

Abstract
Over the past several years, there has been rapidly expanding evidence of epigenetic dysregulation in cancer, in which histone and DNA modification play a critical role in tumor growth and survival. These findings have gained the attention of the drug discovery and development community, and offer the potential for a second generation of cancer epigenetic agents for patients following the approved “first generation” of DNA methylation (e.g., Dacogen, Vidaza) and broad-spectrum HDAC inhibitors (e.g., Vorinostat, Romidepsin). This Review provides an analysis of prospects for discovery and development of novel cancer agents that target epigenetic proteins. We will examine key examples of epigenetic dysregulation in tumors as well as challenges to epigenetic drug discovery with emerging biology and novel classes of drug targets. We will also highlight recent successes in cancer epigenetics drug discovery and consider important factors for clinical success in this burgeoning area.
Epigenetic dysregulation in cancer
Epigenetic information is contained in the cell in multiple forms that include DNA methylation, histone modification (methylation, acetylation, phosphorylation, etc.), nucleosome positioning, and microRNA expression, among others. This combined information constitutes the epigenome. A comprehensive understanding of epigenomic dysregulation in specific cancer types has not been elucidated yet. Currently, there is an understanding of tumor-specific types of epigenetic modifications without a full appreciation of the context of the entire cancer epigenome in the specific tumor.
Cancer epigenetic dysregulation can be categorized into three types: (a) altered DNA or histone modification, (b) somatic alteration in an epigenetic protein, and (c) altered expression of an epigenetic protein. Those types of cancer epigenome dysregulation have been reviewed comprehensively elsewhere (1–3), and only will be referred to here.
The primary types of epigenetic modification that have been targeted by drug discovery efforts in recent years are histone methylation and acetylation. The enzymes that catalyze these histone post-translational modifications, which include histone methyltransferases, histone demethylases, histone acetyltransferases, and histone deacetylases, are considered potentially tractable targets for pharmacological intervention. Stated differently, drug discovery scientists believe that it may be possible to discover and optimize inhibitors to these activated enzyme targets as a direct means of pharmacological targeting of epigenetic dysregulation.
Of the three types of epigenetic dysregulation described above, a higher priority is placed on tumor somatic alterations in epigenetic proteins. There is a higher probability that somatic alterations will be consistent between cultured cancer cells and patient tumor samples as compared to protein expression, histone or DNA modification, since there is some evidence that the latter is different in cell culture (4, 5). Hence, a somatic alteration in a histone-modifying enzyme is, at first analysis, a preferred starting point for drug discovery. (An important exception to this concept, the bromodomain and extra-terminal [BET] inhibitors, is discussed below.)
There is a rapidly expanding list of reported somatic alterations in both hematological and solid tumor types. The somatic alterations span all types of epigenetic genes, but only a subset provide early biological validation for a drug discovery target — those that are recurrent point mutations that activate or alter protein function, e.g., activating mutations in enhancer of zeste homolog 2 (EZH2), a histone methyltransferase that specifically methylates the N-terminal lysine 27 of histone H3 (H3K27). EZH2 point mutations have been reported to be present at Y641 and A677 in germinal center B cell diffuse large B cell lymphomas (DLBCLs) and follicular lymphomas (6). Recombinant enzyme with these point mutations possesses higher catalytic activity for the H3K27 methylation reaction (with important differences among the mutations), and there is a dramatic increase in H3K27 trimethylation (H3K27me3) in DLBCL cells with these point mutations compared to DLBCL cells with the wild-type EZH2 gene (refs. 6–8 and Figure 1A). Similar to the point mutation of V600E BRAF kinase present in a high percentage of malignant melanoma, the EZH2 point mutation provides a means of patient selection and strong evidence of an oncogenic driver role for EZH2 in this selected population.
Figure 1. Epigenetic inhibitor mechanisms of action.
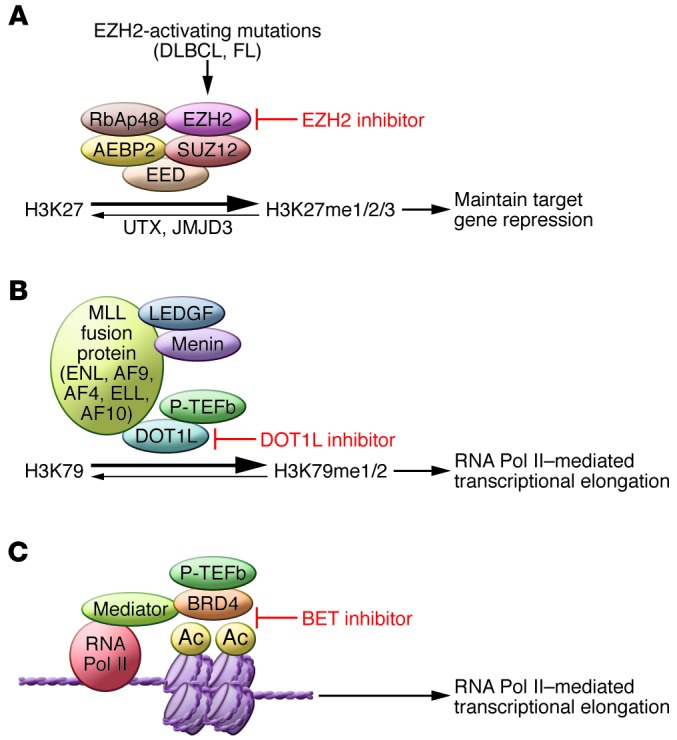
(A) Model of EZH2 mechanism of action in EZH2-mutant DLBCL. PRC2 is a 5-protein complex consisting of EZH2 (the catalytic protein), EED, SUZ12, AEBP2, and RbAp48. EZH2 catalyzes methylation of H3K27 to a mono-, di-, and trimethylated state, and EZH2-activating mutations in DLBCL result in higher K27 trimethylation. An EZH2 inhibitor inhibits the catalytic activity of the enzyme and, combined with the catalytic activity of the H3K27 histone demethylases UTX and JMJD3, decreases methylation at H3K27. In some cells, this results in gene de-repression (increased gene expression). (B) Model of DOT1L mechanism of action in MLL-rearranged leukemias. MLL fusion proteins, including fusions with MLL-ENL, MLL-AF9, MLL-AF4, MLL-ELL, and MLL-AF10, recruit DOT1L, which catalyzes mono- and dimethylation of H3K79, an essential step in RNA Pol II–mediated transcriptional elongation. A DOT1L inhibitor inhibits H3K79 methylation, which inhibits MLL-fusion protein mediated transcription. Adapted from Haematologica (70). (C) Model of BET mechanism of action. Brd4 protein recruits positive transcription elongation factor b (P-TEFb), an essential step in RNA Pol II–mediated transcriptional elongation. A BET inhibitor blocks the binding of BRD4 to acetylated lysines on nucleosome histones, inhibiting RNA Pol II–mediated transcription. Adapted from F1000 Biology Reports (71). Ac, acetylated histone residue.
A second type of somatic alteration in an epigenetic gene that may provide early biological validation for a drug discovery target is a chromosomal translocation that results in protein overexpression or alteration of function. An example of this is found in a subpopulation of multiple myeloma patients with the t(4;14)(p16;q32) translocation, which constitutes 15% of all patients and those with the worst prognosis (9). This translocation results in high over-expression of two genes, FGFR3 and NSD2 (also known as WHSC1 or MMSET). The resulting overexpression of NSD2, an H3K36 histone methyltransferase, in t(4;14)(p16;q32) multiple myeloma leads to increased H3K36me2, genomic disorganization of the H3K36me2, and oncogenic reprogramming (10–12). Additionally, it has been shown that the catalytic activity of this histone methyltransferase is essential for its biological role in the NSD2 translocation subpopulation of multiple myeloma (12). Hence, a small-molecule inhibitor of NSD2 may abrogate the oncogenic role of the epigenetic protein in this subpopulation of multiple myeloma.
Beyond these two types of somatic alterations, there are insertions, deletions, and point mutations of unknown biological consequence in epigenetic genes that occur with high frequency (13). Identifying drug targets from these somatic alterations is more difficult, as some will not be critical for tumor growth and survival. Others will have a critical role in tumor biology, but the somatic alteration will result in an inactivation of gene product function. In these cases, a mechanistic understanding of how inactivation results in epigenetic dysregulation and consequent perturbation of biological function needs to be elucidated. The second requirement is that the inactivation of an epigenetic gene makes the tumor more sensitive to a second druggable target. This is a high requirement for biological validation, but there is early evidence of such an example.
SWI/SNF is a multi-protein chromatin complex that remodels nucleosomes in an ATP-dependent manner, and thereby modulates transcription (14). Somatic alterations have been found in genes encoding the various member proteins of the SWI/SNF complex. These alterations occur with high prevalence in multiple tumor types and they appear to inactivate or alter the chromatin-remodeling function of the complex (14, 15). One of those is the biallelic loss of the tumor suppressor SNF5 (SMARCB1) in malignant rhabdoid tumors (16, 17). Roberts and colleagues have found that SNF5 antagonizes EZH2 in regulating stem cell–associated programs in embryonic stem cells, and that EZH2 is required for the growth of SNF5-deficient malignant rhabdoid tumor cells (18). Consistent with this finding, Pollock and colleagues have reported that a potent selective EZH2 inhibitor induces apoptosis and differentiation in malignant rhabdoid tumor cells, and that dosing of the inhibitor in mice results in regression of xenograft rhabdoid tumors (19). Taken together, an understanding of the interplay between two epigenetic regulators of gene expression (SNF5 and EZH2) was essential to design a targeted pharmacological approach for this tumor population with an inactivating somatic mutation.
Challenges to epigenetic drug discovery
Epigenetic drug discovery may be viewed as being in its infancy, as we are just beginning to understand the complexities of the “histone code” and the effects of pharmacological intervention. Indeed, there are quite a number of scientific and pragmatic challenges, many of which are summarized in Table 1. To begin, few biological tools (antibodies to epigenetic proteins and/or histone marks) or chemical tools (selective inhibitors or activators) are available to probe this biology, build assays for compound screening, and provide starting points for medicinal chemistry. In the absence of these tools, drug discovery scientists are primarily using genetic association (as described above) combined with molecular tools (siRNA, shRNA, overexpression, catalytic-inactive mutants) to identify relevant disease targets. However, each of these methods has caveats and is best confirmed with selective pharmacological modulators where available. Given this daunting task, private and public consortia have been established and are pooling resources to more rapidly identify chemical probes for epigenetic targets, such as the Structural Genomics Consortium (SGC; http://www.thesgc.org). The SGC has enabled the public dissemination of a substantial number of epigenetic protein crystal structures (helpful for computational approaches in medicinal chemistry) and chemical probes, e.g., G9a/GLP (20), EZH2 (21), DOT1L (22), L3MBTL3 (23), BET (24, 25), and PRMT3 (ref. 26 and Figure 2).
Table 1.
Epigenetic drug discovery challenges
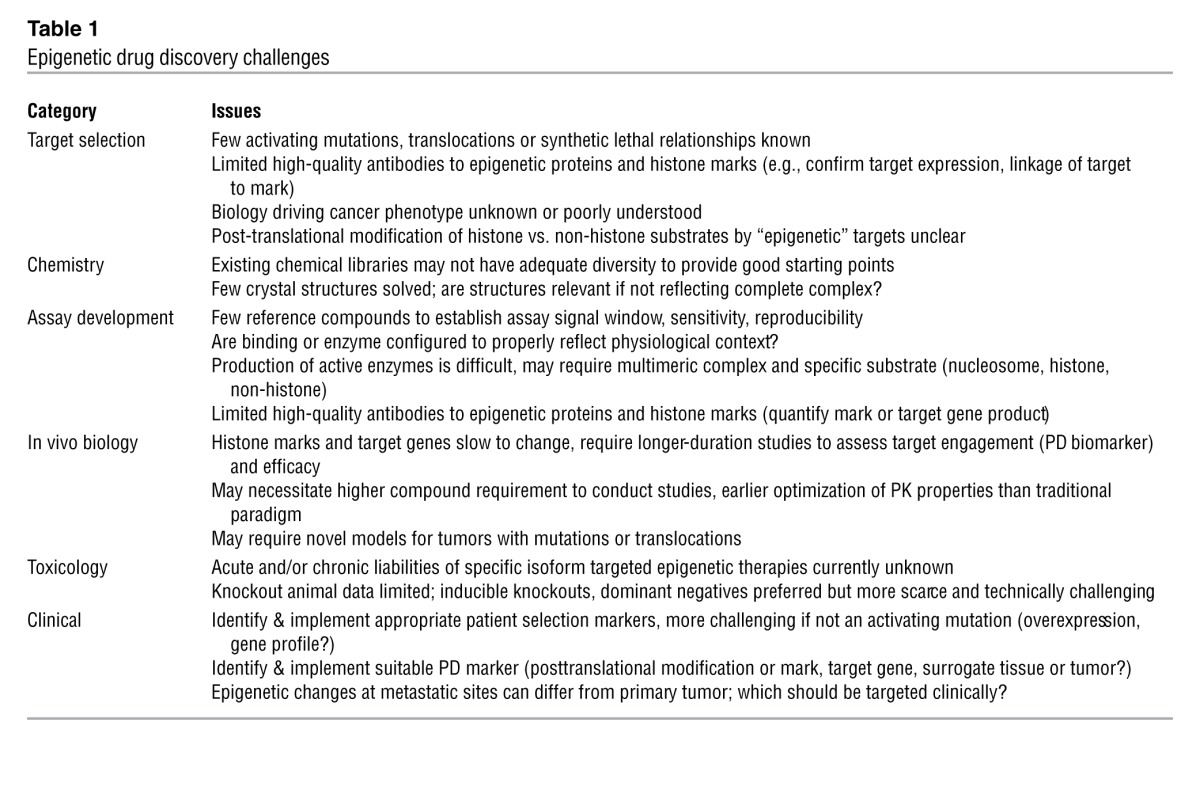
Figure 2. Pharmacological inhibitors of epigenetic proteins.

From a practical standpoint, building assays to screen compound libraries for small molecule modulators of epigenetic enzymes is not straightforward. In most cases, the initial screen is biochemical and ideally should recapitulate the activity in the cellular context. Many epigenetic targets exist as large complexes of proteins (e.g., EZH2 in polycomb repressive complex 2 [PRC2], mixed-lineage leukemia [MLL] in COMPASS), and in some cases these complexes may recruit different proteins to execute specific diverse responses. As such, screens using apo-enzymes may be misleading and not represent the cellular context. Substrates may not be canonical histones, as exemplified by SMYD2, where p53 peptide is preferred over histone as a substrate for methylation (27). To identify an inhibitor that does not compete with the binding of s-adenosyl methionine (SAM) during the enzyme-catalyzed reaction, proper selection of protein/peptide substrate could be critical. Another example of assay development complexity is NSD2, in which the enzyme has been reported to exhibit disparate target preferences based on the nature of the substrate provided, and which is likely best assayed using nucleosomes as substrate (28).
Cellular assays for epigenetic proteins also have proven to be somewhat complex. For many epigenetic targets, effects of knockdown or inhibitors in cell culture often take several days to see histone mark changes or effects on target genes, while phenotypic responses may require up to 7 to 10 days to observe (e.g., EZH2; see refs. 29, 30). Here again, there is a paucity of good antibodies to detect effects of knockdowns on target proteins and even fewer for specific acetylation/methylation sites. Progress in mass spectrometry has allowed for development for LC/MS-based histone mark profiling from cell lysates (12, 31) so this may circumvent some of this need and help confirm antibody-based assay methods. Translating the cell-based activity to traditional xenograft models will require a relevant sensitive tumor and perhaps longer duration studies to see effects (see comments on EZH2 inhibitors below).
Drug discovery examples
EZH2 inhibitors.
EZH2 inhibitor identification efforts at GlaxoSmithKline, among others, were initiated using biochemical assays (32). EZH2 is the catalytic component of PRC2, which selectively methylates H3K27. The PRC2 complex consists of three or five member proteins, and exhibits lysine methyltransferase catalytic activity on peptides, histones, mono-nucleosomes, and oligo-nucleosomes (Figure 1A). Direct EHZ2 inhibitors, those that bind directly to the protein and inhibit its enzyme activity, have been identified through biochemical assays using the PRC2 complex on a variety of substrates (30, 32–35). An additional complexity is that H3K27me3 peptide, the product of the reaction, binds allosterically to PRC2 and increases catalytic activity of the complex. The recognition of H3K27me3 by PRC2 is proposed to maintain repressed chromatin domains by re-establishing H3K27me3 onto naked nucleosomes being incorporated during DNA synthesis (36). Interestingly, some EHZ2 inhibitors have been found to possess greater biochemical potency and a longer enzyme-inhibitor residence time when H3K27me3 peptide is bound to PRC2 (37).
Optimization of GSK EZH2 inhibitors resulted in the development of GSK126 (29, 34), which possesses potent, highly selective biochemical and cellular on-target potency, assessed by decreases in H3K27 trimethylation. GSK126 provides preclinical validation of EZH2 activating mutations as a marker of selectivity for an EZH2 inhibitor in DLBCL, as cell lines with the mutation are more sensitive to growth inhibition by GSK126 than WT DLBCL cell lines (29). Similar EZH2 inhibitor selectivity for EZH2-mutant DLBCL was reported also for the EZH2 inhibitors EPZ005687 (30) and EI1 (33). GSK126 dosed daily or intermittently decreases growth of DLBCL xenografts in mice and improves survival. Tumor growth inhibition continues after cessation of drug dosing for 24 days or longer (29).
It is noteworthy that the three groups have reported EZH2 inhibitors independently, and all three inhibitor structures are strikingly similar (Figure 2). These successful efforts clearly indicate that EZH2 (or PRC2) is a druggable small-molecule target, but the convergent inhibitor structures may indicate that the EZH2 inhibitor-binding pocket (all are competitive with the enzyme cofactor S-adenosyl methionine) is highly stringent.
DOT1L inhibitors.
Another histone methyltransferase, DOT1L, appears to be the sole enzyme responsible for catalyzing the methylation of H3K79 (38). Aberrant DOT1L activity (increased H3K79 mono- or dimethylation) is found in certain MLLs, which possess rearrangements of the mixed-lineage leukemia gene (MLL) located on chromosome 11q23. MLL is a histone-lysine N-methyltransferase that normally catalyzes the methylation of H3K4, but this function is lost when chromosomal translocations occur that produce oncogenic fusion proteins, e.g., MLL-AF4, MLL-AF9, MLL-AF10, MLL-ENL, etc. These MLL translocations occur in approximately 3%–10% of AML patients and 8%–10% of patients with B cell–derived acute lymphoblastic leukemia (ALL) (39). MLL translocations are rarely found in T cell ALL (T-ALL) but may account for up to 80% of all infant acute leukemia cases (39). Accumulated evidence indicates that, in many of these MLL-rearranged leukemias, DOT1L associates with the fusion protein, is activated and drives development and progression of the disease (refs. 40–42 and Figure 1B). Ablation of DOT1L activity by shRNA or small-molecule inhibitors has been demonstrated to inhibit proliferation of various MLL-rearranged leukemia cells in vitro and extend survival in MLL-rearranged tumor xenografts (43–45). This evidence has made DOT1L a compelling drug target for drug discovery, and several small-molecule inhibitors have subsequently been reported (Figure 2 and refs. 46–48). Of these, the DOT1L inhibitor, EPZ-5676 (Epizyme/Celgene), has recently entered Phase I clinical trials as a 21-day i.v. infusion in relapsed/refractory patients with leukemias involving translocation of the MLL gene at 11q23 or advanced hematological malignancies (ClinicalTrials.gov identifier: NCT01684150).
BET inhibitors.
In addition to the epigenetic proteins that “write” the histone code (histone methyltransferases and histone acetyltranferases) and “erase” the histone code (histone demethylases and histone deacetylases), there are epigenetic proteins that “read” the epigenetic code by binding to methylated or acetylated histone residues as part of mechanisms of transcriptional regulation (49). The BET family of bromodomains, which includes BRD2, BRD3, BRD4, and bromodomain testis-specific protein (BRDT), are epigenetic reader proteins that bind acetylated lysine residues on histones. BRD4 binds to the transcription elongation factor P-TEFb and stimulates RNA polymerase II–dependent elongation (refs. 50, 51, and Figure 1C).
The somatic alteration that defines the rare, highly lethal cancer NUT midline carcinoma (NMC) is a t(15;19)(q14;p13.1) chromosomal translocation that results in a fusion protein between the BET proteins BRD3 or BRD4 and the nuclear protein in testis (NUT) (52). Because these proteins are the key oncogenic drivers for this cancer (53), it was hypothesized that an agent that would block the biological activity of BRD3-NUT and/or BRD4-NUT may be an effective therapy for NMC.
The biological action of BET proteins occurs through a protein-protein interaction (BET protein binding to an acetylated histone protein) and, as such, these biochemical activities historically have possessed poor tractability for small molecule drug discovery identification. Hence, Chun-Wa Chung and colleagues (54) did not initiate an oncology drug discovery effort to “drug” the BRD4 protein to target NMC, but rather identified potent, selective BET inhibitors serendipitously. A reporter gene cell-based assay was used to perform a high-throughput screen to identify molecules that result in upregulation of APOA1 as an approach to identify molecules for potential use in atherosclerosis. Through the use of chemoproteomics, siRNA, biophysical assays, and X-ray crystallography, it was determined that the ApoA1 upregulators identified were potent, selective binders to BET proteins BRD2, BRD3, BRD4, and BRDT (54). In independent efforts over the same time period, Mitsubishi Tanabe scientists reported the discovery and development of thienotriazolodiazepine as BET inhibitors (55), which were reported to inhibit CD28 costimulatory effects on T cells (56). Building on these findings, Bradner and colleagues synthesized a thienotriazolodiazepine, JQ1, which is highly selective for binding to BET over non-BET bromodomain proteins (24).
In fact, the initial presumption of low tractability of the BET proteins has proven not to be the case, as multiple investigators have identified potent, selective BET inhibitors with substantially different chemical structures (Figure 2 and refs. 24, 57–59). The crystal structures of these small molecule inhibitors bound to BRD4 illustrates that the binding pocket, which also binds acetyl-lysine, is small, deep, and hydrophobic.
The BET inhibitor JQ1 promotes differentiation, tumor regression and prolonged survival in murine models of NMC (24), consistent with the role of BRD4-NUT in this rare cancer. Similar pharmacological activity has been observed with GSK I-BET762, which is currently in clinical trials for NMC and other cancers (ClinicalTrials.gov identifier: NCT01587703). Both BET inhibitors JQ1 and I-BET151 down regulate the expression of multiple oncogenes in cancer cells, and have been shown to silence MYC and induce cytotoxicity in a range of hematological malignancies that include multiple myeloma and acute myeloid leukemia (58, 60, 61). BET inhibitors also possess broad cytotoxic activity in solid tumor cell lines; therefore, identification of predictive biomarkers that can be used for patient selection would increase the likelihood of clinical success of the agents. Two groups recently reported that BET inhibitors silence MYCN expression in neuroblastoma and that MYCN amplification is a marker of sensitivity in this tumor type (62, 63). MYCN amplification is being employed as a prospective marker for patient selection in an I-BET762 clinical trial (ClinicalTrials.gov identifier: NCT01587703).
The finding that BET proteins, a subclass of epigenetic readers, are amenable to small-molecule drug discovery and that those inhibitors have profound direct effects on transcription is an important breakthrough. For quite some time, a major goal of oncology drug discovery has been the identification of agents that directly and specifically silence oncogenes or increase expression of tumor suppressors. The BET inhibitors dramatically decrease expression of multiple oncogenes, though likely without the ideal specificity desired as they can downregulate more than 100 genes in a cell line. An in-depth understanding of the tumor-specific gene silencing activity profile of a BET inhibitor in tumor type subclasses is needed to apply a precision medicine approach to the use of these agents. Moreover, these advances necessitate determining how many epigenetic readers are druggable, and if targeting these proteins allows for targeted modulation of gene expression with a set of reader protein inhibitors (64). It is preliminary to address the question of whether the success of targeting BET proteins will be common for other epigenetic readers or the exception.
Implications for drug discovery
Great progress has been made in developing second-generation small-molecule inhibitors of epigenetic proteins. Inhibitors of DOT1L, BET, and EZH2 are now entering clinical trials. This clearly demonstrates that many epigenetic targets are druggable, albeit with substantial effort. Progress is being made in developing tools to identify epigenetic drivers of cancer and prosecute epigenetic-targeted drug discovery. Despite these advances, it is evident that epigenetic biology is more complex than initially imagined. For example, there are now several selective inhibitors of EZH2 that are potently anti-proliferative in lymphomas bearing EZH2 activating mutations, yet it is not completely understood why some tumor lines with EZH2 activating mutations are weakly sensitive and why some with WT EZH2 are highly sensitive (29). Deletion of UTX, an H3K27me3 demethylase, does not explain sensitivity in these WT EZH2 tumor lines. Evidently, there are underlying genetics and/or epigenetics that predispose some tumors to EZH2 enzyme inhibition. Moreover, the role of EZH2 (and other epigenetic proteins) may be contextual, acting as oncogenes in one cancer and tumor suppressors in another (myelodysplastic syndrome, ref. 65). Cross-talk or interaction between histone modifications affects protein recruitment and biological response, so the existing histone milieu may need to be considered when selecting patients/tumor type. In addition, many enzymes thought to have an epigenetic role may function in cancer via non-epigenetic mechanisms, i.e., methylation of cytosolic substrates. The presence or absence of SWI/SNF components may contribute to drug sensitivity, as demonstrated by the pronounced effect of EZH2 inhibitors on SMARCB1/SNF5-deficient rhabdhoid tumors (19).
Challenges for existing and future clinical trials with epigenetic modifiers include understanding the utility of pharmacodynamic (PD) (e.g., histone/DNA mark, target gene signature) and patient stratification markers (somatic mutation) to optimize response to these agents. To enable these PD and patient-tailoring biomarkers, clinically approved prognostic and/or diagnostic assays will be required. There is also the longer term prospect of identifying and employing specific histone methyl or acetyl marks for tumor subtype identification and patient stratification. DNA methylation status in colon cancer patients (CpG island methylator phenotype) currently is employed as an important subclassification of this tumor type (66, 67) and the same may be possible using histone marks. The safety profiles in human are as yet unknown and it will be interesting to see if there are commonalities with first generation epigenetic drugs, such as neutropenia or thrombocytopenia. If well tolerated, there is great potential for combination therapies, especially in cases of resistance to existing standards of care and/or refractory states (decitabine in cisplatinum-resistant ovarian cancer: ref. 68; azacytidine combined with etinostat in refractory advanced non–small-cell lung cancer: ref. 69). The proven clinical utility of DNMT inhibitors and pan-HDAC inhibitors, as well as the rapid preclinical advancement of the second generation of epigenetic modulators (e.g., EZH2, DOT1L, BET), lends optimism for future epigenetic drug discovery and development. More clinical experience with these agents will serve to guide strategies for therapeutic application in targeted patient populations.
Acknowledgments
We thank Frederick Derheimer and Neil Johnson for assistance in the preparation of this manuscript.
Footnotes
Conflict of interest: Robert M. Campbell owns equity in and is an employee of Eli Lilly and Company. Peter J. Tummino owns equity in and is an employee of GlaxoSmithKline.
Citation for this article: J Clin Invest. 2014;124(1):64–69. doi:10.1172/JCI71605.
References
- 1.Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011;1815(1):75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Baylin SB, Jones PA. A decade of exploring the cancer epigenome — biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10(10):704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smiraglia DJ, et al. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum Mol Genet. 2001;10(13):1413–1419. doi: 10.1093/hmg/10.13.1413. [DOI] [PubMed] [Google Scholar]
- 5.Paz MF, et al. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003;63(5):1114–1121. [PubMed] [Google Scholar]
- 6.Sneeringer CJ, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107(49):20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yap DB, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCabe MT, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A. 2012;109(8):2989–2994. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keats JJ, Reiman T, Belch AR, Pilarski LM. Ten years and counting: so what do we know about t(4;14)(p16;q32) multiple myeloma. Leuk Lymphoma. 2006;47(11):2289–2300. doi: 10.1080/10428190600822128. [DOI] [PubMed] [Google Scholar]
- 10.Lauring J, et al. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood. 2008;111(2):856–864. doi: 10.1182/blood-2007-05-088674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Garcia E, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117(1):211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo AJ, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44(4):609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 15.Romero OA, Sanchez-Cespedes M. Oncogene. The SWI/SNF genetic blockade: effects in cell differentiation, cancer and developmental diseases [published online ahead of print June 10, 2013]. doi: 10.1038/onc.2013.227 . [DOI] [PubMed] [Google Scholar]
- 16.Versteege I, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 17.Jackson EM, et al. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res. 2009;15(6):1923–1930. doi: 10.1158/1078-0432.CCR-08-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson BG, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knutson SK, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vedadi M, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7(8):566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konze KD, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu W, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 23.James LI, et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol. 2013;9(3):184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fish PV, et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J Med Chem. 2012;55(22):9831–9837. doi: 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu F, et al. Exploiting an allosteric binding site of PRMT3 yields potent and selective inhibitors. J Med Chem. 2013;56(5):2110–2124. doi: 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, et al. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry. 2011;50(29):6488–6497. doi: 10.1021/bi200725p. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem. 2009;284(49):34283–34295. doi: 10.1074/jbc.M109.034462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCabe MT, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 30.Knutson SK, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 31.Jung HR, Pasini D, Helin K, Jensen ON. Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol Cell Proteomics. 2010;9(5):838–850. doi: 10.1074/mcp.M900489-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diaz E, et al. Development and validation of reagents and assays for EZH2 peptide and nucleosome high-throughput screens. J. Biomol. Screen. 2012;17(10):1279–1292. doi: 10.1177/1087057112453765. [DOI] [PubMed] [Google Scholar]
- 33.Qi W, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109(52):21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma SK, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3(12):1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Albrecht BK, Audia JE, Gagnon A, Harmange JC, Naveschuk CG, inventors. Constellation Pharmaceuticals, assignee. Modulators of methyl modifying enzymes, compositions uses thereof. US patent application PCT/US2012/065796. November 19, 2012. [Google Scholar]
- 36.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Aller GS, et al. ACS Chem Biol. Long residence time inhibition of EZH2 in activated polycomb repressive complex 2 [published online ahead of print December 4, 2013]. doi: 10.1021/cb4008748 . [DOI] [PubMed] [Google Scholar]
- 38.Feng Q, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 39.Aplan PD. Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA Repair (Amst). 2006;5(9–10):1265–1272. doi: 10.1016/j.dnarep.2006.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16(1):92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 41.Mueller D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110(13):4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7(11):e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia. 2013;27(4):813–822. doi: 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao Y, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133(42):16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basavapathruni A, et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80(6):971–980. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- 48.Yu W, et al. Bromo-deaza-SAH: a potent and selective DOT1L inhibitor. Bioorg Med Chem. 2013;21(7):1787–1794. doi: 10.1016/j.bmc.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 50.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19(4):535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 52.French CA. Demystified molecular pathology of NUT midline carcinomas. J Clin Pathol. 2010;63(6):492–496. doi: 10.1136/jcp.2007.052902. [DOI] [PubMed] [Google Scholar]
- 53.French CA, et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27(15):2237–2242. doi: 10.1038/sj.onc.1210852. [DOI] [PubMed] [Google Scholar]
- 54.Chung C-W, et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54(11):3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 55. Miyoshi S, Ooike S, Iwata K, Hikawa K, Sugahara K, inventors. Mitsubishi Tanabe Pharma Corporation, assignee. Antitumor agent. International patent PCT/JP2008/073864. September 7, 2009. [Google Scholar]
- 56. Adachi K, et al. Thienotriazolodiazepine compound medicinal use thereof. International patent PCT/JP2006/310709. July 12, 2006. [Google Scholar]
- 57.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dawson MA, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Picaud S, et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013;73(11):3336–3346. doi: 10.1158/0008-5472.CAN-12-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mertz JA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Puissant A, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3(3):308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wyce A, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013;8(8):e72967. doi: 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dawson MA, Kouzarides T, Huntly BJ. Targeting epigenetic readers in cancer. N Engl J Med. 2012;367(7):647–657. doi: 10.1056/NEJMra1112635. [DOI] [PubMed] [Google Scholar]
- 65.Nikoloski G, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 66.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96(15):8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weisenberger DJ, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 68.Matei D, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9):2197–2205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94(7):984–993. doi: 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiang CM. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol Rep. 2009;1:98. doi: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]