

Abstract
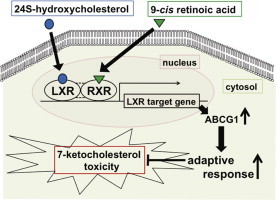
Lipid peroxidation products have been known to induce cellular adaptive responses and enhance tolerance against subsequent oxidative stress through up-regulation of antioxidant compounds and enzymes. 24S-hydroxycholesterol (24SOHC) which is endogenously produced oxysterol in the brain plays an important role in maintaining brain cholesterol homeostasis. In this study, we evaluated adaptive responses induced by brain-specific oxysterol 24SOHC in human neuroblastoma SH-SY5Y cells. Cells treated with 24SOHC at sub-lethal concentrations showed significant reduction in cell death induced by subsequent treatment with 7-ketocholesterol (7KC) in both undifferentiated and retinoic acid-differentiated SH-SY5Y cells. These adaptive responses were also induced by other oxysterols such as 25-hydroxycholesterol and 27-hydroxycholesterol which are known to be ligands of liver X receptor (LXR). Co-treatment of 24SOHC with 9-cis retinoic acid, a retinoid X receptor ligand, enhanced the adaptive responses. Knockdown of LXRβ by siRNA diminished the adaptive responses induced by 24SOHC almost completely. The treatment with 24SOHC induced the expression of LXR target genes, such as ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1). The 24SOHC-induced adaptive responses were significantly attenuated by siRNA for ABCG1 but not by siRNA for ABCA1. Taken together, these results strongly suggest that 24SOHC at sub-lethal concentrations induces adaptive responses via transcriptional activation of LXR signaling pathway, thereby protecting neuronal cells from subsequent 7KC-induced cytotoxicity.
Abbreviations: ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; AD, Alzheimer's disease; atRA, all-trans retinoic acid; CYP46A1, cholesterol 24-hydroxylase; FITC, fluorescein isothiocyanate; HDL, high-density lipoprotein; LDH, lactate dehydrogenase; LXR, liver X receptor; MAP2, microtubule-associated protein 2; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NC, negative control; PI, propidium iodide; RXR, retinoid X receptor; 24SOHC, 24S-hydroxycholesterol; 7KC, 7-ketocholesterol; 9cRA, 9-cis retinoic acid
Keywords: Cell death, Adaptive responses, Liver X receptor, 24S-hydroxycholesterol, 7-ketocholesterol, ATP-binding cassette transporter G1
Graphical abstract
Highlights
-
•
24SOHC induces adaptive responses against 7KC-induced cell death in neuronal cells.
-
•
Co-treatment of 24SOHC with 9cRA, an RXR ligand enhances adaptive responses.
-
•
Knockdown of LXRβ suppresses 24SOHC-induced adaptive responses.
-
•
ABCG1 is involved in LXR-mediated adaptive responses by 24SOHC.
Introduction
The oxidative modification of biomolecules (proteins, lipids, nucleic acids) under oxidative stress has been implicated in a variety of pathological events such as atherosclerosis, cancer and neurodegenerative diseases [1–3]. These oxidation products have been suggested to act as biomarkers providing evidence of elevated oxidative stress in our bodies [4–6]. Some of these products are so active that they enhance further oxidative stress, resulting in development of diseases and aging. It has also been known that many oxidation products, especially lipid peroxidation products possess a variety of physiological functions [7]. Such lipid peroxidation products include oxysterols, which are oxidized forms of cholesterol [8]. Oxysterols have diverse functions not possessed by cholesterol. While oxysterols play important roles in signal transduction and lipid homeostasis [9], their pro-oxidative and pro-inflammatory activities resulting in cell death have been implicated in the pathogenesis of a variety of diseases [10].
Enhanced oxidative stress has been suggested to play an important role in the pathogenesis of neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease [11,12]. Oxidation of cholesterol proceeds enzymatically and non-enzymatically in the brain [9,13]. When cholesterol is attacked by reactive oxygen species at the 7-position, oxysterols such as 7-ketocholesterol (7KC) and 7β-hydroxycholesterol are generated. Since the highly cytotoxic potential of 7KC has been shown in neuronal cells [14–16], 7KC is suspected to be involved in the pathogenesis of neurodegenerative diseases.
Cholesterol is also oxidized enzymatically to produce oxysterols such as 24S-hydroxycholesterol (24SOHC), 25-hydroxycholesterol (25OHC), and 27-hydroxycholesterol (27OHC). Of these, 24SOHC is the most abundant oxysterol in the brain. It has been reported that free 24SOHC is present at high concentrations (about 30 μM) in human brain homogenate [17]. Cholesterol 24-hydroxylase (CYP46A1), which is predominantly expressed in the brain, catalyzes the conversion of cholesterol to 24SOHC [13,18]. Since cholesterol does not penetrate the blood–brain barrier, excess amounts of cholesterol in the brain are converted to 24SOHC, which readily crosses the blood–brain barrier to be eliminated [19]. 24SOHC is also known to act as an endogenous ligand of liver X receptor (LXR), a transcription factor regulating cholesterol efflux. Therefore, CYP46A1 and 24SOHC play pivotal roles in brain cholesterol homeostasis. While 24SOHC is an important physiological molecule, several lines of evidence suggest that 24SOHC is linked to the development of AD [20–22]. Higher concentrations of 24SOHC were detected in plasma and cerebrospinal fluid of patients with AD and mild cognitive impairment compared to healthy donors. Furthermore, neurotoxicity of 24SOHC has been reported [23]. We have recently reported that high concentrations of 24SOHC (50 μM) induce necroptosis, a form of programmed necrosis in neuronal cells [24]. In contrast, we have also reported that low concentrations of 24SOHC (<10 μM) suppress the production of amyloid-β [25]. These current data show the diverse functions of 24SOHC which may be explained by concentration-dependent effects.
It has been demonstrated that certain lipid peroxidation products exhibit not only cytotoxic effects but also cytoprotective effects as a result of induction of adaptive responses [7]. Sub-lethal concentrations of 7α(β)-hydroxycholesterol, a free radical-mediated oxidation product of cholesterol, significantly increase cellular glutathione levels and enhance cell tolerance against the subsequent oxidative stress in rat pheochromocytoma PC12 cells [26]. Other lipid peroxidation products such as 15-deoxy-Δ12,14-prostaglandin J2 and 4-hydroxynonenal at low concentrations have also been found to activate the NF-E2-related factor 2 pathway and increase antioxidant capacity [27–30]. In this study, we found that stimulation of human neuroblastoma SH-SY5Y cells with sub-lethal concentrations of brain-specific oxysterol 24SOHC induced adaptive responses and protected the cells against subsequent cytotoxic stress induced by 7KC. Moreover, we found that the activation of the LXR pathway played a key role in 24SOHC-induced adaptive responses.
Material and methods
Materials
Dulbecco's modified Eagle's medium/nutrient mixture Ham's F-12 (DMEM/F-12) was from Invitrogen. FBS was from Hyclone, Thermo Scientific. 24SOHC was obtained from Biomol International. 7KC was from Steraloids. 9-cis retinoic acid (9cRA) and all-trans retinoic acid (atRA) was from Wako Pure Chemical Industries. Hoechst 33258 was from Dojindo. TO901317 was from Cayman. 27OHC was from Avanti. Oxysterols, 9cRA and atRA were dissolved in ethanol and stored at −20 °C. The following antibodies were from commercial sources: ABCA1 (catalog no. NB400-105) and ABCG1 (catalog no. NB400-132), NOVUS Biologicals; Liver X Receptor β (catalog no. K8917), Perseus Proteomics; β-actin (catalog no. A5441) and microtubule-associated protein 2 (MAP2) (catalog no. M4403), SIGMA. All other chemicals, of analytical grade, were from Sigma-Aldrich or Wako.
Cell culture
SH-SY5Y human neuroblastoma cells (American Type Culture Collection) were routinely maintained in DMEM/F-12 medium with 10% heat-inactivated FBS and antibiotics (100 units/ml penicillin, 100 µg/ml streptomycin; Invitrogen) at 37 °C under an atmosphere of 95% air and 5% CO2. SH-SY5Y cells were differentiated by treatment with 10 μM atRA for 5 days.
Immunofluorescence staining
The cells grown on glass cover slips were fixed with 4% paraformaldehyde/PBS for 10 min, permeabilized with 0.5% Triton X-100/PBS for 10 min and then blocked with 2% BSA/PBS for 30 min. The cover slips were incubated with anti-MAP2 antibody and Alexa Fluor-conjugated secondary antibody. The nuclei were stained with Hoechst. Confocal fluorescence image was obtained using a Zeiss LSM710 confocal microscope lasers with oil objective lens and accompanying software (LSM Software ZEN 2009).
Determination of cell viability
To examine the toxicity of 7KC, SH-SY5Y cells were grown on plates at a density of 4.5×105 cells/ml. After the cells were attached (16–18 h), they were treated with 24SOHC, or with 24SOHC and 9cRA, at different concentrations for 24 h. The culture medium was thereafter replaced with fresh medium containing indicated concentrations of 7KC or vehicle alone. For determination of cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and lactate dehydrogenase (LDH) release assay were conducted for the indicated periods, as described previously [24]. WST-8 assay was performed using Cell Counting Kit-8 according to the manufacturer's instructions (Dojindo). Phosphatidylserine exposure and propidium iodide (PI) staining were analyzed as described previously [24]. Briefly, control and treated cells were labeled with annexin V-fluorescein isothiocyanate (FITC) and PI, followed by analysis with a FACSAriaTM II Cell Sorter (BD Biosciences) with a 488-nm argon laser.
Immunoblotting
Whole cell lysates were prepared in lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 2 mM EDTA) plus protease inhibitor cocktail (nacalai tesque). Cell debris was removed by centrifugation. Protein samples were subjected to SDS-PAGE and immunoblotting by using appropriate antibodies.
Knockdown of LXRβ, ABCA1 and ABCG1 in SH-SY5Y cells by small interfering RNA
Stealth siRNA targeting human LXRβ (5′-UCAAGAAGGUGAUACACUCUGUCUC-3′), human ABCA1 (5′-CACUUCCUCCGAGUCAAGAAGUUAA-3′), and human ABCG1 (5′-UUCAGACCCAAAUCCCUCAAAUAUG-3′), as well as Stealth RNAi Negative Control (NC), were obtained from Invitrogen. Cells grown in 6-well or 48-well plates were transfected with 20 nM of siRNA/well by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. After 24 h incubation, the cells were subjected to 24SOHC pretreatment, followed by 7KC treatments.
RNA isolation, RT-PCR and real-time PCR
Total RNA was isolated with FastPure RNA kit (TAKARA) according to the manufacturer's instructions. Samples were reverse-transcribed using random hexamer and ReverTra Ace (TOYOBO). Synthesized cDNA was subjected to real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) using the following protocol: 10 min at 95 °C, 40 cycles of 15 s at 95 °C, and 1 min at 60 °C, and analyzed with a 7900HT Fast Real Time PCR system (Applied Biosystems). The GAPDH gene was used for the normalization of each reaction. The following primers were used: ABCA1 forward, 5′-GAGCACCATCCGGCAGAA-3′; reverse, 5′-CTCCGCCTTCACGTGCTT-3′; ABCG1 forward, 5′-GCCTCACGCAGTTCTGCAT-3′; reverse, 5′-GGACCGAGTCCCTCATGATG-3′; GAPDH forward, 5′-TTTGGCTACAGCAACAGGGTGGTG-3′; reverse, 5′-ATGGTACATGACAAGGTGCCGCTC-3′.
Statistical analysis
Data are reported as mean±SD of at least three independent experiments unless otherwise indicated. Statistical comparisons were made by an analysis of variance using Tukey's test for multiple comparisons. The difference was considered significant when the P value was less than 0.05.
Results
Effect of 7KC and 24SOHC on viability of neuronal cells
To evaluate the cytotoxicity of 7KC, human neuroblastoma SH-SY5Y cells were treated with several concentrations of 7KC for 24 h and the viability was measured by MTT assay. Since 7KC significantly decreased cell viability at concentrations higher than 30 μM (Fig. 1A), we chose 50 or 75 µM 7KC to induce cell death. The 50% cell death (LD50) occurred at 37 μM 7KC. The LD50 of 7KC in neuronal cells are similar to doses described elsewhere [14–16]. We also evaluated the cytotoxicity of 24SOHC at several concentrations by using WST-8 assay. The results showed that 24SOHC significantly decreased cell viability at concentrations above 10 µM (Fig. 1B). Our previous paper showed that 24SOHC at concentrations higher than 10 µM exhibited significant neurotoxicity in SH-SY5Y cells as evaluated using MTT assay and LDH assay [24]. It has been reported that SH-SY5Y contains CYP46A1 mRNA but does not synthesize 24SOHC [31]. Therefore, we chose 1–10 μM 24SOHC as a range of sub-lethal concentrations at which 24SOHC did not show appreciable cytotoxicity and are within the range observed in human brain [32].
Fig. 1.
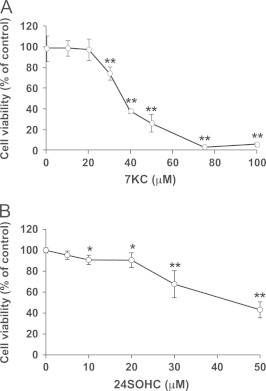
Cell death induced by 7KC or 24SOHC in SH-SY5Y cells. SH-SY5Y cells were treated with variable concentrations of 7KC (A) or 24SOHC (B) for 24 h, and cell viability was measured by MTT assay (A) and by WST-8 assay (B). Each bar represents mean±SD (n=3). ⁎P<0.05, ⁎⁎P<0.01, when compared with vehicle control.
24SOHC induces adaptive responses in SH-SY5Y cells
To evaluate ability of 24SOHC to prevent cell death caused by 7KC, SH-SY5Y cells were pretreated with 0–10 μM 24SOHC for 24 h and followed by subsequent treatment with 7KC. Cell viability was measured by MTT assay (Fig. 2A) and LDH release assay (Fig. 2B). Pretreatment with 24SOHC at 5 μM significantly reduced cell death caused by 50–75 μM 7KC. To further evaluate the protective effects of 24SOHC on 7KC-induced cell death, we analyzed the types of cell death induced by 7KC. Using flow cytometry, apoptosis and necrosis were assessed by annexin V-FITC staining (Fig. 2C) and PI staining (Fig. 2D), respectively. 7KC treatment increased both annexin V-FITC-positive apoptotic cells and PI-positive necrotic cells. Pretreatment of cells with 24SOHC significantly reduced apoptotic cell death and tended to decrease necrotic cell death. These results showed that 24SOHC induced adaptive responses in neuronal cells which resulted in increased resistance to cell death caused by 7KC.
Fig. 2.
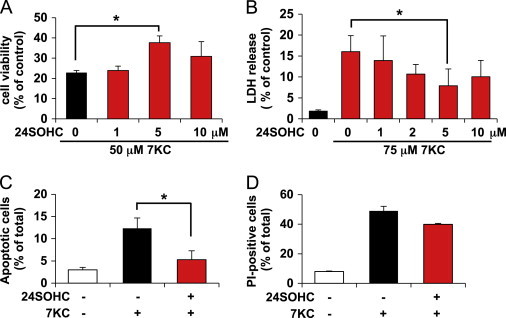
Pretreatment with sub-lethal 24SOHC induces adaptive responses in SH-SY5Y cells. (A) SH-SY5Y cells treated with 0–10 µM 24SOHC for 24 h then treated with 50 μM 7KC for 24 h. The cell viability was measured by MTT assay. (B) The cells were treated with 0–10 µM 24SOHC then treated with 75 μM 7KC for 24 h. The cytotoxicity was measured by LDH assay. (C and D) The cells were treated with 5 µM 24SOHC for 24 h then treated with 60 μM 7KC for 21 h. The cell samples were then treated with annexin V-FITC and PI and subjected to flow cytometry. The percentage of cells exhibiting annexin V-FITC-positive and PI-negative apoptotic (C) and PI-positive (D) cells are shown. Each bar represents mean±SD (n=3). ⁎P<0.05.
Activation of LXR pathway reduces 7KC-induced cell death
Since 24SOHC has been known to act as a ligand of LXR, we investigated the ability of other LXR ligands to induce adaptive responses. TO901317 is a synthetic LXR ligand. Other enzymatically produced oxysterols, 25-hydroxycholesterol (25OHC) and 27-hydroxycholesterol (27OHC), are also known as endogenous LXR ligands. As was the case with 24SOHC, pretreatment of SH-SY5Y cells with 5 μM of TO901317, 25OHC, or 27OHC showed protective effects against 7KC-induced cell death (Fig. 3A).
Fig. 3.
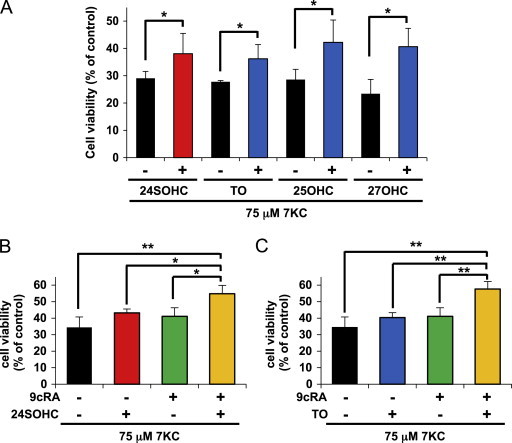
Activation of LXR pathway reduces 7KC-induced cell death. (A) The cells were treated with 5 μM 24SOHC, TO901317, 25OHC or 27OHC, then treated with 75 μM 7KC for 24 h. The cell viability was measured by MTT assay. (B and C) The cells were treated with 5 μM 24SOHC (B) or TO901317 (C) in the presence or absence of 10 μM 9cRA for 24 h. After the treatment with 75 μM 7KC, the cell viability was measured by MTT assay. Each bar represents mean±SD (n=3). ⁎P<0.05, ⁎⁎P<0.01.
LXR forms a functional heterodimer with retinoid X receptor (RXR) and regulates transcription of several genes involved in the cholesterol efflux pathway [33]. We therefore assessed whether co-treatment of cells with 24SOHC and RXR ligand 9-cis retinoic acid (9cRA) enhanced the adaptive responses to prevent 7KC-induced cell death. Co-treatment with 24SOHC and 9cRA significantly enhanced adaptive responses to prevent 7KC-induced cell death (Fig. 3B). The same experiments using TO901317 and 9cRA revealed that the adaptive responses were enhanced by co-treatment of cells with TO901317 and 9cRA (Fig. 3C). Together, these results suggest that activation of LXR pathway by oxysterols is involved in suppression of 7KC-induced cytotoxicity
24SOHC induces adaptive responses in differentiated SH-SY5Y cells
We conducted the experiments using differentiated SH-SY5Y cells by treatment with atRA for 5 days. The differentiation of cells was confirmed by observation of neurite extention and immunofluorescence staining with a marker of differentiated neuron MAP2 by using a fluorescence microscopy (Fig. 4A). Since the viability of the differentiated cells was decreased with concentration dependent manner of 7KC (Fig. 4B) as observed in the undifferentiated cells (Fig. 1A), we chose 60 µM 7KC to induce cell death. Pretreatment with either 5 μM 24SOHC or 10 μM 9cRA, or their co-treatment significantly reduced cell death caused by 60 μM 7KC (Fig. 4C), as observed in undifferentiated SH-SY5Y cells (Fig. 3B).
Fig. 4.
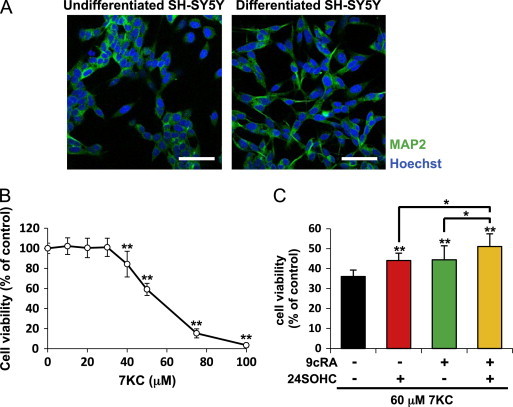
Pretreatment with sub-lethal 24SOHC induces adaptive responses in differentiated SH-SY5Y cells. (A–C) SH-SY5Y cells were treated with 10 µM atRA for 5 days. (A) The cells grown on coverslips were processed for immunofluorescence staining with antibody against MAP2. Blue: Hoechst, Green: MAP2. Representative confocal images are shown. Bar, 20 μm. (B) Differentiated SH-SY5Y cells were treated with variable concentrations of 7KC for 24 h. The cell viability was measured by WST-8 assay. (C) Differentiated SH-SY5Y cells were treated with 5 μM 24SOHC in the presence or absence of 10 μM 9cRA for 24 h. After the treatment with 60 μM 7KC, the cell viability was measured by WST-8 assay. Each bar represents mean±SD (n=3). ⁎P<0.05, ⁎⁎P<0.01, when compared with vehicle control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Knockdown of LXRβ reduces 24SOHC-induced adaptive responses
For further confirmation of the involvement of LXR protein in the adaptive responses triggered by 24SOHC, we performed siRNA knockdown of LXRβ. The LXR family consists of two subtypes, LXRα and LXRβ. LXRβ has a relatively higher level of expression in the brain than LXRα [34]. Immunoblot analysis showed that LXRβ siRNA successfully suppressed expression of LXRβ protein in both cells with and without 24SOHC treatment (Fig. 5A). The cytoprotective effects of 24SOHC disappeared in LXRβ-knockdowned cells (Fig. 5B), suggesting that LXRβ played a key role in 24SOHC-induced adaptive responses.
Fig. 5.
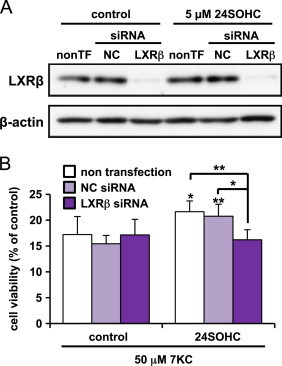
Knockdown of LXRβ reduces 24SOHC-induced adaptive responses in SH-SY5Y cells. (A and B) SH-SY5Y cells were transfected with negative control (NC) or LXRβ siRNA and incubated for 24 h then treated with 5 μM 24SOHC for 24 h. (A) Whole cell lysates were immunoblotted with appropriate antibodies as indicated. (B) The cells were challenged with 50 μM 7KC for 24 h. The cell viability was measured by WST-8 assay. Each bar represents mean±SD (n=3). ⁎P<0.05, ⁎⁎P<0.01, when compared with vehicle control.
Target genes of LXRβ responsible for 24SOHC-induced adaptive responses
Next, we measured mRNA expression levels of LXR target genes ABCA1, ABCG1, and ApoE by real-time PCR and measured protein levels of ABCA1 and ABCG1 by immunoblotting. Without stimulation by 24SOHC, mRNA levels as well as protein levels of ABCA1 and ABCG1 in SH-SY5Y cells are under detectable. Treatment with either 24SOHC or TO901317 increased expression levels of both ABCA1 and ABCG1 mRNA (Fig. 6A and B) but not ApoE mRNA (Fig. S1). Treatment with 7KC did not change expression levels of ABCA1 and ABCG1 mRNA. Up-regulation of ABCA1 and ABCG1 protein levels by 24SOHC or TO901317 was also confirmed by immunoblotting (Fig. 6C and D). When cells were co-treated with 9cRA and either 24SOHC or TO901317, the protein levels of ABCA1 and ABCG1 were further increased (Fig. 6E). The increase in protein levels of ABCA1 and ABCG1 was also observed in differentiated SH-SY5Y cells treated with 24SOHC in the absence or presence of 9cRA but not in cells treated with 7KC (Fig. 6F).
Fig. 6.
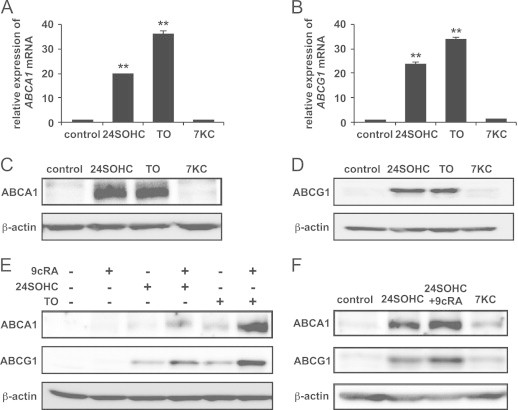
Up-regulation of LXRβ target genes in 24SOHC-treated SH-SY5Y cells. (A–D) The cells were treated with 5 μM of 24SOHC, TO901317 or 7KC for 24 h. ABCA1 (A) and ABCG1 (B) mRNA levels were quantified by real-time PCR, and the relative expression levels are shown. Each bar represents mean±SD (n=3). ⁎⁎P<0.01, when compared with vehicle control. (C and D) Whole cell lysates were immunoblotted with using an antibody against ABCA1 (C) or ABCG1 (D). (E) The cells were treated with 5 μM 24SOHC or TO901317 in the presence or absence of 10 μM 9cRA for 24 h. Whole cell lysates were immunoblotted with appropriate antibodies as indicated. (F) Differentiated SH-SY5Y cells were treated with 5 μM 24SOHC in the presence or absence of 10 μM 9cRA, or 7KC for 24 h as Fig. 4. Whole cell lysates were immunoblotted with using an antibody against ABCA1 or ABCG1.
ABCG1 rather than ABCA1 is responsible for the adaptive responses induced by 24SOHC
Since expression of ABCA1 and ABCG1 was increased in cells treated with 24SOHC or TO901317, we studied the effects of siRNA knockdown of either ABCA1 or ABCG1 on the adaptive responses. Without stimulation by 24SOHC, ABCA1 and ABCG1 expression were not observed in non-transfected cells (Fig. 7A and B) as observed in Fig. 5. Treatment with 24SOHC increased ABCA1 and ABCG1 expression in both non-transfected and NC siRNA-transfected cells. In ABCA1 siRNA-transfected cells, expression of ABCA1 protein was specifically suppressed without affecting ABCG1 protein levels (Fig. 7A). In ABCG1 siRNA-transfected cells, ABCG1 protein expression by 24SOHC treatment was specifically suppressed without affecting ABCA1 protein levels (Fig. 7B). These results show that siRNA for ABCA1 or ABCG1 successfully diminished 24SOHC-induced expression of ABCA1 and ABCG1, respectively. Under these conditions, we evaluated the effects of knockdown of either ABCA1 or ABCG1 on the induction of adaptive responses by 24SOHC. The knockdown of ABCA1 did not affect 24SOHC-induced adaptive responses (Fig. 7C). In contrast, in ABCG1 siRNA-transfected cells, 24SOHC still induced adaptive response, but the protective effect was significantly diminished (Fig. 7D). Knockdown of ABCG1 also reduced TO901317-induced adaptive responses (Fig. S2). Together, these results suggest that ABCG1 but not ABCA1 is involved in LXRβ-mediated adaptive responses induced by 24SOHC.
Fig. 7.
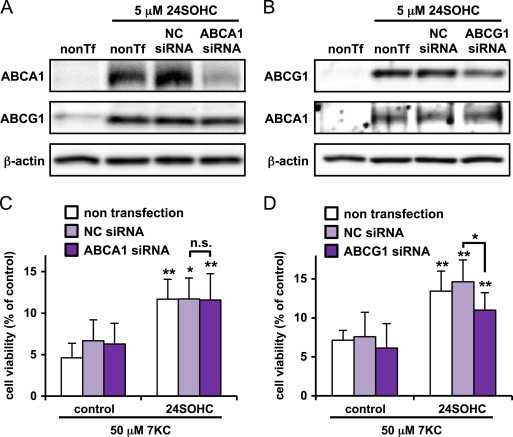
Knockdown of ABCG1 but not ABCA1 reduces 24SOHC-induced adaptive responses in SH-SY5Y cells. (A–D) SH-SY5Y cells were transfected with NC or ABCA1 siRNA (A and C) or ABCG1 siRNA (B and D) and incubated for 24 h then treated with 5 μM 24SOHC for 24 h. (A and B) Whole cell lysates were immunoblotted with appropriate antibodies as indicated. (C and D) The cells were challenged with 50 μM 7KC for 24 h. The cell viability was measured by WST-8 assay. Each bar represents mean±SD (n=3). ⁎P<0.05, ⁎⁎P<0.01, when compared with vehicle control. n.s.: not significant.
Discussion
It has been reported that lipid peroxidation products can induce adaptive responses against subsequent further oxidative stress [7]. Under the induction of adaptive responses by lipid peroxidation products, cell death caused by oxidative stress was reduced significantly but not completely [26–30]. In this study, we showed that pretreatment of human neuronal cells with brain-specific oxysterol 24SOHC at sub-lethal concentrations significantly reduced cell death induced by subsequent treatment with 7KC. It has been thought that the major function of CYP46A1 is to maintain the cholesterol levels in the brain by converting excess amount of cholesterol to 24SOHC [13]. Our findings suggest that CYP46A1 and 24SOHC play important roles not only in brain cholesterol homeostasis but also in protection of neuronal cells against cytotoxic oxidative stress.
The present data suggest that up-regulation of LXR target genes play a pivotal role in 24SOHC-induced adaptive responses. We found that among the well-known LXR target genes, ABCG1 but not ABCA1 was involved in 24SOHC-induced adaptive responses. ABCA1 and ABCG1 in the plasma membrane play crucial roles in cholesterol efflux to high-density lipoprotein (HDL) and/or apolipoprotein A-I and prevent cells from cholesterol accumulation [35]. Terasaka et al. reported that ABCG1 but not ABCA1 promoted efflux of 7KC from macrophages onto HDL and protected macrophages against 7KC-induced cytotoxicity [36]. Another report in the literature showed that trophoblast cells transfected with ABCG1 siRNA were more sensitive to 7KC [37]. Matsuda et al. reported that in the presence of HDL, the efflux of 24SOHC from differentiated SH-SY5Y cells was stimulated when the expression of ABCA1 and ABCG1 was induced via activation of LXR/RXR pathway, and knockdown of ABCA1 but not ABCG1 suppressed 24SOHC efflux [38]. All together with our present data and these literatures suggests that LXR/RXR-regulating efflux of oxysterols is dependent on cell types and oxysterol species. In our preliminary experiments, in addition to 7KC, we have investigated the protective effect of 24SOHC against several oxidative stress including 7β-hydroxycholesterol, 13S-hydroperoxy-9Z,11E-octadecadienoic acid, 6-hydroxydopamine, or 1-methyl-4-phenylpyridinium. 24SOHC did not show adaptive responses against other oxidative stress (data not shown). Such varied effects of 24SOHC on cytotoxicity of different oxidative stress species may be explained by the difference in their efflux mechanisms and/or metabolic pathways.
Although ABCG1 had been considered to localize in plasma membranes and contribute to cholesterol efflux to HDL, it has been indicated that ABCG1 localizes to endosome and facilitates the redistribution of intracellular sterol from the endoplasmic reticulum to plasma membranes [39]. It has also been reported that incorporation of 7KC to lipid raft domains of plasma membranes triggers apoptotic signaling, and α-tocopherol, a lipophilic antioxidant, reduced cytotoxicity of 7KC by inhibiting distribution of 7KC into the lipid raft domains [40,41]. It has recently been reported that ABCG1 mediates the generation of cell surface associated extracellular cholesterol microdomain which serves for reverse cholesterol transport by HDL [42]. Therefore, there is a possibility that 24SOHC induces adaptive responses via ABCG1-mediated intracellular and extracellular redistribution of 7KC. It might be noteworthy that while siRNA for LXRβ completely suppressed 24SOHC-induced adaptive responses, ABCG1 siRNA significantly but not completely abolished them. Although the efficacy of siRNA for ABCG1 was not good enough to inhibit its expression, it is likely that other LXR target genes may also contribute to 24SOHC-induced adaptive responses.
Oxidative damage has been implicated in neurodegenerative diseases [7,8]. Oxidative insult in the brain leads to oxidation of lipid and may contribute to the pathogenesis of neurodegenerative diseases. As had previously been reported in the literatures [14–16], we showed that 7KC induces apoptosis in neuronal cells. The current data therefore suggests that in addition to the role of 24SOHC for cholesterol homeostasis in the brain, its cytoprotective function should also be important for keeping the brain in good health. In line with our findings, synthetic LXR ligands could become candidates for therapeutic drug design against neurodegenerative diseases such as AD and multiple sclerosis [43].
Conclusions
Taken together, these results demonstrate that brain specific-generated oxysterol, 24SOHC protects neuronal cells from 7KC-induced cell death via induction of adaptive responses. We attribute the molecular mechanisms of 24SOHC-induced adaptive responses against 7KC cytotoxicity to up-regulation of LXR-target genes.
Acknowledgments
This work was supported, in part, by KAKENHI Grant-in-Aid for Young Scientists (B) 23700428, 25830041 to Y.U. from JSPS and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities in Japan for years 2012–2016.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Yasuomi Urano, Email: yurano@mail.doshisha.ac.jp.
Noriko Noguchi, Email: nnoguchi@mail.doshisha.ac.jp.
Appendix A. Supplementary material
Supplementary materials associated with this article can be found in the online version at doi:10.1016/j.redox.2013.11.007.
Appendix A. Supplementary materials
Supplementary materials
References
- 1.Halliwell B., Gutteridge J.M.C. fourth ed. Clarendon; Oxford, UK: 2007. Free Radicals in Biology and Medicine. [Google Scholar]
- 2.Chung H.S., Wang S.B., Venkatraman V., Murray C.I., Van Eyk J.E. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ. Res. 2013;112:382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sultana R., Butterfield D.A. Oxidative modification of brain proteins in Alzheimer's disease: perspective on future studies based on results of redox proteomics studies. J. Alzheimers Dis. 2013;33(Suppl. 1):S243–S251. doi: 10.3233/JAD-2012-129018. [DOI] [PubMed] [Google Scholar]
- 4.Il'yasova D., Scarbrough P., Spasojevic I. Urinary biomarkers of oxidative status. Clin. Chim. Acta. 2012;413:1446–1453. doi: 10.1016/j.cca.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jungnickel H., Tentschert J., Luch A. Monitoring oxidative stress biomarkers in the lipidome: is there a roadmap for “human inspection”? Curr. Mol. Med. 2012;12:716–731. doi: 10.2174/156652412800792606. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida Y., Niki E. Bio-markers of lipid peroxidation in vivo: hydroxyoctadecadienoic acid and hydroxycholesterol. Biofactors. 2006;27:195–202. doi: 10.1002/biof.5520270117. [DOI] [PubMed] [Google Scholar]
- 7.Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic. Biol. Med. 2009;47:469–484. doi: 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 8.Murphy R.C., Johnson K.M. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J. Biol. Chem. 2008;283:15521–15525. doi: 10.1074/jbc.R700049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Björkhem I. Five decades with oxysterols. Biochimie. 2013;95:448–454. doi: 10.1016/j.biochi.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 10.Olkkonen V.M. Macrophage oxysterols and their binding proteins: roles in atherosclerosis. Curr. Opin. Lipidol. 2012;23:462–470. doi: 10.1097/MOL.0b013e328356dba0. [DOI] [PubMed] [Google Scholar]
- 11.Simonian N.A., Coyle J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- 12.Jenner P. Oxidative stress in Parkinson's disease. Ann. Neurol. 2003;53(Suppl. 3):S26–S36. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 13.Russell D.W., Halford R.W., Ramirez D.M., Shah R., Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao M.L., Lütjohann D., Ludwig M., Kölsch H. Induction of apoptosis and necrosis in human neuroblastoma cells by cholesterol oxides. Ann. N. Y. Acad. Sci. 1999;893:379–381. doi: 10.1111/j.1749-6632.1999.tb07860.x. [DOI] [PubMed] [Google Scholar]
- 15.Kim D.E., Youn Y.C., Kim Y.K., Hong K.M., Lee C.S. Glycyrrhizin prevents 7-ketocholesterol toxicity against differentiated PC12 cells by suppressing mitochondrial membrane permeability change. Neurochem. Res. 2009;34:1433–1442. doi: 10.1007/s11064-009-9930-y. [DOI] [PubMed] [Google Scholar]
- 16.Jang E.R., Lee C.S. 7-ketocholesterol induces apoptosis in differentiated PC12 cells via reactive oxygen species-dependent activation of NF-κB and Akt pathways. Neurochem. Int. 2011;58:52–59. doi: 10.1016/j.neuint.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Lütjohann D., Breuer O., Ahlborg G., Nennesmo I., Sidén A., Diczfalusy U., Björkhem I. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lund E.G., Guileyardo J.M., Russell D.W. cDNA cloning of cholesterol 24-hydroxylase, a regulator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. USA. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Björkhem I., Lütjohann D., Breuer O., Sakinis A., Wennmalm A. Importance of a novel oxidative mechanism for elimination of brain cholesterol. Turnover of cholesterol and 24(S)-hydroxycholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J. Biol. Chem. 1997;272:30178–30184. doi: 10.1074/jbc.272.48.30178. [DOI] [PubMed] [Google Scholar]
- 20.Jeitner T.M., Voloshyna I., Reiss A.B. Oxysterol derivatives of cholesterol in neurodegenerative disorders. Curr. Med. Chem. 2011;18:1515–1525. doi: 10.2174/092986711795328445. [DOI] [PubMed] [Google Scholar]
- 21.Heverin M., Bogdanovic N., Lutjohann D., Bayer T., Pikuleva I., Bretillon L., Diczfalusy U., Winblad B., Bjorkhem I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer's disease. J. Lipid Res. 2004;45:186–193. doi: 10.1194/jlr.M300320-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Lütjohann D., Papassotiropoulos A., Björkhem I., Locatelli S., Bagli M., Oehring R.D., Schlegel U., Jessen F., Rao M.L., von Bergmann K., Heun R. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 2000;41:195–198. [PubMed] [Google Scholar]
- 23.Kölsch H., Lütjohann D., Tulke A., Björkhem I., Rao M.L. The neurotoxic effect of 24-hydroxycholesterol on SH-SY5Y human neuroblastoma cells. Brain Res. 1999;818:171–175. doi: 10.1016/s0006-8993(98)01274-8. [DOI] [PubMed] [Google Scholar]
- 24.Yamanaka K., Saito Y., Yamamori T., Urano Y., Noguchi N. 24(S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J. Biol. Chem. 2011;286:24666–24673. doi: 10.1074/jbc.M111.236273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urano Y., Ochiai S., Noguchi N. Suppression of amyloid-beta production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013;27:4305–4315. doi: 10.1096/fj.13-231456. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z.H., Saito Y., Yoshida Y., Sekine A., Noguchi N., Niki E. 4-Hydroxynonenal induces adaptive response and enhances PC12 cell tolerance primarily through induction of thioredoxin reductase 1 via transcriptional activation of Nrf2. J. Biol. Chem. 2005;280:41921–41927. doi: 10.1074/jbc.M508556200. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z.H., Yoshida Y., Saito Y., Sekine A., Noguchi N., Niki E. Induction of adaptive response and enhancement of PC12 cell tolerance by 7-hydroxycholesterol and 15-deoxy-delta(12,14)-prostaglandin J2 through up-regulation of cellular glutathione via different mechanisms. J. Biol. Chem. 2006;281:14440–14445. doi: 10.1074/jbc.M600260200. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z.H., Yoshida Y., Saito Y., Noguchi N., Niki E. Adaptive response induced by lipid peroxidation products in cell cultures. FEBS Lett. 2006;580:479–483. doi: 10.1016/j.febslet.2005.12.045. [DOI] [PubMed] [Google Scholar]
- 29.Saito Y., Nishio K., Numakawa Y., Ogawa Y., Yoshida Y., Noguchi N., Niki E. Protective effects of 15-deoxy-Delta12,14-prostaglandin J2 against glutamate-induced cell death in primary cortical neuron cultures: induction of adaptive response and enhancement of cell tolerance primarily through up-regulation of cellular glutathione. J. Neurochem. 2007;102:1625–1634. doi: 10.1111/j.1471-4159.2007.04701.x. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y., Li W., Kong A.N. Anti-oxidative stress regulator NF-E2-related factor 2 mediates the adaptive induction of antioxidant and detoxifying enzymes by lipid peroxidation metabolite 4-hydroxynonenal. Cell Biosci. 2012;2:40. doi: 10.1186/2045-3701-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohyama Y., Meaney S., Heverin M., Ekström L., Brafman A., Brafman A., Shafir M., Andersson U., Olin M., Eggertsen G., Diczfalusy U., Feinstein E., Björkhem I. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatoryaxes. J. Biol. Chem. 2005;281:3810–3820. doi: 10.1074/jbc.M505179200. [DOI] [PubMed] [Google Scholar]
- 32.Lütjohann D., Breuer O., Ahlborg G., Nennesmo I., Sidén A., Diczfalusy U., Björkhem I. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24Shydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willy P.J., Umesono K., Ong E.S., Evans R.M., Heyman R.A., Mangelsdorf D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 34.Steffensen K.R., Neo S.Y., Stulnig T.M., Vega V.B., Rahman S.S., Schuster G.U., Gustafsson J.Å., Liu E.T. Genome-wide expression profiling; a panel of mouse tissues discloses novel biological functions of liver X receptors in adrenals. J. Mol. Endocrinol. 2004;33:609–622. doi: 10.1677/jme.1.01508. [DOI] [PubMed] [Google Scholar]
- 35.Tall A.R., Yvan-Charvet L., Terasaka N., Pagler T., Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Terasaka N., Wang N., Yvan-Charvet L., Tall A.R. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc. Natl. Acad. Sci. USA. 2007;104:15093–15098. doi: 10.1073/pnas.0704602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aye I.L., Waddell B.J., Mark P.J., Keelan J.A. Placental ABCA1 and ABCG1 transporters efflux cholesterol and protect trophoblasts from oxysterol induced toxicity. Biochim. Biophys. Acta. 1801;1013–1024:2010. doi: 10.1016/j.bbalip.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda A., Nagao K., Matsuo M., Kioka N., Ueda K. 24(S)-hydroxycholesterol is actively eliminated from neuronal cells by ABCA1. J. Neurochem. 2013;126:93–101. doi: 10.1111/jnc.12275. [DOI] [PubMed] [Google Scholar]
- 39.Tarling E.J., Edwards P.A. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl. Acad. Sci. USA. 2011;108:19719–19724. doi: 10.1073/pnas.1113021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berthier A., Lemaire-Ewing S., Prunet C., Monier S., Athias A., Bessède G., Pais de Barros J.P., Laubriet A., Gambert P., Lizard G., Néel D. Involvement of a calcium-dependent dephosphorylation of BAD associated with the localization of Trpc-1 within lipid rafts in 7-ketocholesterol-induced THP-1 cell apoptosis. Cell Death Differ. 2004;11:897–905. doi: 10.1038/sj.cdd.4401434. [DOI] [PubMed] [Google Scholar]
- 41.Royer M.C., Lemaire-Ewing S., Desrumaux C., Monier S., Pais de Barros J.P., Athias A., Néel D., Lagrost L. 7-ketocholesterol incorporation into sphingolipid/cholesterol-enriched (lipid raft) domains is impaired by vitamin E: a specific role for alpha-tocopherol with consequences on cell death. J. Biol. Chem. 2009;284:15826–15834. doi: 10.1074/jbc.M808641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.S.R. Freeman, X. Jin, J.J. Anzinger, Q. Xu, S. Purushothaman, M.B. Fessler, L. Addadi, H.S. Kruth, ABCG1-mediated generation of extracellular cholesterol microdomains. J. Lipid Res., 2013 10.1194/jlr.M044552, in press. [DOI] [PMC free article] [PubMed]
- 43.Xu P., Li D., Tang X., Bao X., Huang J., Tang Y., Yang Y., Xu H., Fan X. LXR agonists: new potential therapeutic drug for neurodegenerative diseases. Mol. Neurobiol. 2013;48:715–728. doi: 10.1007/s12035-013-8461-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials