

Abstract
Phenylketonuria (PKU) is a rare genetic condition characterized by an absence or mutation of the PAH enzyme, which is necessary for the metabolism of the amino acid phenylalanine into tyrosine. Recently, sapropterin dihydrochloride, a synthetic form of tetrahydrobiopterin (BH4), has been introduced as a supplemental treatment to dietary phe control for PKU. Very little is known regarding BH4 treatment and its effect on brain and cognition. The present study represents the first examination of potential changes in neural activation in patients with PKU during BH4 treatment. To this end, we utilized an n-back working memory task in conjunction with functional magnetic resonance imaging (fMRI) to evaluate functional brain integrity in a sample of individuals with PKU at three timepoints: Just prior to BH4 treatment, after 4 weeks of treatment, and after 6 months of treatment. Neural activation patterns observed for the PKU treatment group were compared with those of a demographically-matched sample of healthy non-PKU individuals who were assessed at identical time intervals. Consistent with past research, baseline evaluation revealed impaired working memory and atypical brain activation in the PKU group as compared to the non-PKU group. Most importantly, BH4 treatment was associated with improvements in both working memory and brain activation, with neural changes evident earlier (4-week timepoint) than changes in working memory performance (6-month timepoint).
Keywords: Prefrontal cortex, Phenylketonuria, Working memory, fMRI, Executive function, Sapropterin
Highlights
-
•
We examine working memory and neural activation in patients with PKU at baseline.
-
•
We track behavioral and neural changes related to BH4 treatment in the patients.
-
•
BH4 treatment associated with improvement in neural activity at 4-week timepoint.
-
•
BH4 treatment associated with improvement in working memory at 6-month timepoint.
1. Introduction
Phenylketonuria (PKU) is an inborn error of metabolism characterized by a disruption in the ability to metabolize the amino acid phenylalanine (phe) into tyrosine, a precursor for dopamine and other catecholamines. Individuals with PKU have reduced activity of the phenylalanine hydroxylase (PAH) enzyme, which is necessary for this metabolic process to occur. Immediate consequences include decreased tyrosine levels and increased serum levels of phe (Jervis, 1953). The excess phe further competes with the available tyrosine and other large neutral amino acids (LNAAs) to cross the blood–brain barrier. The accumulative consequences of PKU include decreased neurotransmitter production and protein synthesis (Güttler and Lou, 1986; Hughes and Johnson, 1978; Ribas et al., 2011).
Individuals with PKU are typically identified at birth via newborn screening, and dietary intervention aimed at limiting phe intake is begun immediately. Earlier and stricter implementation of treatment is associated with more positive outcome (Koch et al., 2002); however, individuals with early-treated PKU continue to experience neurologic and cognitive impairment (Waisbren et al., 2007; Welsh et al., 1990).
Early-treated PKU is associated with a slight decrease in overall IQ coupled with more significant impairment in particular aspects of cognition, most notably executive function (for review, see Christ et al., 2010a). Executive function refers to higher-order cognitive abilities that facilitate the flexible modification of thought and behavior in response to changing cognitive or environmental demands. Executive function encompasses abilities such as planning, organization, cognitive flexibility, inhibitory control, and working memory. These abilities are considered executive because they require the integration and processing of information across a range of cognitive domains, sensory modalities, and response modalities.
From a neurological standpoint, white matter abnormalities represent the most evident and widely-reported neurologic finding in individuals with PKU (Anderson and Leuzzi, 2010). The extent and severity of such abnormalities appear to be moderated by patient age and dietary adherence (as reflected by blood phe levels), with older age and higher phe levels associated with increased white matter involvement (Antenor-Dorsey et al., 2013; Cleary et al., 1994; Thompson et al., 1993).
Research in our laboratory has begun to also evaluate the functional integrity of the brain and its relationship to the aforementioned executive function impairment associated with PKU. In a recent study (Christ et al., 2010b), we utilized functional magnetic resonance imaging (fMRI) to examine neural activation in a sample of individuals with and without PKU during performance of an n-back working memory task. Group-related differences in neural activity were found in several regions (e.g., bilateral dorsolateral prefrontal cortex, anterior cingulate cortex) known to be important for working memory and executive function (for review, see Owen et al., 2005).
1.1. Tetrahydrobiopterin (BH4) treatment in PKU
Recently, sapropterin dihydrochloride, a synthetic form of tetrahydrobiopterin (BH4), has been introduced as a supplemental treatment to dietary phe control for PKU. BH4 is a naturally-occurring compound that serves as a cofactor for PAH and other enzymes. Kure et al. (1999) and a number of subsequent studies have found that BH4 supplementation is effective in lowering blood serum phe levels in a subset of individuals with PKU (for review, see Hegge et al., 2009). Although the mechanism of action underlying this therapeutic effect is not entirely understood, it is thought that BH4 reduces phe serum levels in some individuals with PKU by improving the folding conformation of certain types of mutant PAH molecules resulting in increased phe catabolism (Erlandsen and Stevens, 2001; Erlandsen et al., 2004).
Not all individuals with PKU experience a decrease in blood phe levels in response to BH4 treatment, and there is considerable variability among those who do show a response (Burton et al., 2007). Multiple factors appear to contribute to responsiveness as evidenced by the fact that even individuals with identical PAH genotype may vary in BH4 responsiveness (Pérez et al., 2005). Within this context, in order to determine whether a given PKU patient is BH4 responsive, a trial period of BH4 treatment is undertaken. Several different protocols for this trial period have been developed, and metabolic clinics tend to use their own guidelines for determining responsiveness (Bélanger-Quintana et al., 2011). Typically, these protocols require that blood phe levels decrease substantially (e.g., ≥ 20%) from baseline during a trial period (e.g., 30 days) for a patient to be considered BH4-responsive.
1.2. The present study
As noted earlier, previous studies have found that improved adherence to dietary phe restrictions (and resulting decreases in serum phe levels) are associated with improvements in cognitive performance and white matter structural integrity in patients with PKU. Much less is known regarding BH4 treatment and its potential impact on brain and cognition in PKU. To date, there are no published studies on BH4 treatment in PKU and its effects on the functional brain integrity, and few existing studies on BH4 treatment and cognitive performance (e.g., Gassió et al., 2010; Lambruschini et al., 2005).
The present study represents the first examination of potential changes in brain activation in patients with PKU during BH4 treatment. To this end, we utilized an n-back working memory task in conjunction with fMRI to evaluate functional brain integrity in a sample of individuals with PKU at three timepoints: Just prior to BH4 treatment, after 4 weeks of treatment, and after 6 months of treatment. Neural activation patterns observed for the PKU treatment group were compared with those of a demographically-matched sample of healthy non-PKU individuals who were assessed at identical time intervals. We hypothesized that BH4 treatment in the PKU group would be associated with improvements in working memory and related neural activity (i.e., neural activity in the PKU group would closer approximate that observed in the non-PKU group).
2. Material and methods
2.1. Study design
A sample of 12 patients with PKU received baseline fMRI evaluations before beginning treatment with BH4. Phe response to BH4 was monitored for 4 weeks. At the end of the 4 weeks, fMRI evaluations were repeated. At this time, phe response to BH4 was also reviewed by the patient's treating physician and a decision was made whether to continue or discontinue treatment. [Note that this decision was made independent of the current authors/research team.] The fMRI evaluations were repeated again at the 6 month mark on those individuals who continued treatment. For comparison purposes, a demographically matched sample of 12 healthy individuals without PKU also received fMRI evaluations at baseline, 4-week, and 6-month timepoints. The non-PKU comparison group did not receive BH4 treatment.
2.2. Participants
A sample of 12 individuals (6 female, 6 male) with PKU ranging in age from 9 to 33 years (M = 23.6; SD = 8.8) participated in the study. Inclusionary criteria for participants were: (1) diagnosis at birth with a blood phe level ≥ 360 μmol/L, (2) recent history of phe levels ≥ 360 μmol/L, (3) ≥ 6 years of age, and (4) intention of their treating physician to prescribe BH4. The PKU participant group represented a sample of convenience and included individuals referred from clinics at University of Missouri (n = 4), University of Florida (n = 3), Kansas City Children's Mercy Hospital (n = 2), Washington University in St. Louis (n = 1), University of Nebraska (n = 1), and University of Kansas Medical Center (n = 1).
For all individuals with PKU, diagnosis was made and phe-restricted dietary treatment was implemented shortly after birth, as indicated by medical records and/or patient report. Unfortunately, complete lifetime records of blood phe level could not be attained for several older participants in the sample. Recent blood phe levels, however, were available for all participants. Although all patients reported that they had continued to maintain phe-restricted dietary treatment, a wide range of blood phe levels was observed in the PKU group. Mean phe levels in the year prior study enrollment ranged from 161 to 1459 μmol/L (group M = 827; SD = 356). Mean phe levels for the month prior to study enrollment were similar (133 to 1459; M = 827, SD = 399).
An age- and gender-matched comparison sample of 12 neurologically uncompromised individuals (7 female, 5 male) without PKU was also recruited. Participants in the non-PKU comparison group ranged in age from 9 to 33 years (M = 24.2; SD = 8.9) and were recruited from the Columbia, Missouri community.
The Wechsler Abbreviated Scale of Intelligence (Psychological Corporation, 1999) was administered to estimate general intellectual ability. For individuals in the PKU group, scores ranged from 73 to 115 (M = 99.7; SD = 11.0). For individuals in the non-PKU group, scores ranged from 102 to 120 (M = 111.0; SD = 7.0). As anticipated based on previous studies (for review, see Brumm and Grant, 2010), we found that scores of the non-PKU group were significantly higher than those of the PKU group, t(22) = 3.02, p < .01.
Consistent with past studies, pre-treatment phe levels for the PKU participants were positively correlated with age [prior month: r = .56, p = .06; prior year: r = .59, p = .05] and negatively correlated with IQ (phe level for prior year: r = −.78, p = .003; Phe level for prior month: r = −.76, p = .004]. Age and IQ, however, were not correlated with each other [r = −.27, p = .39]. Gender was also not related to any of the aforementioned variables [Mann–Whitney U ≥ 11, p > .26 in all instances].
2.3. N-back working memory task
The n-back working memory task represents one of the most widely used paradigms within the cognitive neuroscience field for studying working memory (Smith and Jonides, 1999). The task is well-established and has been used extensively to assess working memory in both healthy (Owen et al., 2005) and clinical populations (Glahn et al., 2005), including individuals with PKU (Christ et al., 2010b).
The n-back working memory task included two conditions: 2-back and 0-back. In the 2-back condition, participants were shown a series of letters one at a time (500 ms presentation time followed by a 1500 ms interstimulus blank period) and were instructed to respond when the current letter was the same as the letter that appeared two items prior. Letter stimuli were randomly drawn from the following stimulus set: B, C, D, F, G, H, J, K, L, M, N, P, Q, R, S, T, V, W, Z. For the 0-back condition, participants were again shown a series of letters; however, in this instance, they were asked to simply respond when they saw the letter X (regardless of what preceded it).
Each functional run included 8 task epochs (4 per condition). Immediately prior to each task epoch, a visual cue (i.e., the text “***2-back***” or “***X-back***”) indicated the forthcoming task condition; this cue was presented in red in the middle of the visual display for 2000 ms followed by a 2000 ms blank interval. Each task epoch lasted 32 s (not including the aforementioned cue) and comprised 4 target and 12 non-target stimuli randomly intermixed. The order of condition administration (2-back vs. 0-back) within each functional run was counterbalanced using an ABBA-BAAB design. Rest epochs (20 s each), in which only a fixation point was shown, were presented at the beginning and end of each functional run and between the task epochs. Each participant completed two functional runs of the task.
2.4. Data acquisition
Prior to entering the MRI scanner, each participant completed a practice block of the working memory task designed to familiarize the participant with the task as well as to control for any potential practice effects that could otherwise contribute to performance.
Scans were obtained on a 3 T Siemens Trio scanner with a standard 8-channel head coil. Stimuli in the working memory task were displayed using an LCD projector, and responses were recorded using a fiber optic switch. For alignment purposes, a set of structural images was collected first using a standard T1-weighted pulse sequence [MP-RAGE sequence: TR = 2400 ms, TE = 3.16 ms, flip angle = 8°, in-plane resolution = 1 × 1 mm, slice thickness = 1 mm, number of slices = 176]. For each functional run, sets of 32 contiguous axial images (TR = 2200 ms, TE = 27, flip angle = 90°, in-plane resolution = 4.0 × 4.0 mm, slice thickness = 4.0 mm) were acquired parallel to the anterior–posterior commissure plane. This procedure offered whole-brain coverage at a high signal-to-noise ratio. As indicated earlier, each participant completed a two functional runs.
2.5. Procedure
The present study was approved by the University of Missouri-Columbia Internal Review Board Study, registered with ClinicalTrials.gov (NCT00964236), and completed in accordance with the Helsinki Declaration. Informed consent was obtained for all participants, and assent was obtained for all participants under 18 years of age in addition to obtaining a parental consent.
2.5.1. Baseline assessment
All participants received a baseline fMRI evaluation. Shortly thereafter (M = 1.9 days, SD = 4.6), participants with PKU began BH4 treatment per standard clinical dosage amounts (20 mg/kg/day) under the supervision of their treating physician. PKU participants were instructed not to alter their usual dietary (protein) intake or initiate/terminate any other treatments during the 4-week BH4 treatment trial period. Participants in the non-PKU comparison group did not receive BH4 treatment.
2.5.2. Four-week assessment
Following four weeks of BH4 treatment (M = 32.9 days, SD = 11.2), participants with PKU received a follow-up fMRI evaluation. Also at this time, patient blood phe responsiveness to treatment was evaluated. Response to BH4 treatment was generally considered positive if a reduction of ≥ 20% in blood phe occurred during the 4-week trial period. Note, however, that the determination of response was made separately by each participant's treating physician and without input from the current authors/research team. As such, the treating physician had latitude to consider other factors beyond phe levels (e.g., anecdotal reports of improved behavior) when determining whether or not to continue BH4 treatment. Following an equivalent time interval (M = 33.3 days, SD = 10.6), participants in the non-PKU comparison group also received a follow-up fMRI evaluation.
2.5.3. Six-month assessment
Evaluations were repeated again following 6 months of treatment (M = 202.7 days, SD = 45.7) for those individuals who were continued on BH4 treatment. [Individuals with PKU who had been discontinued on BH4 treatment by their treating physician at the 4-week mark did not complete this evaluation.] Following an equivalent time interval (M = 217.1 days, SD = 43.2), participants in the non-PKU comparison group also received a follow-up fMRI evaluation.
2.6. Data processing and statistical methods
Functional imaging data were preprocessed and analyzed using BrainVoyager QX software (version 1.10; Brain Innovation, Maastricht, the Netherlands). Preprocessing steps included slice scan time correction, 3D motion correction, transformation to standardized atlas space (Talairach and Tournoux, 1988), and spatial smoothing (6 mm FWHM) to accommodate variations in activation loci across participants. Functional runs with excessive motion (> 3 mm) were omitted from further analysis.
Statistical analysis was conducted using a random effects general linear model approach. For each participant, the two task conditions (2-back and 0-back) were modeled separately using a convolved box-car response function. Confound predictors were also included in the model to account for inter-volume movement (3 predictors for translational movement and 3 predictors for rotational movement), linear trend, and low frequency noise. For all voxel-wise comparisons, statistical and cluster thresholds of p < .05 and contiguous area of > 500 mm3, respectively, were used.
For the non-PKU group, beta values for the contrast of interest [2-back > 0-back] were estimated separately for each participant. A series of voxel-wise comparisons were conducted and confirmed no significant changes in the activation pattern observed between the baseline, 4-week, and 6-month assessments for the non-PKU group. Based on this finding, data for the non-PKU group were collapsed across timepoint for later between-group comparisons. For the PKU group, beta values were estimated separately for each participant and timepoint (baseline, 4-week, and 6-month). We anticipated changes related to BH4 treatment in the PKU group, hence timepoints continued to be modeled separately for this group.
3. Results
The participant flow diagram is shown in Fig. 1. In short, four-week assessments were completed by 11 of 12 participants with PKU that were initially enrolled in the study. Of the 7 PKU participants who continued on BH4 treatment beyond the 4-week timepoint, 5 completed the 6-month assessment. Of the initial 12 non-PKU participants, all 12 completed the 4-week assessment and 8 completed the 6-month assessment.
Fig. 1.
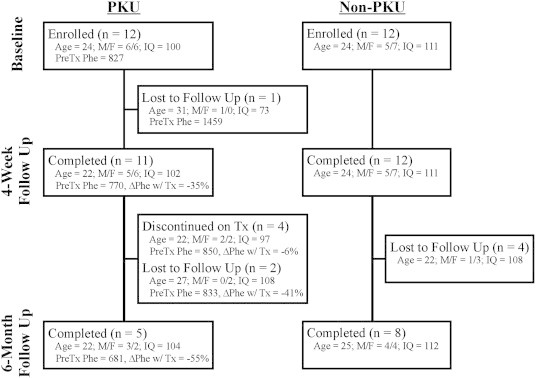
Participant flow diagram. Demographic information [mean age, gender, mean IQ, mean phe level for the year prior study enrollment, and mean blood phe level change (%) between the baseline and 4-week evaluations] are included for each sub-group.
As noted earlier, consistent with past research (Brumm and Grant, 2010), we found a significant difference in IQ between the PKU and non-PKU groups. Both statistical and conceptual concerns, however, precluded us from including IQ as a covariate in our analyses. Given the strong association between IQ and group membership, inclusion of IQ as a covariate would lead to underestimation of the variance in the dependent variable (working memory performance, neural activity) attributable to the independent variable (group membership) thus rendering the analysis results largely uninterpretable (for more extensive treatment of this topic, see Miller and Chapman, 2001). Furthermore, another critical assumption of ANCOVA is that the dependent variable could not have caused group differences in the covariate (Overall and Woodward, 1977; Wildt and Ahtola, 1978). Particularly as it relates to the fMRI data, an argument can be made that PKU causes neural disruption (i.e., atypical neural activation – the dependent variable) that, in turn, underlies the observed group differences in IQ (the potential covariate).
3.1. Working memory performance
Due to technical problems, n-back data were not available for two non-PKU participants at the 4-week timepoint. So as to conceptually parallel the previously described fMRI analysis approach, which focuses on the contrast between the 2-back and 0-back conditions, the primary dependent variable for the behavioral data analysis was the difference in error rate between the 2-back and 0-back conditions. When the analyses described below were repeated with the response time data, no significant effects of group, timepoint, or related interactions were found (p > .05 in all instances). Also note that, due to the small sample sizes involved, non-parametric statistical tests were used for all comparisons among PKU subgroups (e.g., responders vs. non-responders).
3.1.1. Baseline assessment
The PKU group demonstrated significantly poorer working memory performance than the non-PKU group as reflected by the difference score of error rate (%) in the 2-back condition minus that in the 0-back condition (MPKU = 8.8%; MNonPKU = 3.6%) [t(22) = 2.66, p = .01]. Within the PKU group, no significant relationship was found between working memory performance and age [r = −.25, p = .43], IQ scores [r = −.06, p = .86], or recent blood phe levels [prior month: r = −.38, p = .222; prior year: r = −.33, p = .30].
3.1.2. Four-week assessment
Potential changes in working memory performance (as reflected by the difference score of error rate in the 2-back condition minus that in the 0-back condition) were evaluated using a mixed model ANOVA with group (PKU and non-PKU) as the between-subjects factor, and timepoint (baseline and 4-week) as the within-subjects factors. The main effect of group approached significance [F(1,19) = 3.52, p = .08]. However, neither a main effect of timepoint nor group × timepoint interaction were found [F(1,19) < 1.8, p > .20 in both instances].
With the PKU group, the correlation between age and the magnitude of change in working memory performance over the 4-week period approached significance [r = −.57, p = .07], with younger patients showing greater change compared to older patients. No relationship was seen between changes in working memory performance and baseline IQ scores [r = .08, p = .81], or pre-treatment blood phe levels [prior month: r = −.20, p = .56; prior year: r = −.16, p = .65].
For each PKU participant, his/her blood phe change was calculated based on the mean phe level for the month prior to treatment and the final blood phe level recorded during the 4-week trial period [% Phe Change = 100 * (PheWeek4 – PhePreTx)/PhePreTx]. Blood phe changes for the PKU group ranged widely (− 91% to + 36%) with a mean response of − 34.8% (SD = 35.7). Of particular interest, no relationship was seen between the magnitude of phe level change and an individual's change in working memory performance during the same time period (4-week performance minus baseline performance) [r = .06, p = .85]. The magnitude of phe level change was also not correlated with age [r = −.25, p = .46], baseline IQ scores [r = −.47, p = .15], baseline working memory performance [r = −.02, p = .96], or pre-treatment blood phe levels [prior month: r = .48, p = .14; prior year: r = .36, p = .28].
Given that there was not perfect correspondence between the magnitude of phe level change and the designation of participants as ‘responders’ or ‘non-responders’ (treating physicians had the latitude to consider additional factors), additional analyses were conducted comparing these two subgroups. As anticipated, the Responder subgroup (M = − 50.9%, SD = 28.9) showed a larger drop in blood phe levels as compared to the PKU Non-Responder subgroup (M = − 6.4%, SD = 30.0). The two subgroups did not differ significantly in age, IQ scores, or pre-treatment blood phe levels [Mann–Whitney U ≥ 5.0, p > .10 in all instances]. They also did not differ from each other in terms of working memory performance at baseline or the 4-week timepoint, or the magnitude of change in performance between these two timepoints [Mann–Whitney U ≥ 12, p > .78 in all instances].
3.1.3. Six-month assessment
As detailed previously, only those PKU participants who continued on BH4 treatment (as per their treating physician) completed the 6-month follow-up assessment. Data for this PKU ‘Responder’ subgroup and the non-PKU group were entered into a mixed model ANOVA with group and timepoint as factors. No main effect of group was found [F(1,11) = 2.84, p = .12]. The main effect of timepoint was significant [F(1,11) = 7.82, p = .02]. Most importantly, there was also a significant interaction between group and timepoint [F(1,11) = 7.13, p = .03]. As can be seen in Fig. 2, subsequent analysis confirmed that this effect was driven by an improvement between baseline and the 6-month assessment in PKU Responder subgroup's performance (MBaseline = 9.1%; M6Month = 5.0%) [Wilcoxon signed-ranks Z = − 2.02, p = .04]. No relationship was seen between changes in working memory performance at the 6-month timepoint and age [rs = .00, p = 1.00], baseline IQ scores [rs = .10, p = .87], or pre-treatment blood phe levels [prior month: rs = −.10, p = .87; prior year: rs = −.10, p = .87].
Fig. 2.
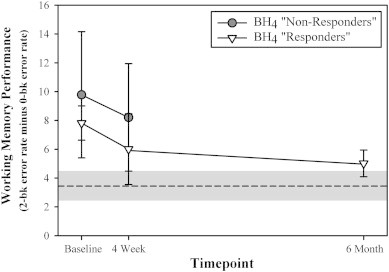
Working memory performance (as reflected as reflected by the difference score of error rate in the 2-back condition minus that in the 0-back condition) shown separately for timepoint (baseline, week 4, and 6 month) and BH4 responsiveness as determined independently at the 4-week timepoint by each participant's treating physician. [Patients deemed “non-responders” were discontinued on BH4 treatment after the 4-week trial period. “Responders” were continued on BH4 treatment and remained in the present study through the 6-month timepoint.] For comparison purposes, the mean and standard error of the behavioral performance observed for the non-PKU group are represented by a dashed line and gray shaded area, respectively.
For each PKU participant, his/her blood phe change during the 6 months was calculated based on the mean phe level for the month prior to treatment and the mean blood phe level for the subsequent 6 months. Blood phe changes for the PKU group ranged from − 57% to + 51% with a mean difference of − 14.6% (SD = 44.4). The correlation between the magnitude of phe level change and an individual's change in task performance during the same time period (6-month performance minus baseline performance) approached significance [rs = −.80, p = .10], with greater drop in phe levels associated with larger improvement in working memory performance. No relationship was evident between the magnitude of phe level change and age [rs = −.10, p = .87], baseline IQ scores [rs = .40, p = .51], or pre-treatment blood phe levels [prior month: rs = −.10, p = .87; prior year: rs = −.10, p = .87].
3.2. fMRI results
Due to excessive head motion, neuroimaging data were unusable for 2 non-PKU participants at the 4-week timepoint and 1 non-PKU participant at the 6-month timepoint.
3.2.1. Baseline assessment
As illustrated in Fig. 3, significant group-related differences in activation were observed for several regions-of-interest (ROIs). The location, size, and statistical values associated with each of these ROIs (n = 15) are further detailed in Table 1.
Fig. 3.
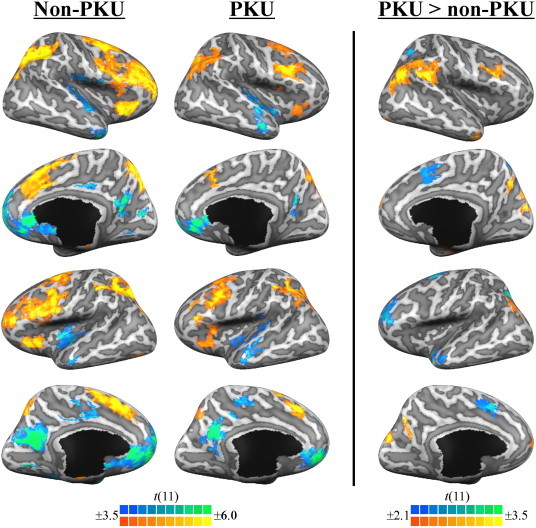
The left and center panels illustrate significant task-related (2-back minus 0-back) activity, shown separately for the non-PKU group and PKU group, respectively. The right panel illustrates regions demonstrating significant group differences in task-related activity. Results are viewed on the inflated surface of an exemplar brain. Statistical maps were thresholded at p < .005 and p < .05, respectively, for illustration purposes.
Table 1.
Regions exhibiting a group (non-PKU and PKU) differences in task-related (2-back minus 0-back) activity.
| Peak activation |
Volume |
Mean signal change (%) |
Effect size |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | Location | BA | x | y | z | (mm3) | non-PKU | PKU | t value⁎ | p value | (d) |
| PKU > non-PKU | |||||||||||
| 0 | Right inferior parietal lobule | 40 | 39 | − 61 | 32 | 5421 | 0.01 | 0.28 | 3.61 | .002 | 1.54 |
| 1 | Right fusiform gyrus | 19 | 24 | − 74 | − 7 | 1665 | − 0.08 | 0.10 | 2.83 | .010 | 1.21 |
| 2 | Bilateral precuneus | 7, 31 | − 1 | − 71 | 28 | 3814 | − 0.11 | 0.18 | 3.58 | .002 | 1.53 |
| 3 | Right middle temporal gyrus | 21 | 40 | 2 | − 22 | 733 | − 0.09 | 0.04 | 3.27 | .003 | 1.39 |
| 4 | Right inferior parietal lobule | 40 | 53 | − 36 | 25 | 1888 | 0.02 | 0.24 | 3.04 | .006 | 1.30 |
| 5 | Right hippocampus | NA | 24 | − 18 | − 9 | 541 | − 0.13 | 0.05 | 2.64 | .015 | 1.13 |
| 6 | Right middle frontal gyrus | 8 | 37 | 9 | 30 | 514 | 0.11 | 0.29 | 2.96 | .007 | 1.26 |
| 7 | Bilateral superior frontal gyrus | 9 | 5 | 57 | 26 | 616 | − 0.41 | − 0.13 | 3.04 | .006 | 1.30 |
| 8 | Left inferior parietal lobule | 7, 19, 40 | − 42 | − 61 | 29 | 973 | − 0.11 | 0.17 | 2.80 | .011 | 1.19 |
| non-PKU > PKU | |||||||||||
| 9 | Left superior frontal gyrus | 6 | − 23 | − 5 | 57 | 1194 | 0.39 | 0.12 | 2.82 | .010 | 1.20 |
| 10 | Left intraparietal sulcus | 7, 40 | − 25 | − 52 | 35 | 781 | 0.25 | 0.12 | 3.37 | .003 | 1.44 |
| 11 | Left middle temporal gryus | 21 | − 45 | 0 | − 11 | 893 | 0.06 | − 0.15 | 3.12 | .005 | 1.33 |
| 12 | Left middle frontal gyrus | 9, 46 | − 34 | 41 | 25 | 1621 | 0.31 | 0.06 | 3.17 | .004 | 1.35 |
| 13 | Bilateral anterior cingulate | 32 | 2 | 2 | 46 | 877 | 0.25 | 0.05 | 2.49 | .021 | 1.06 |
| 14 | Right precuneous | 7 | 17 | − 50 | 44 | 514 | 0.14 | 0.03 | 2.73 | .012 | 1.16 |
Notes. NA: Not applicable. BA: approximate Brodmann's area.
Degrees of freedom = 22.
Within the PKU group, ROIs in right inferior parietal lobule (region 0), right middle frontal gyrus (region 6), and left middle frontal gyrus (region 12) showed significant correlations between the magnitude of activation and recent blood phe levels [r > .61, p < .05 in all instances]. No relationships were seen between ROI activation and age or IQ with a two exceptions: The aforementioned ROI in right inferior parietal lobule (region 0) also showed a marginal correlation between activation and age [r = .49, p = .10]. An ROI in bilateral precuneus (region 2) showed a marginal correlation between activation and IQ [r = −.53, p = .08].
3.2.2. Four-week assessment
The aforementioned ROIs identified at baseline as showing significant group-related differences in activations were assessed for possible changes in activation between baseline and 4-week evaluation in the PKU group. As illustrated in the top two panels of Fig. 4, one-tailed paired samples t tests revealed significant changes in activation between the baseline and 4-week evaluations for regions in left inferior parietal lobule (region 8) [t(10) = 2.27, p = .02] and bilateral anterior cingulate cortex (region 13) [t(10) = 2.05, p = .03]. In both cases, the activation pattern observed at the 4-week evaluation better approximated that observed for the non-PKU group.
Fig. 4.
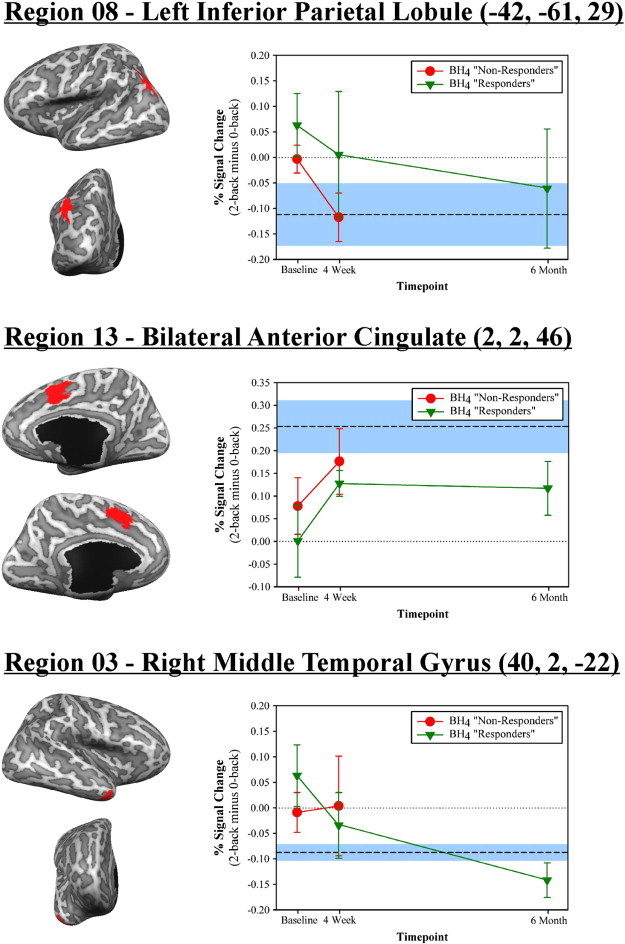
The pattern of activation for three ROIs that demonstrated significant changes in activation over time with BH4 treatment. The upper and middle panels illustrate regions in left inferior parietal lobule (region 08) and bilateral anterior cingulate cortex (region 13) that showed significant changes between baseline and 4-week evaluation (p < .05 in all instances). The bottom panel illustrates a region in right middle temporal gyrus (region 03) that showed significant changes between baseline and 6-month evaluation. Data is graphed separately for timepoint (baseline, week 4, and 6 month) and BH4 responsiveness (responder vs. non-responder) as determined independently at the 4-week timepoint by each participant's treating physician. For comparison purposes, the mean and standard error of the activation levels observed for the non-PKU group are represented by a dashed line and blue shaded area, respectively.
Interestingly, the magnitude of activation change between the baseline and 4-week evaluations in the aforementioned inferior parietal lobule ROI (region 8) was marginally correlated with concurrent changes in blood phe levels [r = −.58, p = .07]. Greater drop in phe levels was associated with less activation (which better approximated the non-PKU group). A similar relationship was not observed for the bilateral anterior cingulate ROI (region 13) [r = .13, p = .70]. Changes in activation of the two ROIs were not related to age [|r| < .28, p > .41], baseline IQ scores [|r| < .15, p > .66], or pre-treatment blood phe levels [prior month: |r| < .47, p > .14; prior year: |r| < .50, p > .11].
In comparing the responder and non-responder PKU subgroups, the two groups did not differ in baseline activation for either ROI [Mann–Whitney U ≥ 5.0, p > .10 in both instances]. The magnitude of activation change observed between baseline and the 4-week timepoint for the two ROIs was also similar for the subgroups [Mann–Whitney U ≥ 10, p > .52 in both instances].
3.2.3. Six-month assessment
For the PKU responder subgroup (i.e., participants who remained on BH4 treatment), the baseline ROIs were assessed for possible activation changes between baseline and 6-month evaluation. As shown in the bottom panel of Fig. 4, a significant change was observed for a region in right middle temporal gyrus (region 3) [t(4) = 2.60, p = .03]. Working memory-related activation better approximated that of the non-PKU comparison group (mean % signal change = −.10) at the 6-month assessment (mean % signal change = −.14) than at baseline (mean % signal change = .06). The magnitude of the observed activation change was not significantly correlated with age, IQ, pre-treatment blood phe levels, or blood phe level change between baseline and the 6-month timepoint [|rs| < .40, p > .23].
4. Discussion
As anticipated based on past behavioral studies (Christ et al., 2010a), we found that the PKU group performed more poorly than the non-PKU comparison group on the working memory task at baseline (without BH4 treatment). In terms of neuroimaging findings, PKU was associated with atypical activation of several regions in prefrontal and parietal cortex (e.g., bilateral dorsolateral prefrontal cortex, anterior cingulate cortex, left intraparietal sulcus) known to be critical for working memory (Owen et al., 2005). The prefrontal cortex finding is generally consistent with the hypothesis that PKU-related depletion in dopamine leads to prefrontal dysfunction which, in turn, contributes to the executive function impairments often seen in this population (Diamond, 1996; Welsh, 1996).
Group-related differences in activation patterns were also observed in areas (e.g., left inferior parietal lobule, right hippocampal area) known to be part of the default mode network, a network of interconnected brain regions that are generally more active when a person is at rest compared with when he/she is performing a cognitive task (for review, see van den Heuvel and Hulshoff Pol, 2010). In the non-PKU group, as expected these regions showed significant deactivation in the 2-back condition as compared to the cognitively less demanding 0-back condition. In the PKU group, however, these regions showed little or no difference in activation between the two conditions. This finding is consistent with our previous two functional neuroimaging studies of PKU (Christ et al., 2010b, 2012), both of which also found PKU-related disruptions in the default mode network.
Curiously, superimposed on the aforementioned patterns of atypical activation, there appeared to also be a trend towards under-activation of left brain areas and over-activation of right brain areas in the PKU group as compared to the non-PKU group. Although speculative, this pattern could be related to impairment in the utilization of verbal-based encoding and strategies (e.g., Banerjee et al., 2011) thus forcing the PKU group to rely more heavily on less optimal, non-verbal strategies supported primarily by the right hemisphere.
Following four weeks of BH4 treatment, changes in neural activation but not working memory performance were observed in the PKU group. Regions showing significant changes included a ‘task positive’ working memory-related region in anterior cingulate cortex and a ‘task negative’ default mode network region in left inferior parietal lobule. Importantly, after 4 weeks of treatment, the activation pattern for the PKU group better approximated that observed for the non-PKU group in both cases. In addition, as can be seen in Fig. 4, this pattern continued to hold for these regions at the 6-month timepoint as well.
Following an additional five months of continued treatment, improvements in working memory performance were now evident. Error rate in the 2-back condition for the PKU group dropped from 10.0% and 10.8% at baseline and the 4-week timepoints, respectively, to 6.9% at the 6-month follow. During the same time period, performance for the non-PKU group remained relatively stable (baseline = 5.7%, 4-week = 4.9%, 6-month = 4.8%). An additional change in neural activity was also observed. A region in lateral right temporal cortex, which represents another default mode network region, showed better approximation to the non-PKU activation pattern with treatment. As illustrated in Fig. 4, a trend was present at the 4-week timepoint but the change did not reach significance until the 6-month timepoint.
The primary mechanism-of-action for BH4 treatment in PKU is believed to be the activation of residual PAH enzyme (Erlandsen and Stevens, 2001). Consistent with this, we found tentative evidence of a relationship between phe level changes during treatment and measurements of neural activity and working memory performance. Specifically, a larger drop in phe levels was associated a greater improvement in neural activity at the 4-week timepoint and working memory performance at the 6-month timepoint. Consistent with the notion that neuroplasticity varies with age (i.e., the “Kennard principle”), the current analysis also revealed a potential relationship between age and the changes in working memory performance observed for PKU participants. Younger PKU participants (as compared to older participants) showed greater gains in working memory performance over the first four weeks of treatment. Additional research with a larger sample size is critical in confirming these findings.
4.1. Limitations and future directions
The present sample size is reflective of the rarity of PKU (1 in 15,000) as well as the general challenge of conducting treatment-related longitudinal research. The sample size provided sufficient statistical power to detect the aforementioned changes in task performance and brain activity. Future research employing larger sample sizes, however, may reveal additional (more subtle) changes in neural activity and task performance that otherwise went undetected in the current study.
A larger sample size would also allow for a proper evaluation of the extent to which factors such as age and prior treatment history (e.g., historic phe levels) may moderate treatment-related changes in cognitive and neural function. Longer-term study is also needed. Additional behavioral and/or neural changes might be apparent if one were to follow patients receiving BH4 treatment beyond the time period (6 months) included in the present study. For example, changes in not only brain activation but also structure (e.g., the severity of white matter abnormalities) might occur.
Another potential limitation relates to the fact that only those PKU participants who were deemed ‘responders’ by their treating physicians were continued on BH4 treatment beyond the 4-week trial period and completed the 6-month evaluation. Although the ‘responding’ and ‘non-responding’ groups in the present study appeared demographically indistinguishable from one another (see Fig. 1), the select nature of this group necessitates that the 6-month findings be interpreted with caution. Future studies in which all participants, regardless of initial blood phe response, are continued on BH4 treatment and complete long term follow-up is needed.
Such research would allow us to explore the possibility that BH4 may have therapeutic benefits in PKU beyond its effect on phe metabolism. BH4 serves as a cofactor for several other metabolic processes including the conversions of tyrosine to L-DOPA, tryptophan to 5-hydroxtryptophan, L-arginine to nitric oxide, and 1-alkyl-sn-glycerol to 1-hydroxyalkyl-sn-glycerol (Fitzpatrick, 1999; Kappock and Caradonna, 1996; Marletta, 1993). Although speculative, it remains possible that BH4 treatment-related changes in these additional processes may have contributed to the neurocognitive changes observed in the current study. Indeed, reports of positive outcome following BH4 treatment in non-PKU clinical populations such as autism and depression confirm these pathways as viable therapeutic targets and sites of action for BH4 treatment (e.g., Bottiglieri et al., 1992; Danfors et al., 2005; Nakamura et al., 2006).
4.2. Summary and conclusions
The current study represents the first examination of changes in neural activation in patients with PKU during BH4 treatment. Consistent with past research, baseline evaluation revealed impaired executive function and atypical brain activation patterns in the PKU group as compared to the non-PKU group. Most importantly, BH4 treatment was associated with improvements in both behavioral performance and brain activation, with the latter evident earlier (4-week timepoint) as compared to the behavioral aspect (6-month timepoint). Future research with larger sample sizes is needed to disentangle the extent to which the observed improvements were due to improved metabolism of phe versus potential secondary therapeutic pathways of BH4.
Acknowledgements
This research was funded by a research grant from BioMarin Pharmaceutical Inc. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors. The authors would also like to thank the participants and their families for their time and participation, which contributes to knowledge to better understand PKU. We would like to thank the faculty and staff of the metabolic clinics at the University of Missouri, University of Florida, Kansas City Children's Mercy Hospital, Washington University in St. Louis, University of Nebraska, and University of Kansas Medical Center for their assistance with participant recruitment and data collection.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- Anderson P.J., Leuzzi V. White matter pathology in phenylketonuria. Mol. Genet. Metab. 2010;99(Suppl. 1):S3–S9. doi: 10.1016/j.ymgme.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Antenor-Dorsey J.A.V., Hershey T., Rutlin J., Shimony J.S., McKinstry R.C., Grange D.K., White D.A. White matter integrity and executive abilities in individuals with phenylketonuria. Mol. Genet. Metab. 2013;109:125–131. doi: 10.1016/j.ymgme.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P., Grange D.K., Steiner R.D., White D.A. Executive strategic processing during verbal fluency performance in children with phenylketonuria. Child Neuropsychol. 2011;17:105–117. doi: 10.1080/09297049.2010.525502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger-Quintana A., Burlina A., Harding C.O., Muntau A.C. Up to date knowledge on different treatment strategies for phenylketonuria. Mol. Genet. Metab. 2011;104:S19–S25. doi: 10.1016/j.ymgme.2011.08.009. (Suppl.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottiglieri T., Hyland K., Laundy M., Godfrey P., Carney M.W., Toone B.K., Reynolds E.H. Folate deficiency, biopterin and monoamine metabolism in depression. Psychol. Med. 1992;22:871–876. doi: 10.1017/s0033291700038447. [DOI] [PubMed] [Google Scholar]
- Brumm V.L., Grant M.L. The role of intelligence in phenylketonuria: a review of research and management. Mol. Genet. Metab. 2010;99(Suppl. 1):S18–S21. doi: 10.1016/j.ymgme.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Burton B.K., Grange D.K., Milanowski A., Vockley G., Feillet F., Crombez E.A., Dorenbaum A. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J. Inherit. Metab. Dis. 2007;30:700–707. doi: 10.1007/s10545-007-0605-z. [DOI] [PubMed] [Google Scholar]
- Christ S.E., Huijbregts S.C.J., de Sonneville L.M.J., White D.A. Executive function in early-treated phenylketonuria: profile and underlying mechanisms. Mol. Genet. Metab. 2010;99(Suppl. 1):S22–S32. doi: 10.1016/j.ymgme.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Christ S.E., Moffitt A.J., Peck D. Disruption of prefrontal function and connectivity in individuals with phenylketonuria. Mol. Genet. Metab. 2010;99(Suppl. 1):S33–S40. doi: 10.1016/j.ymgme.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Christ S.E., Moffitt A.J., Peck D., White D.A., Hilgard J. Decreased functional brain connectivity in individuals with early-treated phenylketonuria: evidence from resting state fMRI. J. Inherit. Metab. Dis. 2012;35:807–816. doi: 10.1007/s10545-011-9439-9. [DOI] [PubMed] [Google Scholar]
- Cleary M.A., Walter J., Wraith J., Jenkins J., Alani S., Tyler K., Whittle D. Magnetic resonance imaging of the brain in phenylketonuria. Lancet. 1994;344:87–90. doi: 10.1016/s0140-6736(94)91281-5. [DOI] [PubMed] [Google Scholar]
- Danfors T., von Knorring A.-L., Hartvig P., Langstrom B., Moulder R., Stromberg B., Eeg-Olofsson O. Tetrahydrobiopterin in the treatment of children with autistic disorder: a double-blind placebo-controlled crossover study. J. Clin. Psychopharmacol. 2005;25:485–489. doi: 10.1097/01.jcp.0000177667.35016.e9. [DOI] [PubMed] [Google Scholar]
- Diamond A. Evidence for the importance of dopamine for prefrontal cortex functions early in life. Philos. Trans. R. Soc. B-Biol. Sci. 1996;351:1483–1494. doi: 10.1098/rstb.1996.0134. [DOI] [PubMed] [Google Scholar]
- Erlandsen H., Pey A.L., Gámez A., Pérez B., Desviat L.R., Aguado C., Stevens R.C. Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16903–16908. doi: 10.1073/pnas.0407256101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandsen H., Stevens R.C. A structural hypothesis for BH4 responsiveness in patients with mild forms of hyperphenylalaninaemia and phenylketonuria. J. Inherit. Metab. Dis. 2001;24:213–230. doi: 10.1023/a:1010371002631. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick P.F. Tetrahydropterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- Gassió R., Vilaseca M.A., Lambruschini N., Boix C., Fusté M.E., Campistol J. Cognitive functions in patients with phenylketonuria in long-term treatment with tetrahydrobiopterin. Mol. Genet. Metab. 2010;99(Suppl. 1):S75–S78. doi: 10.1016/j.ymgme.2009.10.187. [DOI] [PubMed] [Google Scholar]
- Glahn D.C., Ragland J.D., Abramoff A., Barrett J., Laird A.R., Bearden C.E., Velligan D.I. Beyond hypofrontality: a quantitative meta-analysis of functional neuroimaging studies of working memory in schizophrenia. Hum. Brain Mapp. 2005;25:60–69. doi: 10.1002/hbm.20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güttler F., Lou H. Dietary problems of phenylketonuria: effect on CNS transmitters and their possible role in behaviour and neuropsychological function. J. Inherit. Metab. Dis. 1986;9(Suppl. 2):169–177. doi: 10.1007/BF01799701. [DOI] [PubMed] [Google Scholar]
- Hegge K.A., Horning K.K., Peitz G.J., Hegge K. Sapropterin: a new therapeutic agent for phenylketonuria. Ann. Pharmacother. 2009;43:1466–1473. doi: 10.1345/aph.1M050. [DOI] [PubMed] [Google Scholar]
- Hughes J.V., Johnson T.C. Experimentally induced and natural recovery from the effects of phenylalanine on brain protein synthesis. Biochim. Biophys. Acta. 1978;517:473–485. doi: 10.1016/0005-2787(78)90214-9. [DOI] [PubMed] [Google Scholar]
- Jervis G.A. Phenylpyruvic oligophrenia deficiency of phenylalanine-oxidizing system. Proc. Soc. Exp. Biol. Med. 1953;82:514–515. [PubMed] [Google Scholar]
- Kappock T.J., Caradonna J.P. Pterin-dependent amino acid hydroxylases. Chem. Rev. 1996;96:2659–2756. doi: 10.1021/cr9402034. [DOI] [PubMed] [Google Scholar]
- Koch R., Burton B., Hoganson G., Peterson R., Rhead W., Rouse B., Azen C. Phenylketonuria in adulthood: a collaborative study. J. Inherit. Metab. Dis. 2002;25:333–346. doi: 10.1023/a:1020158631102. [DOI] [PubMed] [Google Scholar]
- Kure S., Hou D.C., Ohura T., Iwamoto H., Suzuki S., Sugiyama N., Narisawa K. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J. Pediatr. 1999;135:375–378. doi: 10.1016/s0022-3476(99)70138-1. [DOI] [PubMed] [Google Scholar]
- Lambruschini N., Pérez-Dueñas B., Vilaseca M.A., Mas A., Artuch R., Gassió R., Campistol J. Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol. Genet. Metab. 2005;86(Suppl. 1):S54–60. doi: 10.1016/j.ymgme.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Marletta M.A. Nitric oxide synthase structure and mechanism. J. Biol. Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- Miller G.A., Chapman J.P. Misunderstanding analysis of covariance. J. Abnorm. Psychol. 2001;110:40–48. doi: 10.1037//0021-843x.110.1.40. [DOI] [PubMed] [Google Scholar]
- Nakamura K., Sugawara Y., Sawabe K., Ohashi A., Tsurui H., Xiu Y., Hirose S. Late developmental stage-specific role of tryptophan hydroxylase 1 in brain serotonin levels. J. Neurosci. 2006;26:530–534. doi: 10.1523/JNEUROSCI.1835-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall J.E., Woodward J.A. Nonrandom assignment and the analysis of covariance. Psych. Bull. 1977;84:588–594. [Google Scholar]
- Owen A., McMillan K., Laird A., Bullmore E. N-back working memory paradigm: a meta-analysis of normative functional neuroimaging studies. Hum. Brain Mapp. 2005;25:46–59. doi: 10.1002/hbm.20131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez B., Desviat L.R., Gómez-Puertas P., Martínez A., Stevens R.C., Ugarte M. Kinetic and stability analysis of PKU mutations identified in BH4-responsive patients. Mol. Genet. Metab. 2005;86(Suppl. 1):S11–S16. doi: 10.1016/j.ymgme.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Psychological Corporation . Psychological Corporation; San Antonio, TX: 1999. Wechsler Abbreviated Scale of Intelligence. [Google Scholar]
- Ribas G.S., Sitta A., Wajner M., Vargas C.R. Oxidative stress in phenylketonuria: what is the evidence? Cell. Mol. Neurobiol. 2011;31:653–662. doi: 10.1007/s10571-011-9693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.E., Jonides J. Storage and executive processes in the frontal lobes. Science. 1999;283:1657–1661. doi: 10.1126/science.283.5408.1657. [DOI] [PubMed] [Google Scholar]
- Talairach J., Tournoux P. Thieme; New York: 1988. Co-planar sterotaxic atlas of the human brain. [Google Scholar]
- Thompson A.J., Tillotson S., Smith I., Kendall B., Moore S., Brenton D. Brain MRI changes in phenylketonuria. Associations with dietary status. Brain. 1993;116:811–821. doi: 10.1093/brain/116.4.811. [DOI] [PubMed] [Google Scholar]
- Van den Heuvel M.P., Hulshoff Pol H.E. Exploring the brain network: a review on resting-state fMRI functional connectivity. Eur. Neuropsychopharmacol. 2010;20:519–534. doi: 10.1016/j.euroneuro.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Waisbren S.E., Noel K., Fahrbach K., Cella C., Frame D., Dorenbaum A., Levy H. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol. Genet. Metab. 2007;92:63–70. doi: 10.1016/j.ymgme.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Welsh M. A prefrontal dysfunction model of early-treated phenylketonuria. Eur. J. Pediatr. 1996;155:S87–S89. doi: 10.1007/pl00014259. [DOI] [PubMed] [Google Scholar]
- Welsh M., Pennington B.F., Ozonoff S., Rouse B., McCabe E. Neuropsychology of early-treated phenylketonuria: specific executive function deficits. Child Dev. 1990;61:1697–1713. [PubMed] [Google Scholar]
- Wildt A.R., Ahtola O.T. Sage; Beverly Hills: 1978. Analysis of covariance. [Google Scholar]