated mitogenesis. Fifth, we found that TGF-β1 increased Wnt7b expression, whereas silencing Wnt7b markedly attenuated TGF-β1–induced mitogenesis and TOPFlash activity, confirming the role of Wnt7b in this novel pathway. Mechanistically, TGF-β1 enhanced Wnt7b promoter activity in a Smad4-dependent manner, as we confirmed by using luciferase readout and ChIP assays and by documenting the absence of a response in Smad4-null cells. Thus, loss of RB function converts TGF-β1 from an inhibitor of PCC proliferation that promotes tumor growth through paracrine actions, to a mitogen that also acts directly to stimulate PCC proliferation by activating canonical and noncanonical TGF-β signaling cascades (Figure 10).
Figure 10. TGF-β1 enhances PCC proliferation, invasion, and EMT when RB is inactive.

Phosphorylation of RB, Smad7 overexpression, suppression of RB by miRNA-132 and miRNA-212, and mutations in the RB1 gene (rare) are mechanisms by which RB can be inactivated in PCCs, leading to loss of TGF-β growth inhibition. Inactive RB, combined with TGF-β, increases Wnt7b expression through canonical (Smad4-dependent) TGF-β signaling pathways, which increases PCC proliferation EMT and invasion. TGF-β also enhances PCC proliferation through the activation of noncanonical signaling pathways, including ERK, Src, and PI3K.
Active RB represses the transcription of E2F-regulated genes during the G1 phase of the cell cycle (47), whereas p21Waf1 inactivates cyclin/CDK complexes during G1, S, and G2 phases, inhibits PCNA-dependent DNA synthesis, and inactivates E2F (48). Consequently, under nonpathological conditions, RB and p21Waf1 induce cell cycle arrest. However, p53 and Smad4 are frequently mutated in hPDAC (3), and p53 and TGF-β are major inducers of p21Waf1. Indeed, we found that p21Waf1 expression was attenuated in many hPDAC cases and in KPC, KIC, and KRC mPDACs. Moreover, RB reexpression failed to restore TGF-β1–mediated induction of p21Waf1 (not shown), indicating that RB-independent mechanisms contribute to its attenuated expression. By contrast, p21Waf1 was abundant in KC mPDAC. These observations suggest that attenuated p21Waf1 expression is common in hPDAC and mPDAC whenever oncogenic KRAS is combined with tumor suppressor gene loss. Given that p21Waf1 preferentially inactivates CDK2 (48), that KRC cells had elevated CDK2 levels, and that KRC PanIN and mPDAC exhibited nuclear CDK2 immunoreactivity, our observations also suggest that p21Waf1 dysregulation, combined with increased CDK2 expression, may further enhance PCC proliferation in KRC mPDAC.
KRC cells express high levels of TGF-β2, Wnt7b, and total β-catenin and exhibit strong nuclear p-Smad2 immunoreactivity, raising the possibility that TGF-βs act in an autocrine manner to upregulate deleterious TGF-β and Wnt signaling. In agreement with this conclusion, we found that Wnt7b expression in KRC cells was suppressed by SB505124 and by a TGF-β2–neutralizing antibody. TGF-β1 also increased Wnt reporter activity, consistent with activation of canonical Wnt/β-catenin pathways, and this effect was abrogated by Wnt7b silencing. Similarly, we observed that TGF-β1 enhanced invasion in KRC cells, and this effect was also blocked by Wnt7b silencing. Moreover, our GSEA analysis indicated that the gene expression profile of KRC cells exhibited statistically significant concordance with genes upregulated by TGF-β and Wnt pathway activation (Figure 6, A and B). Although there are no reports of the APC or axin mutations in hPDAC that lead to increased Wnt/β-catenin activity in certain cancers (49, 50), the relevance of the current observations to hPDAC is underscored by the presence of moderate-to-strong Wnt7b immunoreactivity in the cancer cells in 45% of hPDAC tissues, which correlated with the presence of nuclear Smad4. Moreover, hPDACs exhibit aberrant Wnt/β-catenin activation (31, 51), and some cultured human PCCs have high β-catenin activity (36) and express elevated levels of Wnt7b (36–38). Taken together, these data indicate that TGF-βs can act on PCCs in an autocrine manner to enhance Wnt7b expression and to crosstalk with Wnt signaling cascades.
PCCs often exhibit mesenchymal cell features, especially following EMT induction by TGF-β1, leading to enhanced invasion and expression of markers associated with tumor-initiating cells (52). We found that in KRC cells, in addition to enhancing proliferation, TGF-β1 readily induced EMT, evidenced morphologically and by decreased E-cadherin expression and its redistribution to the cytoplasm, as well as by increased N-cadherin, ZEB1, and SNAIL expression. Moreover, in vivo, the administration of SB505124 ten days after PCC injection into the pancreas greatly attenuated PCC proliferation, tumor growth, metastasis, and expression of the mesenchymal markers N-cadherin and ZEB1. We found that SB505124 also markedly decreased Wnt7b expression and suppressed stroma formation in the orthotopic tumors, and targeting the stroma in PDAC may facilitate drug delivery to the cancer cells within the tumor mass (53). Therefore, our findings indicate that targeting TGF-β signaling exerts multiple beneficial effects that prolong survival, disrupt crosstalk between TGF-β and Wnt, inhibit the metastatic process, and may also allow for improved delivery of chemotherapeutic agents.
TGF-β and Wnt ligands have been implicated in maintaining a pool of tumor-initiating cells that exhibit enhanced metastatic potential (39), and it may take 20 years of tumor progression for PDAC to acquire the capacity to form distant metastases (54). Therefore, our findings suggest that combinatorial targeting of TGF-β and Wnt ligands, such as Wnt7b, may be a useful approach for overcoming RB dysfunction, suppressing metastasis formation, and enhancing the efficacy of other therapeutic strategies in PDAC. Given the advanced stage of PDAC at clinical presentation, our findings also indicate that targeting TGF-β is unlikely to interfere with its tumor suppressor functions, since these functions are lost, and TGF-βs are acting as mitogenic tumor promoters in advanced stages of PDAC.
Methods
Mice.
KRC mice, originally described as Rb/K mice, were generated as described (18). Conditional Ink4a/ArfLoxP/LoxP and Smad4LoxP/LoxP mice were provided by N. Bardeesy (Harvard Medical School, Boston, Massachusetts, USA), and Trp53LoxP/LoxP mice were from the NCI Mouse Repository (strain 01XC2). KIC, KSC, and KPC mice were generated as described (15–17). Mouse nomenclature is based on the classification used for other GEMs of PDAC, indicating that they express oncogenic Kras (K) and lack RB (R), INK4a/ARF (I), Smad4 (S), or p53 (P) in the pancreas due to PDX1-driven Cre (C) recombination. Mice were maintained on a mixed FVB, 129, and C57Bl/6 genetic background.
Cell lines.
Murine PCCs were generated using standard cell isolation and culture procedures (18).
Immunostaining.
Paraffin-embedded hPDAC tissues were obtained from the Indiana University Simon Cancer Center Solid Tissue Bank. Pancreata from GEMs and syngeneic orthotopic tumors were processed and stained as described (18). The antibodies used were: phospho-RB-XP(Ser807/Ser811) (Cell Signaling Technology); phospho-Smad2(Ser465/Ser467) (Millipore); Ki67 (Novocastra); phospho-Smad3(Ser423/Ser425) (Abcam); p21, CDK4, and CDK6 (Santa Cruz Biotechnology Inc.); Wnt7b (Aviva Systems Biology); N-cadherin (BD Pharmingen); ZEB1 (Novus Biologicals); CK19 (TromaIII; Developmental Studies Hybridoma Bank, University of Iowa); Smad4 (Leica Biosystems); and CDK2 (Bethyl Laboratories).
Images were acquired using an Olympus BX60 microscope and a QImaging EXi Blue camera. For quantitation, images were acquired from three different fields at ×20 magnification, and cells were counted using ImageProPlus version 7.0 software (Media Cybernetics). For immunofluorescence, all images were acquired at the same exposure.
qPCR.
qPCR was performed as described (18). Tgfb1 mRNA levels were quantified in KC, KIC, KPC, and littermate control pancreata (4 per group). Expression levels were calculated relative to KC controls. For Wnt7b and Rb1 in PCCs, Ct values were normalized to β-actin, and changes were calculated relative to controls.
Cell proliferation assays.
Cell proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay (55). For transfections, KRC cells were transfected with mutant RB (M12), provided by D. Goodrich (Roswell Park Cancer Institute, Buffalo, New York, USA) (25) or control siRNA or Wnt7b-targeting siRNA (ON-TARGETplus; Dharmacon, Thermo Scientific) using Lipofectamine 2000 (Invitrogen). The means for controls were normalized to 100%. For cell cycle analysis, KRC cells were stained using PI/RNase Buffer (BD Pharmingen), and DNA content was determined as described (56). For RB restoration, KRC cells were transduced as described in (56), and proliferation was assessed by MTT assay.
Immunoblotting.
Immunoblots were performed as described (18). The antibodies used were: phospho-Smad2 (Millipore); cyclin D1, SNAIL, phospho- and total AKT, phospho- and total Src, phospho- and total S6K, non–phospho-β-catenin, and phospho- and total GSK3β (all from Cell Signaling Technology); E- and N-cadherin (BD Pharmingen); ZEB1 (Novus Biologicals); Smad2/3, Smad4, p21, p15Ink4b, p27Kip1, CDK2, CDK4, CDK6, and ERK2 (all from Santa Cruz Biotechnology Inc.); PCNA (Leica Biosystems); and tubulin (Sigma-Aldrich).
Immunofluorescence on KRC cells.
Immunofluorescence was performed as described (18). The following antibodies were used: Smad2/3 and Smad4 (Santa Cruz Biotechnology Inc.); and E- and N-cadherin (BD Pharmingen). Phase-contrast images were taken prior to fixation using an inverted Nikon Diaphot 300 microscope.
Luciferase assays.
Transcriptional activity was assessed as previously described (18) using SBE4-Luc (plasmid 16495), p3TP-Lux (plasmid 11767; Addgene), or TOPFlash (provided by M. Waterman, University of California Irvine, Irvine, California, USA). For wild-type Wnt7b, 1.1 kb of the Wnt7b promoter was PCR amplified from KRC cells and inserted between Kpn1/Xho1 sites of pGL3-luciferase (Promega). SBE1 and SBE2 were then mutated (see Supplemental Table 1 for the primer sequences). To control for transfection efficiency, Renilla (pRL-TK; Promega) was cotransfected, and luciferase activity was normalized to Renilla.
3D culture.
KRC cells were grown in 3D culture (28). Four days after plating and every three days thereafter, cells were treated with control media or media with TGF-β1, with or without inhibitors. The final concentration of DMSO in all experiments was 0.05%. For Wnt7b silencing, KRC cells were plated, transfected with control siRNA or Wnt7b-targeting siRNA for 24 hours, then grown in 3D culture. Colony growth was quantified on day 14 using the MTT growth assay (28), and the colony area was determined using ImageProPlus, version 7.0. The area of control-treated colonies was normalized to 100%, and changes in growth were calculated as a percentage of control.
For immunofluorescence, colonies were fixed in 10% formalin, embedded, and frozen (Tissue-Tek O.C.T. Compound). Ten-micrometer sections were prepared using a Leica CM1900 cryostat and stained for phospho-Smad2 and Ki67.
ChIP assays.
KRC cells were plated in 15-cm dishes, serum starved, then treated with TGF-β1 (0.5 nM, 24 hours). ChIP assays were performed with control IgG (Santa Cruz Biotechnology Inc.) or Smad2/3 and 4 antibodies using a Simple ChIP Enzymatic Chromatin IP kit (Cell Signaling Technology) according the manufacturer’s recommendations. The Wnt7b promoter was PCR amplified (Supplemental Table 1). PCR products were run on an agarose gel, bands were quantified with ImageJ software (NIH), and data were normalized to input.
Microarray and GSEA analysis.
Three Agilent whole-mouse genome microarrays with total RNA from three different RNA isolations from KC (Cy3-labeled) and KRC (Cy5-labeled) cells were performed by Miltenyi Biotec. All bioinformatic and statistical analyses were performed by Miltenyi Biotec. Briefly, intensity values were extracted, converted to log2 scale, and locally weighted regression (LOESS) normalization was performed. Heatmaps of each microarray were generated, reflecting normalized intensity values. Unpaired Student’s t tests with equal variance were used to test log2-normalized data for significant differences. P values were subjected to multiple testing (Benjamini-Hochberg) correction to reduce the false discovery rate (FDR). P < 0.001 was considered statistically significant. Microarray data are available at the European Bioinformatics Institute (accession number E-MEXP-3988).
For GSEA, normalized, log2-transformed data were prepared in GSEA format (57). A custom chip file that mapped probesets from this array to HUGO gene symbols was generated. GSEA was run and compared with gene sets upregulated by TGF-β1 (58) or TGF-β1 and Wnt3a (59) using version 2.0.13 of the command line jar application on collections from the Molecular Signatures Database, version 4.0 (57, 60). Significant gene sets with a family-wise error (FWER) less than 0.05 were then identified.
Invasion assays.
Invasion assays were performed as described (55).
Orthotopic model.
KRC cells (200,000 cells) were injected into the pancreas of 28 (20 males, 8 females) 10-week-old syngeneic mice using KRC1017 and KRC1022-4 cells (10 males, 4 females per cell line). On day 10, mice were randomized to vehicle and SB505124 treatment groups (7 mice per group; 5 males and 2 females in each). Before treatment began, tumors were imaged using Vevo2100 high-resolution ultrasound (VisualSonics). Mice were injected i.p. daily with vehicle (75% DMSO, 25% saline) or 10 mg/kg SB505124, and 7 days later tumors were imaged. 3D abdominal scans were acquired on days 10 (pretreatment) and 17 (1 week after treatment), and tumor volumes were calculated using Vevo2100 System software. Daily injections continued up to day 50, and mice were sacrificed as they became moribund. For immunofluorescence, 5 of 7 tumors per group were stained as described above. Masson’s trichrome staining was performed using a Chromaview kit (Richard-Allan Scientific). Images from four different fields of each tumor were acquired at ×20 magnification.
Statistics.
One-way ANOVA with Tukey’s post-hoc test and 1-tailed Student’s t tests were used to test for significant differences using Sigma Plot version 11.0 software (Systat Software). All statistics were calculated on triplicate experiments. For the syngeneic orthotopic model, Sigma Plot was used for log-rank survival analysis, and 1 mouse from the SB505124 group was censored from the survival analysis since it died on day 10, shortly after the first injection. For all statistics, P < 0.05 was considered statistically significant.
Study approval.
Approval for the acquisition of human tissues was granted by the Office of Research Administration of Indiana University. All mouse studies were approved by the IACUC of Indiana University.
Supplementary Material
Acknowledgments
This manuscript is dedicated to the fond memory of Patricia Lorenzo, PhD, a former member of the Molecular Oncogenesis NIH study section and the University of Hawaii Cancer Center, who brightened deliberations with her compassion, affability, and dedication. Her remarkable courage in the face of terminal cancer has been inspirational. This work was supported by National Cancer Institute grant CA-R37-075059 (to M. Korc). We thank the Indiana University Simon Cancer Center Solid Tissue Bank for pancreatic cancer tissues and David Goodrich (Roswell Park Cancer Institute) for the mutant RB construct.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(1):338–352. doi:10.1172/JCI71526.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Xiong HQ, Carr K, Abbruzzese JL. Cytotoxic chemotherapy for pancreatic cancer: advances to date and future directions. Drugs. 2006;66(8):1059–1072. doi: 10.2165/00003495-200666080-00003. [DOI] [PubMed] [Google Scholar]
- 3.Kern SE. Molecular genetic alterations in ductal pancreatic adenocarcinomas. Med Clin North Am. 2000;84(3):691–695. doi: 10.1016/s0025-7125(05)70251-0. [DOI] [PubMed] [Google Scholar]
- 4.Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194(4 suppl):S84–S86. doi: 10.1016/j.amjsurg.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCleary-Wheeler AL, et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013;328(2):212–221. doi: 10.1016/j.canlet.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Preis M, Korc M. Kinase signaling pathways as targets for intervention in pancreatic cancer. Cancer Biol Ther. 2010;9(10):754–763. doi: 10.4161/cbt.9.10.11534. [DOI] [PubMed] [Google Scholar]
- 7.Ottenhof NA, de Wilde RF, Maitra A, Hruban RH, Offerhaus GJ. Molecular characteristics of pancreatic ductal adenocarcinoma. Patholog Res Int. 2011;2011:620601. doi: 10.4061/2011/620601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCleary-Wheeler AL, McWilliams R, Fernandez-Zapico ME. Aberrant signaling pathways in pancreatic cancer: A two compartment view. Mol Carcinog. 2012;51(1):25–39. doi: 10.1002/mc.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10(10):683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friess H, et al. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105(6):1846–1856. doi: 10.1016/0016-5085(93)91084-u. [DOI] [PubMed] [Google Scholar]
- 11.Rowland-Goldsmith MA, Maruyama H, Kusama T, Ralli S, Korc M. Soluble type II transforming growth factor-beta (TGF-β) receptor inhibits TGF-β signaling in COLO-357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin Cancer Res. 2001;7(9):2931–2940. [PubMed] [Google Scholar]
- 12.Melisi D, et al. LY2109761, a novel transforming growth factor-beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7(4):829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hezel AF, et al. TGF-β and αvβ6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 2012;72(18):4840–4845. doi: 10.1158/0008-5472.CAN-12-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 15.Bardeesy N, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20(22):3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bardeesy N, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103(15):5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguirre AJ, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrière C, et al. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterology. 2011;141(3):1091–1101. doi: 10.1053/j.gastro.2011.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu W, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2(11):862–873. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang WS, et al. Genome-wide characterization of pancreatic adenocarcinoma patients using next generation sequencing. PLoS One. 2012;7(10):e43192. doi: 10.1371/journal.pone.0043192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derynck R, Feng X-H. TGF-β receptor signaling. Biochim Biophys Acta. 1997;1333(2):F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- 22.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 23.Boyer Arnold N, Korc M. Smad7 abrogates transforming growth factor-β1-mediated growth inhibition in COLO-357 cells through functional inactivation of the retinoblastoma protein. J Biol Chem. 2005;280(23):21858–21866. doi: 10.1074/jbc.M500583200. [DOI] [PubMed] [Google Scholar]
- 24.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrientes S, Cooke C, Goodrich DW. Glutamic acid mutagenesis of retinoblastoma protein phosphorylation sites has diverse effects on function. Oncogene. 2000;19(4):562–570. doi: 10.1038/sj.onc.1203332. [DOI] [PubMed] [Google Scholar]
- 26.Herrera RE, Makela TP, Weinberg RA. TGFβ-induced growth inhibition in primary fibroblasts requires the retinoblastoma protein. Mol Biol Cell. 1996;7(9):1335–1342. doi: 10.1091/mbc.7.9.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko TC, Sheng HM, Reisman D, Thompson EA, Beauchamp RD. Transforming growth factor-β 1 inhibits cyclin D1 expression in intestinal epithelial cells. Oncogene. 1995;10(1):177–184. [PubMed] [Google Scholar]
- 28.Sempere LF, Gunn JR, Korc M. A novel 3-dimensional culture system uncovers growth stimulatory actions by TGFβ in pancreatic cancer cells. Cancer Biol Ther. 2011;12(3):198–207. doi: 10.4161/cbt.12.3.15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DaCosta Byfield S, Major C, Laping NJ, Roberts AB. SB-505124 is a selective inhibitor of transforming growth factor-β type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2004;65(3):744–752. doi: 10.1124/mol.65.3.744. [DOI] [PubMed] [Google Scholar]
- 30.Attisano L, Labbe E. TGFβ and Wnt pathway cross-talk. Cancer Metastasis Rev. 2004;23(1–2):53–61. doi: 10.1023/a:1025811012690. [DOI] [PubMed] [Google Scholar]
- 31.Pasca di Magliano M, et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS One. 2007;2(11):e1155. doi: 10.1371/journal.pone.0001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu M, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487(7408):510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen B, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Üren A, et al. Secreted frizzled-related protein-1 binds directly to wingless and is a biphasic modulator of Wnt signaling. J Biol Chem. 2000;275(6):4374–4382. doi: 10.1074/jbc.275.6.4374. [DOI] [PubMed] [Google Scholar]
- 35.Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development. 2002;129(20):4831–4842. doi: 10.1242/dev.129.20.4831. [DOI] [PubMed] [Google Scholar]
- 36.Arensman MD, et al. Oncogene. WNT7B mediates autocrine Wnt/[beta]-catenin signaling and anchorage-independent growth in pancreatic adenocarcinoma [published online ahead of print February 18, 2013]. doi: 10.1038/onc.2013.23 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner KW, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13(9):1070–1077. doi: 10.1038/nm1627. [DOI] [PubMed] [Google Scholar]
- 38.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–307. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 40.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schutte M, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–3130. [PubMed] [Google Scholar]
- 43.Park J-K, et al. miR-132 and miR-212 are increased in pancreatic cancer and target the retinoblastoma tumor suppressor. Biochem Biophys Res Commun. 2011;406(4):518–523. doi: 10.1016/j.bbrc.2011.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park BJ, Park JI, Byun DS, Park JH, Chi SG. Mitogenic conversion of transforming growth factor-beta1 effect by oncogenic Ha-Ras-induced activation of the mitogen-activated protein kinase signaling pathway in human prostate cancer. Cancer Res. 2000;60(11):3031–3038. [PubMed] [Google Scholar]
- 45.Galliher-Beckley AJ, Schiemann WP. Grb2 binding to Tyr284 in TβR-II is essential for mammary tumor growth and metastasis stimulated by TGF-β. Carcinogenesis. 2008;29(2):244–251. doi: 10.1093/carcin/bgm245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gatza CE, et al. Type III TGF-β receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia. 2011;13(8):758–770. doi: 10.1593/neo.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995. 81 3 323 – 330. 10.1016/0092-8674(95)90385-2 [DOI] [PubMed] [Google Scholar]
- 48.Warfel NA, El-Deiry WS. p21WAF1 and tumourigenesis: 20 years after. Curr Opin Oncol. 2013;25(1):52–58. doi: 10.1097/CCO.0b013e32835b639e. [DOI] [PubMed] [Google Scholar]
- 49.Furuuchi K, et al. Somatic mutations of the APC gene in primary breast cancers. Am J Pathol. 2000;156(6):1997–2005. doi: 10.1016/s0002-9440(10)65072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satoh S, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24(3):245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 51.Wang L, et al. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and β-catenin stabilization. Cancer Cell. 2009;15(3):207–219. doi: 10.1016/j.ccr.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gordon KJ, Dong M, Chislock EM, Fields TA, Blobe GC. Loss of type III transforming growth factor β receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis. 2008;29(2):252–262. doi: 10.1093/carcin/bgm249. [DOI] [PubMed] [Google Scholar]
- 53.Olive KP, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neupane D, Korc M. 14-3-3sigma Modulates pancreatic cancer cell survival and invasiveness. Clin Can Res. 2008;14(23):7614–7623. doi: 10.1158/1078-0432.CCR-08-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu F, Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012;11(10):2138–2148. doi: 10.1158/1535-7163.MCT-12-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plasari G, et al. Nuclear factor I-C links platelet-derived growth factor and transforming growth factor β1 signaling to skin wound healing progression. Mol Cell Biol. 2009;29(22):6006–6017. doi: 10.1128/MCB.01921-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labbé E, et al. Transcriptional cooperation between the transforming growth factor-β and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res. 2007;67(1):75–84. doi: 10.1158/0008-5472.CAN-06-2559. [DOI] [PubMed] [Google Scholar]
- 60.Mootha VK, et al. PGC-1[alpha]- responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.