

Abstract
The ubiquitin proteasome system (UPS) is an essential metabolic constituent of cellular physiology that tightly regulates cellular protein concentrations with specificity and precision to optimize cellular function. Inhibition of the proteasome has proven very effective in the treatment of multiple myeloma, and this approach is being tested for utility in other malignancies. New pharmaceuticals targeting the proteasome itself or specific proximal pathways of the UPS are in development as antiproliferatives or immunomodulatory agents. In this article, we discuss the biology of UPS-targeting drugs, their use as therapy for neoplasia, and the state of clinical and preclinical development for emerging therapeutics.
Introduction
A new era in myeloma therapy.
At the turn of the millennium, targeted molecular therapeutics against hematologic malignancies initiated a shift in our perspective on treatment, prognosis, and survival for patients with some of the most aggressive and fatal neoplasms. Bortezomib (branded and marketed as Velcade by Millennium Pharmaceuticals) entered the armamentarium of new antiproliferative therapies with approval in May 2003 for the hematologic malignancy multiple myeloma (MM), in which B cell–derived plasma cells clonally proliferate and produce large quantities of monoclonal antibody. MM can be a devastating disease, with renal failure, blood hyperviscosity, and bone marrow invasion seen clinically. Before 2000, there were few life-prolonging therapies for the disease. Bortezomib blocks the proteolytic activity of the 26S proteasome, a cellular structure whose role in cell metabolism has now been meticulously characterized; indeed, bortezomib is the first agent available for use in humans that inhibits the activity of this system. Bortezomib quickly proved effective in refractory MM (1), and its inclusion in initial MM treatment was superior to the conventional cytotoxic chemotherapy regimen alone (2).
During this time thalidomide, an agent that produced deformities in infants of mothers prescribed the drug during pregnancy, further suppressed myeloma plasma cell proliferation when added to the regimen. A decade of careful clinical trials since these first breakthrough observations has revealed that therapeutic combinations including bortezomib with thalidomide or related compounds (collectively called immunomodulatory drugs, or IMiDs) and steroids confer a very favorable prognosis compared with historic therapy, greatly prolonging the median survival time from diagnosis over this period (3). When this therapy is implemented in conjunction with autologous bone marrow stem cell transplant, recent clinical trials show a three-year progression-free survival of 60% and overall survival of 90% for patients eligible for stem cell transplant (4), compared with only 48% three-year relative survival for patients diagnosed in 1999 (5). Multiple clinical trials for this new generation of MM molecular therapies are underway, with median survival projected by some to exceed 10 years in the post-bortezomib era. Moreover, new proteasome inhibitor and IMiD agents have recently been approved (6, 7). Further, personalization of therapy based on cancer genotype will likely enhance efficacy in treatment of MM and cancer in general. In this review, we highlight the molecular physiology and pharmacology of the proteasome and its upstream ubiquitin (Ub) system, with special attention to existing and promising modulators of this essential component of cellular physiology.
The ubiquitin proteasome system.
The ubiquitin proteasome system (UPS) is an elaborate and highly regulated cellular system that mediates selective protein degradation to control the abundance and therefore activity of the majority of proteins in the cell. The expression of most proteins is controlled by the proximal activity of the UPS through the activity of specific proteins and enzyme complexes, including the 76-amino-acid protein Ub, the E1, E2, and E3 Ub ligase machinery, and deubiquitinating enzymes (DUBs). Ub monomers are attached through a series of steps. The human genome encodes one or two human E1 activating enzymes that in an ATP-dependent fashion transfer Ub to one of the 38 E2-conjugating enzymes. Ub is then covalently bound to specific target proteins through selective action of any one of several hundred E3 ligases that tag a target protein (or substrate) for quality control, cellular sorting, or degradation by the proteasome or lysosome (8). Though there are multiple forms of ubiquitination, the most important scheme for the purposes of this discussion is polyubiquitination of a substrate protein whereby an E2-E3 ligase combination mediates the bond between an acceptor lysine (K) residue on the substrate and the terminal glycine residue of Ub; then subsequent Ub subunits are added by their K48 residues to form a chain of polyubiquitin, which brands that substrate protein for proteasomal degradation by the 26S proteasome (Figure 1). Other ubiquitination schemas are critical for signaling and cellular compartmentalization. K63 ubiquitination and deubiquitination often control membrane protein processing by directing endocytosis and trafficking of proteins between cellular compartments (8, 9). “Linear ubiquitination” is a recently described enhancer of TNF signaling, whereby substrates are joined to the Ub NH2-terminal methionine (M1) residue by a distinct E3 ligase complex that mediates end-to-end polyubiquitination, increasing the substrate’s availability and affinity for its targets (10).
Figure 1. A schematic of the UPS.
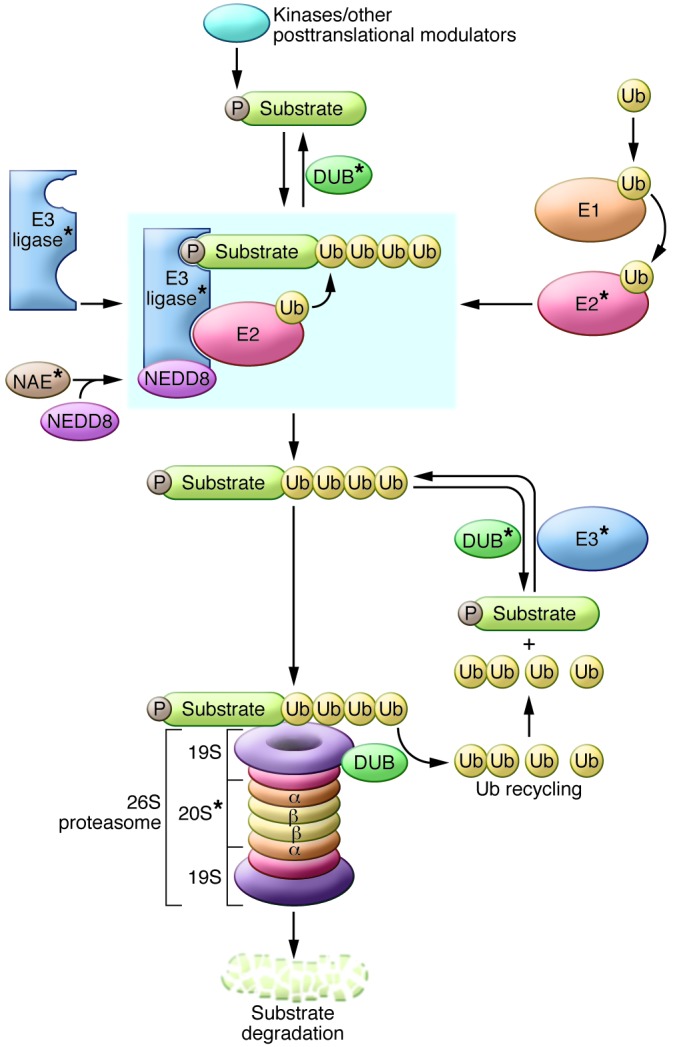
Ub is loaded onto the E1 activating enzyme in an ATP-dependent fashion and then transferred to an E2 Ub-conjugating enzyme. For the case of cullin-RING E3 ligases, the E3 is primed by neddylation by the NEDD8-activating enzyme (NAE). E3 ligases may be single proteins or multi-subunit enzyme complexes that mediate Ub transfer from an E2 conjugating enzyme to the substrate through interaction of a degron motif (usually a posttranslationally modified molecular recognition signature such as phosphorylation [P]) within the substrate and the binding domain of the E3. Ub monomers are covalently added to the substrate protein, and the polyubiquitinated protein is recognized and bound by the 19S subunit and degraded by the 20S subunit of the 26S proteasome. DUB enzymes are capable of “rescuing” Ub substrates from degradation, while other DUBs mediate cleavage and recycling of the Ub monomers. Components that are potential therapeutic targets are indicated by asterisks.
When substrates are K48 polyubiquitinated, they gain affinity for the 26S proteasome, a multimeric barrel-shaped protein structure containing a central 20S and one or two 19S cap subunits, each composed of several component proteins. The 20S subunit is composed of two pairs of seven-membered rings of proteins, wherein α and β rings stack onto each other, and β rings (which contain the protease catalytic domains) interface with other β rings to form the active central portion of the proteasome chamber. The 19S subunit binds the exterior α rings and serves as filter and portal for protein degradation, recognizing polyubiquitinated proteins with four or more Ub proteins in the chain. Proteasome-associated deubiquitinase (DUB) enzymes cleave the Ub chain and recycle it to the free pool of Ub for reuse, while the substrate proteins are unfolded in an ATP-dependent fashion as they are fed into the 20S pore of the proteasome. Protease domains of β1, β2, and β5 subunits, respectively, exert caspase-, trypsin-, and chymotrypsin-like activities on the incoming substrate protein, resulting in thorough and nonspecific proteolysis (11). Thus, the degradation activity of the UPS finely calibrates cellular abundance of proteins not only for homeostatic regulation, but also to mediate critical changes in metabolism in response to an external signal or for cell cycle progression.
E3 ligases and DUBs regulate protein turnover and cellular function.
E3 ligases usually bind and ubiquitinate their targets when the substrate has some structural modification, which can be misfolding in the case of quality control at the level of the ER or Golgi, or a posttranslational modification such as phosphorylation. Molecular signatures that E3 ligases recognize for ubiquitination are referred to as degrons, and the diversity of known degron motifs is expanding as the field advances. The two main E3 ligase families, the HECT (homologous to the E6-AP carboxyl terminus) and cullin-RING E3 ligases, possess conserved E2-association domains, but display wide diversity in their substrate-binding domain structures. Many of the E3 ligases thus far characterized are multi-subunit complexes, which contain a Ub-loaded E2 conjugation enzyme and an interchangeable substrate-binding protein (sometimes with other linker proteins) through which they mediate K48 ubiquitination and subsequent selective degradation (12). It should also be noted that priming of the ubiquitination machinery often requires multiple other protein activities, such as adduction of NEDD8 to cullin by NEDD8-activating enzyme for the function of the large family of cullin-RING E3 ligases.
The 79 identified DUB enzymes oppose the activity of E3 ligases by removing Ub from substrates to refine sorting and processing; thus, DUBs can rescue substrates from proteasomal degradation (13). DUB-mediated reversal of ubiquitination adds another layer of regulatory complexity to the performance of the UPS. There are 5 DUB families, with varying specificities. Some DUBs directly bind the substrate and cleave the attached Ub chain, while others bind and cleave specific Ub linkage types (e.g., K48 versus K63), and still others trim Ub monomers from the end of a chain. Sometimes E3 ligases and DUBs are complexed together for constitutive regulation or editing of the ubiquitination process and regulation of E3 ligase autoubiquitination (13). DUBs are also temporally and spatially regulated, and their varied roles in protein signaling and stability are a current area of investigation (13).
E3 ligases’ central role in “choosing” which proteins are degraded has been heavily studied to elaborate how aberrant signaling at this level impacts cell behavior and disease. For example, the von Hippel–Lindau protein (vHL) is an E3 ligase that mediates degradation of hydroxylated HIF, a transcriptional activator for multiple cytoproliferative processes. In von Hippel–Lindau disease, mutant vHL fails to regulate HIF, and patients display cytoproliferation leading to spontaneous tumor development (14). Itch, an E3 ligase first identified in mice that degrades proinflammatory transcription factors (15), is mutated in a cohort from an Amish family, with affected individuals suffering from autoimmunity, inflammatory lung disease, and premature death (16). CYLD is a DUB that regulates NF-κB activity in epidermal cells (17–19) and deubiquitinates multiple upstream second messenger proteins including TRAF2 and NEMO. Mutations of CYLD that result in the loss of DUB activity increase the activity of NF-κB transcriptional activation (20). Such mutations cause the autosomal disease familial cylindromatosis, which is characterized by the formation of deforming tumors of the skin. The field of E3 ligase and DUB biology is advancing rapidly, with new E3 ligase–substrate relationships being characterized nearly every month and our global understanding of this system developing at an accelerated pace.
Therapeutics
Targets for UPS therapy.
Like any hierarchical biologic system, the UPS displays multiple levels of regulation for a final biologic outcome. Inhibition of the proteasome only targets the last step in the process, with a resultant accumulation of the upstream ubiquitinated proteins as a secondary effect. It has been postulated that these Ub-laden proteins aggregate in the dorsal root ganglia, causing the well-documented side effect of bortezomib-induced peripheral neuropathy (21). This is often a cause for dose reduction or discontinuation of therapy and could represent a class-wide dose-limiting side effect of all proteasome inhibitors. Thus, efforts have been made to develop agents targeting other steps in UPS protein processing, including E2, E3 ligase, and DUB activities, though no agents developed as such are currently FDA approved. In this section, we discuss proteasome inhibitors as well as many of the preclinical and investigational drugs targeting the UPS, with attention to molecular targeting and disease applications. We also discuss the newly characterized mechanistic activity for thalidomide and the other IMiDs as modulators of the UPS.
Bortezomib is a unique first-in-class compound that has demonstrated tangible success in the molecular targeting of disease. It was designed specifically to block the proteasome due to its specific charge targeting boron-based design and dipeptidyl structure that docks the drug into the active proteolytic site of the chymotrypsin portion of the 20S proteasomal subunit (22). Bortezomib’s main cytotoxic effect on myeloma cells is through the induction of apoptosis after accumulation of excessive protein (23), as these immunoglobulin-laden cells display activation of apoptotic caspases, cell cycle arrest, DNA fragmentation, and cell death. Additionally, bortezomib suppresses inflammatory signals including IL-6 and TNF secretion, and blunts activity of the master regulator transcription factor NF-κB. Because of the selective toxicity seen for cells producing high amounts of immunoglobulin for MM, bortezomib has been tested in other malignancies, with favorable results and is now FDA approved for treatment of mantle cell lymphoma.
The newest agent approved for relapsed or refractory MM is another proteasome inhibitor, carfilzomib. It is a molecule functionally similar to bortezomib, with selective inhibition of the chymotrypsin activity of the 20S proteasome, and is also based on a peptide structure. Unlike that of bortezomib, carfilzomib’s activity is irreversible. Carfilzomib was well tolerated in phase I and II clinical trials as therapy for MM and shows promising efficacy, with near complete clinical response in a majority of previously untreated MM patients when combined with lenalidomide and dexamethasone (6). Studies in refractory/relapsed MM show responses to carfilzomib in bortezomib-treated or naive individuals (24, 25). Further, cell and animal studies show osteoclast suppression and improvement in bone health with proteasome inhibitors, generating optimism that carfilzomib may secondarily prevent some of the bone-destructive processes common to MM (26). These early results indicate that carfilzomib will enhance the arsenal of effective therapies for treatment of MM, and a large phase III trial is underway (27).
Other proteasome inhibitors.
Recently, several clinical trials have been undertaken on two promising agents that may join the list of FDA-approved proteasome inhibitors (Table 1). The first is marizomib (also known as NPI-0052), a highly potent proteasome inhibitor that affects chymotrypsin, trypsin, and caspase activities of the 20S proteasome and is derived from a marine bacterium. The activity of this novel drug is slightly distinct from that of bortezomib, with resultant tumoricidal synergy of the two agents in vitro (28). Marizomib has undergone phase I trials, with excellent efficacy in proteasome inhibition. Side effects were limited to gastrointestinal symptoms without neuropathy or other significant systemic toxicity observed with earlier agents (29). Clinical outcomes appear promising, but additional studies are needed. A second drug in clinical development is the orally available proteasome inhibitor MLN9708, a boron-containing, peptidic agent with structural and functional similarity to bortezomib. MLN9708 has been tested in cancer patients and was reasonably tolerated in phase I studies, with chemotherapeutic side effects of fatigue, nausea, anemia, and thrombocytopenia (30). Additional phase I and II trials are planned (ref. 31; clinical trials NCT01454076, NCT01939899, and others).
Table 1.
Pharmacologic agents targeting the ubiquitin proteasome system
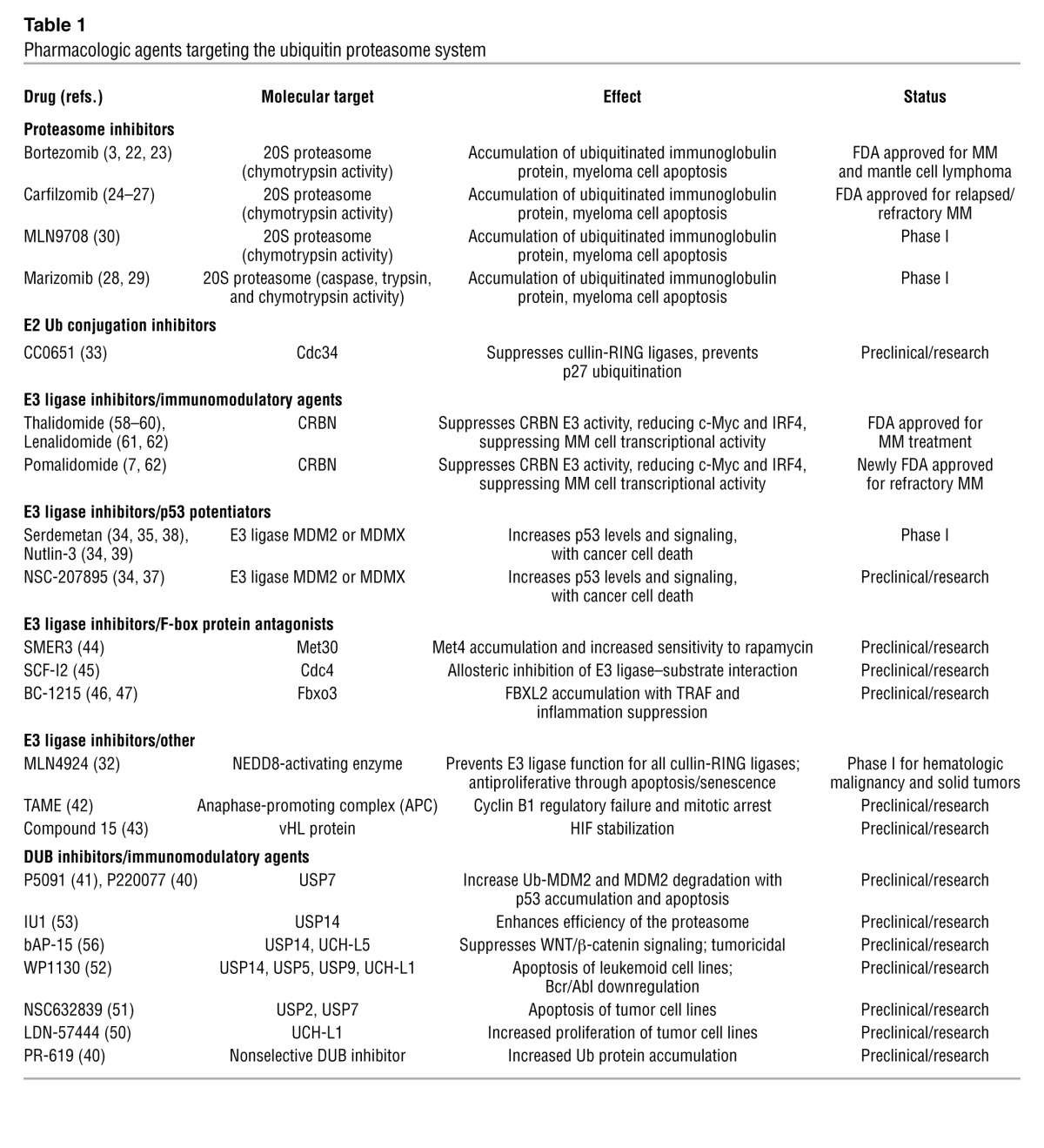
Emerging and preclinical drugs.
Because the field of Ub biology is still burgeoning, many of the intermolecular interactions between specific Ub enzymes and their cognate substrates are either newly characterized or unknown. While a few drugs have been developed to specifically antagonize E2-conjugating enzymes, E3 ligases, and DUBs, none of these have yet entered advanced clinical trials (Table 1). The ubiquitination activity of some E3 ligases requires the activity of other proteins. In particular, the cullin-RING E3 ligases require covalent binding of the Ub-like protein NEDD8 to the cullin component of the E3 ligase for proper function. The compound MLN4924 is a potent inhibitor of NEDD8 activation, and the drug has been shown in multiple preclinical models to effectively block neoplastic cell proliferation (32). Phase I trials of this agent have been completed for non-hematologic malignancies, and other trials are underway or planned for the use of this drug in an array of hematologic malignancies and solid tumors (NCT00677170, NCT00911066).
Cdc34 is a Ub-conjugating enzyme for cullin-RING E3 ligases whose activity mediates degradation of a very large number of cellular proteins, including the tumor suppressor p27. CC0651 is a small molecule that targets Cdc34 and suppresses p27 ubiquitination, but it has not been pursued for development as a therapy (33).
There has been considerable interest in targeting the E3 ligases MDM2 and MDMX, both of which mediate degradation of the tumor suppressor p53. Agents suppressing the interaction of p53 with these E3 ligases result in accumulation of p53, triggering apoptotic cancer cell death, making them prime drug design candidates (34). Many compounds, including serdemetan, nutlin-3 (RO5045337), and NSC-207895 all demonstrate in vitro anticancer activity (35–37). Serdemetan was tested in a phase I trial, with p53 induction seen, but cardiac conduction defects were observed (38). Nutlin-3 may be a promising agent and is registered for phase I trials for an array of malignancies (NCT00559533, NCT00623870), but these studies have not been published. Targeting of nutlin-3 to B cell leukemia has been attempted by conjugating the drug to the B cell–specific antibody rituximab and has shown promising results in preclinical studies (39). Another method of targeting this pathway has been developed using inhibitors of the DUB USP7, which stabilizes cellular concentrations of MDM2 by removing Ub. Two small molecule DUB inhibitors, p5091 and p220077, specifically targeting USP7 have been developed and tested in vitro. Treatment enhanced ubiquitination and degradation of MDM2 and caused accumulation of p53 and apoptosis in cancer cells and myeloma cell lines (40, 41). Hence, modulation of cell survival targets such as p53 via manipulation of the Ub system remains an attractive approach.
Tosyl-l-arginine methyl ester (TAME) inhibits the anaphase-promoting complex E3 ligase, which is required for mitotic division by depletion of cyclin B1 and potently induces mitotic arrest in rapidly dividing cells (42). Another small molecule was recently designed to interfere with the binding between the transcriptional activator HIF and the vHL E3 ligase protein (43). This may be an attractive therapeutic strategy for anemia and ischemia, although additional studies are needed to determine the side effect profile before further drug development can proceed.
Small molecules designed to target particular substrate-specifying F-box proteins that are components in the multi-subunit SKP1/cullin/F-box containing complex (SCF) E3 ligases are now emerging as distinct pharmacologic entities. One example is SMER3, detected from a screen for potentiators of the antiproliferative drug rapamycin. SMER3 potently blocks the F-box protein Met30 to prevent degradation of Met4, an antiproliferative transcriptional activator (44). Another small molecule screening approach revealed the compound SCF-I2, which allosterically blocks activity of the yeast F-box protein Cdc4, but not its human ortholog FBXW7 (45). BC-1215, a small molecule inhibitor of F-box protein Fbxo3, was recently synthesized to block SCF E3 ligase degradation of another F-box (Fbxl2), which in turn degrades the TNF receptor–associated factor (TRAF) adaptor proteins; thus, BC-1215 decreases TRAF proteins and blunts NF-κB activation and inflammation through the TNF signaling axis (46, 47). Because Fbxl2 also targets proteins within the cell cycle (48, 49), drugs targeting the Fbxo3/Fbxl2 axis may also be anti-neoplastic.
Aside from the two small molecule inhibitors of USP7 mentioned above, there has been further compound development for both nonspecific and selective DUB inhibitors, with interesting results. The compound PR-619 is a nonselective DUB inhibitor developed in parallel with the USP7 inhibitor p220077 (40). Another group used high-throughput assays to characterize the DUB inhibitor LDN-57444, which suppresses activity of the DUB UCH-L1 and results in increased cell proliferation in tumor cell lines (50). Other broad-spectrum DUB inhibitors include NSC 632839 (which targets USP2 and USP7, among others) and WP1130 (blocking USP5 and USP14), which both trigger apoptotic cell death in cancer cell lines (51, 52).
The inhibitor IU1 was characterized for specific activity against the proteasome-associated DUB USP14 after chemical library screening, showing global enhancement of proteasome activity by curtailing the Ub chain–shortening activity of USP14 (53). IU1 treatment enhanced proteasome efficiency in vitro, with resultant degradation of the tau protein in initial studies. The clinical utility of a specific USP14 inhibitor may merit more attention, as new studies indicate that USP14 activity is necessary for WNT/β-catenin signaling that is active in some cancers, and that high levels of USP14 correlate with gastrointestinal cancer severity and lung cancer mortality (54, 55). To this end, bAP-15 is a recently developed compound that antagonizes USP14 and another proteasome-associated DUB, USP5, and displays antiproliferative activity in vitro and tumoricidal activity in animal models (56).
Immunomodulatory agents as E3 ligase antagonists.
The immunomodulatory agents thalidomide and lenalidomide are now standard of care therapy for MM (57), with a mechanism of action that was previously only partially characterized. Thalidomide was initially developed in the mid-twentieth century as a sedative and was used to combat hyperemesis gravidarum in pregnant women. The drug caused catastrophic birth defects, with limb deformities in infants exposed to the drug in utero. Although an FDA ban ensued, its serendipitous off-label use in patients with erythema nodosum leprosum (ENL) led to international clinical trials to assess and characterize its immunomodulatory effects (58). Thalidomide regained FDA approval in 1998 for the treatment of ENL and was entered into clinical trials for MM with a greater clinical efficacy than other available agents (59). Although there was a wealth of data displaying hearty suppression of cytokines and growth factors, thalidomide’s specific mechanism of action was essentially unknown.
As the use of thalidomide for myeloma grew, a second IMiD compound, lenalidomide, was developed for optimal pharmacology against MM. Further studies revealed that the specific molecular target for thalidomide teratogenicity was the E3 ligase cereblon (CRBN) (60), whose substrate proteins were unknown. This revelation prompted assessment of the immunomodulators’ activity on CRBN in myeloma cells (61, 62), where it was determined that CRBN was the principal target for the tested anti-myeloma activity of the IMiDs. In vitro, RNA depletion of CRBN caused MM cell death, concordant with the pharmacotoxicity of IMiDs, but a subpopulation of surviving cells tolerated low CRBN conditions. These CRBN-downregulated MM cells were resistant to killing by thalidomide, lenalidomide, and the newly developed pomalidomide. Genotypic analysis of MM cells chronically challenged with lenalidomide revealed that resistant cells had acquired a CRBN knockdown phenotype with lower RNA and protein levels of the E3 ligase. The pharmacology of these effects requires intact E3 ligase activity of CRBN, which has been linked to degradation of a calcium-activated chloride channel and AMPK through ubiquitination, along with stabilization of oncogenic c-Myc and IRF4 (a myeloma-promoting transcription factor). Predictably, IMiD treatment causes accumulation of the chloride channels and AMPK, with depletion of c-Myc and IRF4, though resistant cells have restored IRF4 levels despite downregulation of CRBN. To translate these findings, plasma cell CRBN levels assayed in cells from subjects with MM were decreased in a vast majority of clones that had developed resistance to IMiD therapy (62). Further, high CRBN levels correlated strongly with prolonged survival for MM patients on thalidomide, but not bortezomib, maintenance therapy (63).
The overall impact of these findings is quite compelling, and they have three important implications. First, routine analysis of CRBN levels in patients with MM may soon lead to a personalized approach to the selection of therapeutic regimens. Second, the two drug classes that are most effective against MM both target the activity of the UPS to cause apoptosis of cancer cells. Third, IMiDs appear to act as E3 ligase antagonists, providing target validation for drug design of future pharmaceuticals. Newer small molecules targeting human E3 ligases may deliver very selective activity with less nonspecific systemic toxicity.
Future disease indications and clinical promise
In the current era of drug design and clinical investigation, cancer therapeutics has been the most fertile ground for pharmaceutical development. Although these systems are very complex, neoplastic cells are defined by their hyperproliferative nature, greatly facilitating cell-based research and high-throughput screening of small molecules. Another key to the great progress in cancer therapy with these or any agents lies in the exceptionally well-organized clinical trials network. Looking forward, there may be broad application for proteasome inhibitors or other drugs that target the UPS in treatment of other cancer states or non-neoplastic disease, with off-label trials of bortezomib ongoing for multiple hematologic malignancies and solid tumors, as well as amyloidosis and IgA nephropathy, among others (NCT00027898, NCT00298766, NCT01103778, and others). The rheumatologic maladies, especially antibody-mediated autoimmune conditions, may be the most easily targeted by proteasome inhibitors or emerging UPS-targeting drugs, and case reports of successful treatment of SLE and cryoglobulinemia with off-label bortezomib are beginning to appear (64, 65). Given the suppression of NF-κB activation and inflammatory cytokines by proteasome inhibitors (66) and IMiDs, using these agents for their antiinflammatory traits seems justifiable in refractory disease.
While there are currently no specific, validated applications for UPS-targeting agents outside the above-mentioned fields of medicine, there are proposals discussing the importance of this system in the development and disease processes of other organ systems (67–69), and UPS-targeting agents may soon be considered for many disease states other than malignancies. In particular, inflammatory diseases with common signaling biology seem to manifest in multiple organ systems together or independently, and there are still limited choices for effective, steroid-sparing immunomodulatory agents. Although it is tempting to hail such new agents as “magic bullet” therapeutics, we must proceed cautiously. So few E3 ligase/substrate relationships have been characterized that the potential for unintended off-target effects is high, and these new agents must be carefully tested to insure that we avoid tragic outcomes like those seen from thalidomide toxicity.
The proliferation of pharmaceutical development in this emergent area of human biology is both exciting and transformative. The fruits from these labors may be harvested over decades, as new drugs with a known mechanism of action are approved for one indication, then used by astute practitioners off-label for different diseases, with more rigorous evaluation for promising therapeutic applications. Additionally, the NIH and Pharma have embarked on a coordinated effort to “repurpose” abandoned drugs proven safe in phase I trials to accelerate therapeutic development (70). There are already examples of successful identification of relevant novel drug activity through this initiative (71, 72). Indeed there may be many compounds already in existence with unrecognized activity against the UPS (as was the case for the IMiDs). As our understanding of protein regulation through Ub biology advances and structural information on protein interactions expands, the ability to generate or repurpose new candidate molecules to target very specific biologic phenomena is becoming increasingly refined. We have reason for optimism that the UPS will present many reachable targets for continued progress in the treatment of human disease.
Acknowledgments
This material is based upon work supported, in part, by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. This work was supported by a Merit Review Award from the US Department of Veterans Affairs, NIH R01 grants HL096376, HL097376, and HL098174, and NIH P01 grant HL114453 (to R.K. Mallampalli).
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(1):6–12. doi:10.1172/JCI71602.
References
- 1.Richardson PG, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 2.San Miguel JF, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 3.Kumar SK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavo M, et al. Bortezomib-thalidomide-dexamethasone is superior to thalidomide-dexamethasone as consolidation therapy after autologous hematopoietic stem cell transplantation in patients with newly diagnosed multiple myeloma. Blood. 2012;120(1):9–19. doi: 10.1182/blood-2012-02-408898. [DOI] [PubMed] [Google Scholar]
- 5. Howlader N, et al. SEER Cancer Statistics Review (CSR) 1975–2010. National Cancer Institute Web Site. http://seer.cancer.gov/csr/1975_2010/ . Updated June 14, 2013. Accessed November 15, 2013.
- 6.Jakubowiak AJ, et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood. 2012;120(9):1801–1809. doi: 10.1182/blood-2012-04-422683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elkinson S, McCormack PL. Pomalidomide: first global approval. Drugs. 2013;73(6):595–604. doi: 10.1007/s40265-013-0047-x. [DOI] [PubMed] [Google Scholar]
- 8.MacGurn JA, Hsu PC, Emr SD. Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem. 2012;81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 9.Clague MJ, Liu H, Urbe S. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev Cell. 2012;23(3):457–467. doi: 10.1016/j.devcel.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Rieser E, Cordier SM, Walczak H. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem Sci. 2013;38(2):94–102. doi: 10.1016/j.tibs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5(3):177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 12.Cardozo T, Pagano M. The scf ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5(9):739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 13.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 14.Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–173. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 15.Perry WL, Hustad CM, Swing DA, O’Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet. 1998;18(2):143–146. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- 16.Lohr NJ, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet. 2010;86(3):447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature. 2003;424(6950):797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 18.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. 2003;424(6950):801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 19.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424(6950):793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 20.Sun SC. Cyld: a tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death Differ. 2010;17:25–34. doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richardson PG, et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J Clin Oncol. 2006;24(19):3113–3120. doi: 10.1200/JCO.2005.04.7779. [DOI] [PubMed] [Google Scholar]
- 22.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14(3):451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011;10(11):2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vij R, et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol. 2012;158(6):739–748. doi: 10.1111/j.1365-2141.2012.09232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vij R, et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood. 2012;119(24):5661–5670. doi: 10.1182/blood-2012-03-414359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurchla MA, et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia. 2013;27:430–440. doi: 10.1038/leu.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hájek R, Bryce R, Ro S, Klencke B, Ludwig H. Design and rationale of focus (PX-171-011): a randomized, open-label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM). BMC Cancer. 2012;12:415. doi: 10.1186/1471-2407-12-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauhan D, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell. 2005;8(5):407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Millward M, et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest New Drugs. 2012;30(6):2303–2317. doi: 10.1007/s10637-011-9766-6. [DOI] [PubMed] [Google Scholar]
- 30.Rodler E, et al. First-in-human, phase I dose-escalation study of investigational drug MLN9708, a second-generation proteasome inhibitor, in advanced nonhematologic malignancies. J Clin Oncol. 2010;28:15s. (suppl; abstr 3071). [Google Scholar]
- 31. ClinicalTrials.gov. NIH Web Site. http://www.clinicaltrials.gov . Accessed November 15, 2013.
- 32.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 33.Ceccarelli DF, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145(7):1075–1087. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 34.Khoury K, Domling A. P53 Mdm2 inhibitors. Curr Pharm Des. 2012;18(30):4668–4678. doi: 10.2174/138161212802651580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chargari C, et al. Preclinical assessment of JNJ-26854165 (Serdemetan), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 2011;312(2):209–218. doi: 10.1016/j.canlet.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther. 2011;10(1):69–79. doi: 10.1158/1535-7163.MCT-10-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabernero J, et al. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin Cancer Res. 2011;17(19):6313–6321. doi: 10.1158/1078-0432.CCR-11-1101. [DOI] [PubMed] [Google Scholar]
- 39.Voltan R, et al. Nanoparticles engineered with rituximab and loaded with Nutlin-3 show promising therapeutic activity in B-leukemic xenografts. Clin Cancer Res. 2013;19(14):3871–3880. doi: 10.1158/1078-0432.CCR-13-0015. [DOI] [PubMed] [Google Scholar]
- 40.Altun M, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem Biol. 2011;18(11):1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 41.Chauhan D, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22(3):345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng X, et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell. 2010;18(4):382–395. doi: 10.1016/j.ccr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buckley DL, et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc. 2012;134(10):4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aghajan M, et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotechnol. 2010;28(7):738–742. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orlicky S, Tang X, Neduva V, Elowe N, Brown ED, Sicheri F, Tyers M. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol. 2010;28(7):733–737. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen BB, et al. A combinatorial F box protein directed pathway controls traf adaptor stability to regulate inflammation. Nat Immunol. 2013;14(5):470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mallampalli RK, et al. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J Immunol. 2013;191(10):5247–5255. doi: 10.4049/jimmunol.1300456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen BB, Glasser JR, Coon TA, Mallampalli RK. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene. 2012;31:2566–2579. doi: 10.1038/onc.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen BB, Glasser JR, Coon TA, Mallampalli RK. Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates aurora B to inhibit tumorigenesis. Cell Death Dis. 2013;4:e759. doi: 10.1038/cddis.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, et al. Discovery of inhibitors that elucidate the role of UCH-l1 activity in the H1299 lung cancer cell line. Chem Biol. 2003;10(9):837–846. doi: 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 51.Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 2006;66(18):9235–9244. doi: 10.1158/0008-5472.CAN-06-0702. [DOI] [PubMed] [Google Scholar]
- 52.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70(22):9265–9276. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- 53.Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467(7312):179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shinji S, et al. Ubiquitin-specific protease 14 expression in colorectal cancer is associated with liver and lymph node metastases. Oncol Rep. 2006;15(3):539–543. [PubMed] [Google Scholar]
- 55.Wu N, Liu C, Bai C, Han YP, Cho WC, Li Q. Over-expression of deubiquitinating enzyme USP14 in lung adenocarcinoma promotes proliferation through the accumulation of β-catenin. Int J Mol Sci. 2013;14(6):10749–10760. doi: 10.3390/ijms140610749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.D’Arcy P, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17(12):1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 57.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 58.Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its imid derivatives as anticancer agents. Nat Rev Cancer. 2004;4(4):314–322. doi: 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- 59.Palumbo A, et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood. 2008;111(8):3968–3977. doi: 10.1182/blood-2007-10-117457. [DOI] [PubMed] [Google Scholar]
- 60.Ito T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 61.Lopez-Girona A, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326–2335. doi: 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu YX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–4779. doi: 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broyl A, et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood. 2013;121(4):624–627. doi: 10.1182/blood-2012-06-438101. [DOI] [PubMed] [Google Scholar]
- 64.Hiepe F, et al. Refractory SLE patients respond to the proteasome inhibitor bortezomib. Ann Rheum Dis. 2012;71:A15–A16. [Google Scholar]
- 65.Quartuccio L, Rupolo M, Michieli M, De Vita S. Rheumatology (Oxford). Efficacy tolerability of repeated cycles of a once-weekly regimen of bortezomib in lupus [published online ahead of print August 20, 2013]. doi: 10.1093/rheumatology/ket284 . [DOI] [PubMed] [Google Scholar]
- 66.van der Heijden JW, Oerlemans R, Lems WF, Scheper RJ, Dijkmans BA, Jansen G. The proteasome inhibitor bortezomib inhibits the release of NFκB-inducible cytokines and induces apoptosis of activated t cells from rheumatoid arthritis patients. Clin Exp Rheumatol. 2009;27(1):92–98. [PubMed] [Google Scholar]
- 67. Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003. 40 (2)427– 446 [DOI] [PubMed] [Google Scholar]
- 68.Weathington NM, Sznajder JI, Mallampalli RK. The emerging role of the ubiquitin proteasome in pulmonary biology disease. Am J Respir Crit Care Med. 2013;188(5):530–537. doi: 10.1164/rccm.201304-0754PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106(3):463–478. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang R, et al. The ncgc pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3(80):80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun W, et al. Rapid identification of antifungal compounds against exserohilum rostratum using high throughput drug repurposing screens. PLoS One. 2013;8(8):e70506. doi: 10.1371/journal.pone.0070506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia M, et al. Identification of repurposed small molecule drugs for chordoma therapy. Cancer Biol Ther. 2013;14(7):638–647. doi: 10.4161/cbt.24596. [DOI] [PMC free article] [PubMed] [Google Scholar]