

Abstract
Hibernating mammals need to be insensitive to acid in order to cope with conditions of high CO2; however, the molecular basis of acid tolerance remains largely unknown. The African naked mole-rat (Heterocephalus glaber) and hibernating mammals share similar environments and physiological features. In the naked mole-rat, acid insensitivity has been shown to be conferred by the functional motif of the sodium ion channel NaV1.7. There is now an opportunity to evaluate acid insensitivity in other taxa. In this study, we tested for functional convergence of NaV1.7 in 71 species of mammals, including 22 species that hibernate. Our analyses revealed a functional convergence of amino acid sequences, which occurred at least six times independently in mammals that hibernate. Evolutionary analyses determined that the convergence results from both parallel and divergent evolution of residues in the functional motif. Our findings not only identify the functional molecules responsible for acid insensitivity in hibernating mammals, but also open new avenues to elucidate the molecular underpinnings of acid insensitivity in mammals.
Keywords: SCN9A, adaptation, convergent evolution, acid insensitivity
1. Introduction
Acid insensitivity is crucial for the survival of animals chronically exposed to high levels of CO2, because acid is potentially lethal. Two main types of mammals live in high-CO2 conditions: subterranean and hibernating mammals. Subterranean mammals live in congested, poorly ventilated chambers or burrows, where CO2 builds up to high levels [1,2]. Similarly, hibernating mammals also experience decreasing pH, caused both by cramped living conditions and by reduced metabolic rates [3].
The social subterranean African naked mole-rat (Heterocephalus glaber) is insensitive to acid. In this animal, there is a genetic basis for acid tolerance, namely, the sodium ion channel NaV1.7 [4], which is encoded by SCN9A. This gene largely determines the threshold for the action potential in nociceptors [5]. Acid insensitivity is functionally driven by negatively charged amino acids (EKE, –+–; amino acids 1718–1720 in human NaV1.7) in the P-loop region of the NaV1.7 domain IV [4]. In other mammals, the homologous residues in the motif are highly conserved and positively charged (KKV: ++0). If residue KKV of human NaV1.7 mutates into EKE (i.e. the condition in the naked mole-rat), acid (pH 6.0) significantly inhibits the activity level of NaV1.7 to a level similar to that of the naked mole-rat [4].
Given the similar physiological features and the high-CO2 environment between the naked mole-rat and hibernating mammals, we hypothesized that the two groups have comparable functional motifs in NaV1.7. Consequently, we investigated the possibility that there might be convergent evolution in the P-loop region of NaV1.7 domain IV using 71 species of mammals, including 22 species that hibernate. Evolutionary and structural modelling analyses revealed repeated functional convergences in NaV1.7 between groups that hibernate, which resulted from the parallel and divergent changes of amino acids.
2. Material and methods
(a). Species, PCR amplification and sequencing
Our dataset contained 71 mammalian species (see the electronic supplementary material, table S1), including 41 species from the ENSEMBL database using previously described methods [6–9] and 30 species from the molecular experiments. The genomic DNA of 30 mammalian species was isolated from muscle tissue. Using the primers designed from the conserved sequences of SCN9A, we performed PCR to amplify a target region (TR) that included a 525 bp fragment of the entire P-loop region with partial fragments of the flanking transmembrane regions in domain IV. We also amplified a 492 bp fragment with similar evolutionary rates and selection pressures to the TRs as a control region (CR). The CR was selected by using a sliding window analysis [10] along the full-length coding sequence of NaV1.7 from 14 mammals (see the electronic supplementary material, figure S2). All primer sequences are shown in the electronic supplementary material, table S2. PCR products were isolated and sequenced as per our previous methods [6]. For a positive control, we amplified the TR of mouse NaV1.7 because the quality of the mouse genome was high and the full-length sequence of mouse NaV1.7 was available. All amino acid sequences of NaV1.7 from 71 mammalian species were aligned using MUSCLE [11], and then checked visually (see the electronic supplementary material, figure S1). All de novo sequences were deposited in GenBank (accession numbers: JX171245–JX171280 and KF052669–KF052698).
(b). Predicting the tertiary structure of NaV1.7
We used the protein sequence of human NaV1.7 as a query for searching the Protein DataBank (PDB) database [12]. NaV1.7 was discovered to have four homologous transmembrane domains that hit the same template (PDB ID: 3RVY) of a voltage-gated sodium channel [13] with an E-value of <10−14. MODELLER [14] was used to construct the three-dimensional structure of these domains and assemble them into a complex using the crystal structure of 3RVY as a template. We constructed three-dimensional structures of mutant human NaV1.7, in which we changed KKV residues of the functional motif into EKE, DKD, EIE, EKD and EKV, respectively. We also estimated the pKa values of the ionizable residues and pI values of the proteins using PROPKA3 [15] to determine proton affinity. This widely used approach, which was shown to agree satisfactorily with experimental data [15,16], was used to predict pKa values of the proteins.
(c). Construction of ancestral sequences and tests for parallel changes
We inferred the ancestral protein sequences of the TR for each interior node of the tree for all 71 species, using the maximum-likelihood and maximum-parsimony methods [17]. Ancestral reconstructions appeared to be reliable because both methods gave identical results, and nodal support values exceeded 99%. We then looked for parallel or convergent changes by comparing ancestral and extant protein sequences [6,8]. Parallel changes were required to have the same descendant amino acids from the same ancestral amino acids, and convergence was considered to have occurred when identical amino acids were derived from different ancestral amino acids. To exclude the effects of random changes, we calculated the probability that the observed number of parallel or convergent sites exceeded that expected by chance [18].
(d). Selection tests
Maximum-likelihood estimates were generated using site models in codeml from PAML v. 4 [17] to determine if the P-loop region of NaV1.7 domain IV experienced positive selection. This method was used to identify residues that evolved under positive selection by comparing the ratio of non-synonymous to synonymous substitution rates, termed ω. The site models allowed ω to vary between residuals, and then identified specific sites according to their ω values. We also calculated the mean selection pressures on different branches of the tree using a branch-model analysis based on the phylogeny (figure 1). First, we estimated ω across the tree under a one-ratio model. Next, we estimated an independent ω value for each branch under the free-ratio model. Finally, we used the two-ratio ‘branch model’ to compare the estimated ratio on specific foreground branches in the phylogeny with the background ratio.
Figure 1.

Phylogeny of mammals used in this study. Species shaded orange can hibernate, and the yellow box indicates the African naked mole-rat. Branches labelled a–g denote functional convergences. The residues in the functional motif (amino acids 1718 to 1720) of NaV1.7 domain IV are shown. ‘+’, ‘–’ and ‘0’ indicate positive, negative and neutral charge of residues, respectively. Residuals on the interior nodes of the tree are the ancestral states of the functional motif.
3. Results and discussion
(a). Repeated convergent patterns of NaV1.7 protein sequences in hibernating mammals
We obtained TRs from a total of 71 mammals, using sequencing and data mining. Several methods were used to identify and exclude other sodium ion channels. First, we amplified the TR from the mouse genome DNA and verified that its best hit was NaV1.7 by blasting the mouse genome (data not shown). Second, we used species' protein sequences as queries to blast against the non-redundant database of NCBI (http://www.ncbi.nlm.nih.gov/) to confirm that the best hits were orthologues of previously annotated NaV1.7. Finally, we identified diagnostic sites of NaV1.7 in TRs to exclude other members of the sodium ion channel gene family. For example, sites 1676, 1739 and 1771 of NaV1.7 contained asparagine, tyrosine and threonine in most mammalian NaV1.7 sequences, but not in other types of NaV ion channels (see the electronic supplementary material, figure S1). After filtering by these strict criteria, the species used in this study spanned the phylogeny of mammals, covering 16 orders and 40 families (figure 1). There are 22 species that hibernate in 11 families with distant phylogenetic relationships, which included nearly all of the families that contained hibernating species [19–21]. By comparison, we found that four branches of the phylogeny, including the hedgehog family and three families of bats, exhibited the same pattern of amino acid charge found in naked mole-rats (figure 1; branches b–e). The three families of bats were not sister taxa, and thus gains of the amino acid charge pattern (–+–) probably arose four times independently: once in each family of bats and the fourth time in hedgehogs. In detail, the Rhinopomatidae shared the amino acid motif EKD with Myotis lucifugus (family Vespertilionidae), whereas the Emballonuridae, its sister group, possessed the motif DKD. Smith et al. [4] also reported negatively charged amino acids in the functional motif of NaV1.7 in M. lucifugus. Hedgehogs that hibernate possessed the pattern EKD, whereas the hedgehogs that do not hibernate had the positively charged pattern as seen in other mammals that do not hibernate (figure 1).
In addition to the amino acid charge pattern EKD (–+–), we identified two other patterns (−0 +; EKV) and (−0 –; EIE) in 10 other hibernating mammals. The former pattern occurred in the ancestral branch of marsupials, whereas the latter developed in the ancestral branch of the Rhinolophidae and Hipposideridae bats. It is obvious that these two gains evolved independently. Thus, we reasoned that NaV1.7 in hibernating mammals might also function for acid insensitivity, as happens in naked mole-rats. The negatively charged residues in the functional motif inhibit the generation of action potentials, and consequently block the acid-sense because of their high affinities for protons [4,22].
(b). Functional convergence of NaV1.7 in hibernating mammals
Next, we predicted the three-dimensional structure of NaV1.7 to test whether the four kinds of functional motifs we identified can influence proton affinity by replacements of negatively charged residues. We calculated the pKa values of ionizable sites in the functional motif as well as the pI values of whole proteins. The pKa value, a logarithmic measure of the acid dissociation constant, was used to examine the proton affinity of charged residues and the pI value of proteins was calculated from the mean of the pKa of charged residues of the proteins [23].
Using human NaV1.7 as the wild-type, we modelled its three-dimensional structure and estimated that the pI value of this protein was 5.69 and the pKa values of ionizable sites of K1718 and K1719 were 10.69 and 8.22 (figure 2), respectively. Next, we artificially mutated the three residues of KKV into EKV, EIE, DKD, EKE and EKD in the human NaV1.7 protein sequence. We then predicted the three-dimensional structures of these mutant types and estimated corresponding pI and pKa values of each based on their own structures. The pKa and pI values of these mutants shifted in the acid direction (figure 2). Smaller values for whole proteins indicated correspondingly stronger affinities with protons. Thus, our results suggested that the four similar amino acid charge patterns functioned for acid insensitivity, resulting from negatively charged residues in the functional motif, as reported in the naked mole-rat.
Figure 2.

Predicted three-dimensional structure of human NaV1.7. The box indicates the P-loop region of human NaV1.7 domain IV. Structures of P-loop regions of different mutants are shown on the top panel. The pKa values of the functional motif residues and pI values of proteins are shown.
Functional convergence of NaV1.7 for acid insensitivity occurred independently at least six times in hibernating mammals. The number of families with negatively charged residues in the functional motif that hibernate was significantly greater than the number of families without these residues that did not hibernate (p < 0.001, Fisher's exact test). This discovery constituted the first documentation of multiple functional convergences at the molecular level in distantly related mammals.
Three species of hibernating mammals do not display convergence for acid insensitivity in the functional motif of NaV1.7. There are two possible explanations for this finding: first, differences in microclimates, physiological characteristics and behaviours may result in inconsistent evolutionary patterns. For example, ground squirrels store food before winter, wake up for 12–20 h every week and may expel the accumulated CO2 from their bodies [24]. Second, besides NaV1.7, other molecular elements may play important roles for acid insensitivity because high-CO2 conditions during hibernation produce a systemic effect on organisms [25,26].
(c). Positive selection drive the functional convergence for acid insensitivity of hibernating mammals
Convergent, parallel and divergent sequence evolution can drive identical physiological function. To determine which mechanism is responsible for independent incidences of acid insensitivity, we inferred the ancestral states of amino acids in the motif on all interior nodes of the species tree. KKV, the inferred ancestral sequence, occurred in most of the species (figure 1). Functional convergence in seven branches owed to complex evolutionary processes at the level of amino acid sequences. For example, site 1718 experienced parallel rather than convergent evolution among branches a–c and e–g after excluding possible random changes p < 0.05), and after multiple testing (see the electronic supplementary material, table S3) [18]. By contrast, branch d experienced divergent evolution. Similarly, site 1720 contained glutamic acid on branches a and f, and aspartic acid on branches b–e. This site experienced independent parallel evolution on branches a and f, but within branches b–f divergent evolution occurred. Thus, both parallel and divergent evolution of amino acids appear to drive the functional convergence for acid insensitivity among hibernating mammals.
Mammals that do and do not hibernate may be under different evolutionary pressures on the P-loop region of domain IV in NaV1.7. We calculated ω using the maximum-likelihood method to estimate the evolutionary rates of TR. We defined different models for the branches. First, assuming a uniform ω for all branches of the 71 mammalian species (Model A, table 1), ω was estimated to be 0.05, which was significantly less than 1 (p < 0.0001; Model B, table 1), suggesting that the P-loop region was under an overall strong purifying selection. Second, we applied a model in which every branch had its own ω (Model C). This model was significantly more effective than Model A (p < 0.001; table 1), suggesting that ω varied between the different lineages. These results might indicate different evolutionary histories of mammals that do and do not hibernate. Consequently, we examined the evolutionary rates of the two groups of mammals by developing a model that allowed for a different ω for each group (Model D). This model was a significantly better fit to the data than Model A (p = 0.0058, table 1). To exclude false positive results, we repeated the same evolutionary analyses with that of TRs based on the CRs (see electronic supplementary material, figure S2). However, we cannot detect the different evolutionary rates for the two groups (table 1). Thus, our evolutionary analyses determined that the TRs of mammals that do and do not hibernate were subjected to different evolutionary rates.
Table 1.
Likelihood ratio tests for positive selection of target and CRs.
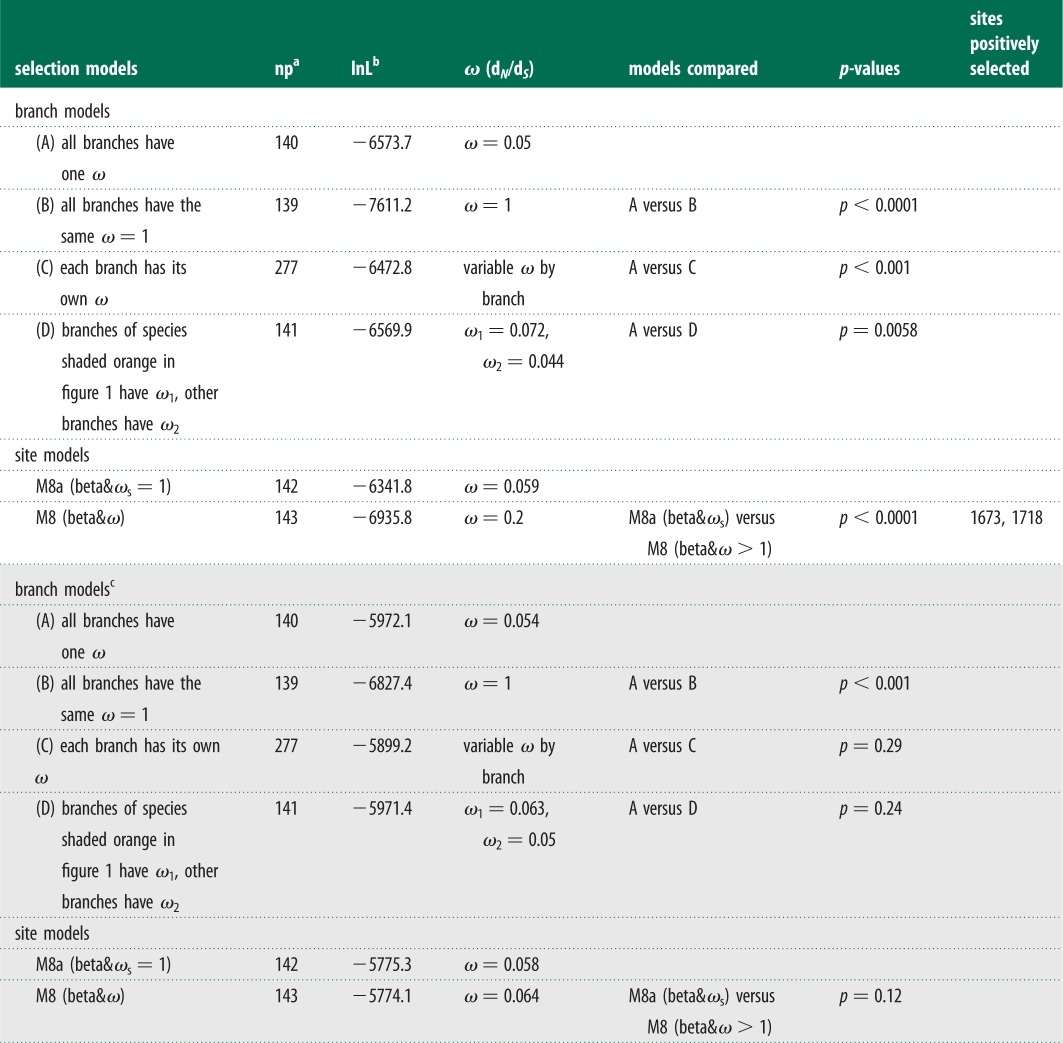 |
aNumber of parameters.
bThe natural logarithm of the likelihood value.
cThe results of selection test for CRs are shaded grey.
To further identify the evolutionary forces that best explain differences in evolutionary rates between hibernating and non-hibernating mammals, we calculated site-by-site ω values by comparing model M8a (beta&ωs = 1) versus M8 (beta&ω) [17]. This approach was more robust than other by-site model tests (e.g. M1a versus M2a; M7 versus M8) [27,28]. Our analysis showed that the P-loop regions of domain IV were under positive selection, and two residues had high posterior probabilities with ω > 1 (table 1), of which site 1718 was located in the functional motif for acid insensitivity. Thus, positive selection had resulted in the different evolutionary rates. These results further supported that functional convergence for acid insensitivity is an adaptation to high-CO2 environments in the hibernating mammals.
4. Conclusion
Our evolutionary analyses provide novel molecular evidence for the evolution of acid insensitivity in hibernating mammals. Our findings, taken together with recent reports about sodium ion channels in electric fish [29] and snakes [30], highlight the usefulness of convergent and parallel evolution for identifying the functional genes underlying phenotypic effects. Such analyses contribute to the ongoing resolution of phenotype–genotype relationships. Lastly, our example has the largest number of functional convergent events shown in mammals to date. The results extend the current understanding of the molecular mechanisms of phenotypic convergences on a large evolutionary scale. A functional analysis of the parallel and divergent amino acid substitutes identified here, as described by Smith et al. [4], may provide deeper insight into the molecular mechanisms for acid insensitivity of mammals.
Acknowledgements
We thank Prof. Jianzhi Zhang (University of Michigan) for valuable comments and Andrew Willden (Kunming Institute of Zoology) for early revisions to this manuscript. Z.L., T.Z.-Z. and P.S. managed the project. Z.L., W.W., T.-Z.Z, K.H. and X.-L.J prepared the DNA sample. Z.L. and W.W. performed molecular experiments. Z.L. and P.S. designed analyses. Z.L., G.-H.L. and J.-F.H. performed structural modelling. Z.L., T.Z.-Z. and P.S. performed evolutionary analyses. Z.L., R.W.M. and P.S. wrote the paper.
All work with live animals followed Animal Use Protocols approved by the Kunming Institute of Zoology Animal Care and Ethics Committee.
Funding statement
This work was supported by a start-up fund of the ‘Hundreds-Talent Program’ from the Chinese Academy of Sciences and by grants from the National Natural Science Foundation of China (30930015, 31325013) to P.S. Manuscript preparation was supported by a fellowship for Senior International Scientists and, in part, by a Visiting Professorship for Senior International Scientists from the Chinese Academy of Sciences and the Natural Sciences and Engineering Research Council of Canada (Discovery grant no. 3148).
References
- 1.Darden TR. 1972. Respiratory adaptations of a fossorial mammal, the pocket gopher (Thomomys bottae). J. Comp. Physiol. A 78, 121–137 (doi:10.1007/BF00693609) [Google Scholar]
- 2.Zeng J, Wang Z, Shi Z. 1984. Metabolic characteristics and some physiological parameters of the mole rat (Myospalax baileyi) in an alpine area. Acta Biol. Plat. Sin. 3, 163–171 [Google Scholar]
- 3.Malan A, Mioskowski E. 1988. pH–temperature interactions on protein function and hibernation: GDP binding to brown adipose tissue mitochondria. J. Comp. Physiol. B 158, 487–493 (doi:10.1007/BF00691146) [DOI] [PubMed] [Google Scholar]
- 4.Smith ES, Omerbasic D, Lechner SG, Anirudhan G, Lapatsina L, Lewin GR. 2011. The molecular basis of acid insensitivity in the African naked mole-rat. Science 334, 1557–1560 (doi:10.1126/science.1213760) [DOI] [PubMed] [Google Scholar]
- 5.Rush AM, Cummins TR, Waxman SG. 2007. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14 (doi:10.1113/jphysiol.2006.121483) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Z, Li S, Wang W, Xu D, Murphy RW, Shi P. 2011. Parallel evolution of KCNQ4 in echolocating bats. PLoS ONE 6, e26618 (doi:10.1371/journal.pone.0026618) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Shi P, Zhang YP, Zhang J. 2005. Composition and evolution of the V2r vomeronasal receptor gene repertoire in mice and rats. Genomics 86, 306–315 (doi:10.1016/j.ygeno.2005.05.012) [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Liu Z, Shi P, Zhang J. 2010. The hearing gene Prestin unites echolocating bats and whales. Curr. Biol. 20, R55–R56 (doi:10.1016/j.cub.2009.11.042) [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Li GH, Huang JF, Murphy RW, Shi P. 2012. Hearing aid for vertebrates via multiple episodic adaptive events on prestin genes. Mol. Biol. Evol. 29, 2187–2198 (doi:10.1093/molbev/mss087) [DOI] [PubMed] [Google Scholar]
- 10.Pride DT. 2006. Swaap 1.0.3: a tool for analyzing substitutions and similarity in multiple alignments See http://asiago.stanford.edu/SWAAP/SwaapPage.htm. [Google Scholar]
- 11.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rose PW, et al. 2010. The RCSB protein data bank: redesigned web site and web services. Nucleic Acids Res. 39, D392–D401 (doi:10.1093/nar/gkq1021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Payandeh J, Scheuer T, Zheng N, Catterall WA. 2011. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (doi:10.1038/nature10238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 15, 5.6.1–5.6.30 (doi:10.1002/0471250953.bi0506s15) [DOI] [PubMed] [Google Scholar]
- 15.Olsson MH, Søndergaard CR, Rostkowski M, Jensen JH. 2011. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 (doi:10.1021/ct100578z) [DOI] [PubMed] [Google Scholar]
- 16.Rostkowski M, Olsson MH, Sondergaard CR, Jensen JH. 2011. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 11, 6 (doi:10.1186/1472-6807-11-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Z. 2007. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (doi:10.1093/molbev/msm088) [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Kumar S. 1997. Detection of convergent and parallel evolution at the amino acid sequence level. Mol. Biol. Evol. 14, 527–536 (doi:10.1093/oxfordjournals.molbev.a025789) [DOI] [PubMed] [Google Scholar]
- 19.Geiser F. 1994. Hibernation and daily torpor in marsupials: a review. Aust. J. Zool. 42, 1–92 (doi:10.1071/ZO9940001) [Google Scholar]
- 20.Geiser F, Stawski C. 2011. Hibernation and torpor in tropical and subtropical bats in relation to energetics, extinctions, and the evolution of endothermy. Integr. Comp. Biol. 51, 337–348 (doi:10.1093/icb/icr042) [DOI] [PubMed] [Google Scholar]
- 21.Carey HV, Andrews MT, Martin SL. 2003. Mammalian hibernation: cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 83, 1153–1181 (doi:10.1152/physrev.00008.2003) [DOI] [PubMed] [Google Scholar]
- 22.Khan A, Romantseva L, Lam A, Lipkind G, Fozzard HA. 2002. Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J. Physiol. 543, 71–84 (doi:10.1113/jphysiol.2002.021014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miessler G, Tarr D. 1991. Acid–base and donor–acceptor chemistry: inorganic chemistry, 2nd edn Englewood Cliffs, NJ: Prentice Hall [Google Scholar]
- 24.Pengelley E, Fisher K. 1961. Rhythmical arousal from hibernation in the golden-mantled ground squirrel, Citellus lateralis tescorum. Can. J. Zool. 36, 105–120 (doi:10.1139/z61-013) [Google Scholar]
- 25.Malan A. 1988. pH and hypometabolism in mammalian hibernation. Can. J. Zool. 66, 95–98 (doi:10.1139/z88-013) [Google Scholar]
- 26.Park TJ, et al. 2008. Selective inflammatory pain insensitivity in the African naked mole-rat (Heterocephalus glaber). PLoS Biol. 6, e13 (doi:10.1371/journal.pbio.0060013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swanson WJ, Nielsen R, Yang Q. 2003. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 20, 18–20 (doi:10.1093/oxfordjournals.molbev.a004233) [DOI] [PubMed] [Google Scholar]
- 28.Wong WS, Yang Z, Goldman N, Nielsen R. 2004. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168, 1041–1051 (doi:10.1534/genetics.104.031153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zakon HH, Lu Y, Zwickl DJ, Hillis DM. 2006. Sodium channel genes and the evolution of diversity in communication signals of electric fishes: convergent molecular evolution. Proc. Natl Acad. Sci. USA 103, 3675–3680 (doi:10.1073/pnas.0600160103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldman CR, Brodie ED, Jr, Brodie ED, 3rd, Pfrender ME. 2012. Constraint shapes convergence in tetrodotoxin-resistant sodium channels of snakes. Proc. Natl Acad. Sci. USA 109, 4556–4561 (doi:10.1073/pnas.1113468109) [DOI] [PMC free article] [PubMed] [Google Scholar]