

Abstract
The roles of Type I interferon (IFN) in human immunodeficiency virus Type 1 (HIV-1) neuropathogenesis are poorly understood; both protective and deleterious effects of IFN signaling have been described. We used genetically modified mice deficient in the Type I IFN receptor (IFNRKO) to analyze the progress of HIV-1 brain infection and neuropathogenesis in the absence of IFN signaling. IFNRKO and wild-type (WT) mice on the 129xSv/Ev or C57BL/6 strain backgrounds were infected systemically with EcoHIV, a chimeric HIV-1 that productively infects mice. IFNRKO mice showed higher HIV-1 expression in spleen and peritoneal macrophages and greater virus infiltration into the brain compared to WT mice. Neuropathogenesis was studied by histopathological, immunohistochemical, immunofluorescence, and polymerase chain reaction analyses of brain tissues after the virus was inoculated into the brain by stereotaxic intracerebral injection. Both IFNRKO and WT mice showed readily detectable HIV-1 and brain lesions, including microglial activation, astrocytosis, and increased expression of genes coding for inflammatory cytokines and chemokines typical of human HIV-1 brain disease. Parameters of HIV-1 neuropathogenesis, including HIV-1 expression in microglia/macrophages, were significantly greater in IFNRKO than in WT mice. Our results show unequivocally that Type I IFN signaling and responses limit HIV-1 infection and pathogenesis in the brains of mice.
Key Words: HIV-1, Interferon α/β receptor, IFN signaling, Neuropathogenesis, Receptor knockout mice, Stereotaxic injection, Type I interferon
INTRODUCTION
Human immunodeficiency virus Type 1 (HIV-1) infection causes a range of nervous system impairments known in aggregate as HIV-1–associated neurocognitive disorders or HAND (1). The prevalence of individual diagnostic categories encompassed in HAND reflects the cumulative effect of combination antiretroviral therapy (cART) in populations with access to treatment (1). HIV-1–associated dementia, which before the advent of cART was a frequent occurrence, has been responsive to treatment, with its prevalence diminishing to approximately 1% (2). In contrast, mild HAND manifestations including mild neurocognitive disorder and asymptomatic neurocognitive impairment have been largely refractory to cART, even in people with sustained virus suppression and immunological improvement (3–6). These CNS manifestations have emerged as major complications of HIV-1 infection in populations on cART, with a combined prevalence of approximately 50% (3, 6, 7).
We are interested in the mechanism of mild HIV-1 neuropathogenesis seen in many individuals on cART. Recent clinical studies indicate that nadir CD4 is the strongest predictor of HAND in such individuals (8–10), suggesting that the pathophysiological processes underlying HAND originate early after infection (9). These processes likely include early HIV-1 entry into the CNS (11–14) and persistent infection of infiltrating macrophages and glial cells, the main HIV-1 reservoirs in the brain (15, 16). Indeed, some infected individuals tested during the early/acute stage of infection show mild metabolic changes in the brain that indicate cell activation and inflammation and some have symptoms of neurocognitive disease (13, 17–20); however, the majority develop clinically apparent HAND years after primary infection (8–10). These findings point to a substantial period of disease latency between HAND initiation at the molecular level and the appearance of clinical HAND. Understanding the mechanisms controlling this latency may help to prevent or mitigate HAND.
Type I interferons (IFNs) may contribute to the imposition of HIV latency. The innate immune responses mediated by Type I IFNs serve as the first line of defense against viral infections including HIV-1 (21–23). These responses are triggered by pattern-specific viral “sensors,” including Toll-like and RIG-1-like receptors, which in turn lead to activation of interferon regulatory factors IRF3 and IRF7 and induction of IFNs (24). Type I IFNs, including IFN-α and IFN-β, act through a common cell surface receptor, IFN-α/β receptor or IFNAR1 (21, 25, 26), to initiate a cascade of cellular signaling pathways that culminates in induction of interferon-stimulated genes with antiviral, proapoptotic, and ubiquitination-modifying properties (27).
Both HIV-1 and the closely related simian immunodeficiency virus (SIV) elicit strong Type I IFN responses in their respective hosts as indicated by broad activation of IFN-related genes in peripheral tissues and brain (28–33). These responses might be particularly important in the imposition of latency upon HIV-1 in the brain because Type I IFN has been shown to reduce HIV DNA transcription in monocytes and macrophages (34, 35) and persistent expression of HIV-1 and neuroinflammatory products by these cells is believed to mediate HAND pathogenesis (15, 16). Indeed, work in SIV-infected macaques (36) suggested the role of IFN-β in the establishment of transient SIV transcriptional latency in the brain during the early stages of disease in these animals (37, 38). Of interest, recent analysis of brain tissues from HIV-1–positive presymptomatic patients found a preponderance of HIV-1 DNA compared to viral RNA in glial cells, although the role of Type I IFN in this process was not determined (39). In longer-term infection, native Type I IFN responses are insufficient for the stable suppression of SIV or HIV-1 after the onset of immunodeficiency (36, 39, 40). There is some evidence that they contribute to disease through chronic immune activation and IFN-α neurotoxicity (41–43). The antiviral efficacy of IFN in vivo might be reduced by the capacity of HIV-1 to antagonize innate immune effectors (44–48). The balance of beneficial versus harmful effects of Type I IFN responses in early HIV brain disease has not been conclusively determined.
The present study was designed to investigate directly the potential role of IFN responses in the control of both early HIV-1 infection in the brain and of neuropathogenesis. To that end, we took advantage of 2 experimental systems, i.e. genetically modified IFN-α/β receptor-negative (IFNRKO) mice lacking the IFN receptor and thus rendering the mice incapable of mounting IFN responses (21, 22), and chimeric HIV-1 (EcoHIV) that expresses gp80 of ecotropic murine leukemia virus in place of gp120 to permit infection of mice (49–53). Similar to the acute phase of HIV-1 infection in humans (14, 40), conventional mice infected with EcoHIV mount adaptive immune responses that limit virus replication but do not prevent HIV-1 transmission (50) or HIV-1 neuroinvasion and induction of pathogenic events in the brain (52, 53). We report that IFNRKO mice permit EcoHIV expression at higher levels, with more extensive changes in host cell gene expression in the brain and more florid brain abnormalities than are found in WT mice. These findings show directly that Type I IFN responses can exert significant control over HIV-1 expression and pathological effects in the brain.
MATERIALS AND METHODS
Mice
Breeding pairs of IFN-α/βR−/− (IFNRKO) mice on the 129Sv/Ev background (22) were kindly provided by Dr Joan Durbin, Ohio State University, Columbus, OH, and were bred in our colony. To generate IFNRKO mice on the background of the better-breeding C57BL/6 strain, the region containing the insertionally mutated IFN-α/βR gene (22) was bred into C57BL/6 mice by crossing 129Sv/Ev/IFNRKO with C57BL/6; progeny mice were screened for mutated IFN-α/β receptor and homozygotes were back-crossed to C57BL/6 with sequential progeny screening for 6 generations. The WT IFN-α/β receptor gene was distinguished from the interrupted gene containing a neo gene insert (22) by a custom-designed reverse transcription–polymerase chain reaction (RT-PCR) amplification protocol yielding a 571-bp RNA product for intact and a 689-bp RNA product for mutant receptor gene allele. Total cellular RNA was isolated from peritoneal cells obtained by lavage and reverse transcribed to complementary DNA (cDNA) using the SuperScript RT-PCR kit (Life Technologies, Grand Island, NY). The cDNAs were subjected to PCR using GoTaq DNA polymerase (Promega, Madison, WI) and custom-designed primers IF5 (IFNARI forward primer) 5′-AAT ATC GAA CAA AAG ACG AGG CGA AG-3′, IF3 (IFNARI reverse primer) 5′-GAC GCA ATG TAG TCC CAT TTC AGG AC-3′, and Neo3 (Neo reverse primer) 5′-TCG GCA GGA GCA AGG TGA GAT GAC-3′. The expected mouse screening results: +/+ 571 bp; +/− 571 bp/689 bp; and −/− 689 bp. The C57BL/6 and 129x1/Sv mouse strains used as controls were obtained from Jackson Laboratory (Bar Harbor, ME). All studies using mice were conducted in full compliance with the National Institutes of Health guidelines and St. Luke’s-Roosevelt IACUC approval.
Responses to IFN-α in Tissue Culture
Bone marrow cells were isolated and differentiated to macrophages as described (54). Adherent macrophages were or were not activated with recombinant murine IFN-α2 (1,000 U/ml) at 37°C for 24 hours, and cells were harvested for amplification of cellular transcripts by quantitative real-time PCR (qPCR). Responses were reported as fold change in the number of copies of a transcript in IFN-α activated compared to control cells.
EcoHIV Plasmids and Infectious Virus Stocks
We used chimeric EcoHIV clones on the background of either HIV-1/NDK (EcoHIV/NDK) or HIV-1/NL4-3 (EcoHIV/NL4-3) (49, 53). The infectious viruses generated from these clones are referred to as EcoHIV, and specific clones used are indicated in legends or as appropriate. Chimeric HIV-1 expressing indicator enhanced green fluorescence protein (EGFP) (EcoHIV/NL4-3-GFP) was constructed in 2 steps. First, the EGFP coding region and the internal ribosome entry site (IRES) region were inserted into the HIV-1 clone NL4-3 immediately after the termination codon in gp41 and immediately preceding the initiation codon of Nef, generating NL4-3-GFP. The insert consisted of EGFP, GenBank number U-55761 nt 97-816, a linker region containing the NotI restriction site (5′-AGCGGCCGCACTAGTGATAATTCCG-3′), and an IRES region, GenBank number U-89673 nt 1299-1873. EGFP and IRES clones were obtained from Clontech (Mountain View, CA). In the second step, EcoHIV/NL4-3-EGFP was constructed by replacing the gp120 coding region in NL4-3-EGFP with the gp80 coding region of MuLV, exactly as described for EcoHIV/NL4-3 (53). Infectious EcoHIV stocks were prepared and titered for their p24 HIV-1 core antigen by p24 Elisa (PerkinElmer, Waltham, MA), as described (49, 52, 53).
Infection of Mice
To initiate systemic infection, groups of mice were injected intraperitoneally with 106 pg of p24 each of sterile suspension of virus stock, as described (49). To deliver virus directly to the brain, stereotaxic intracranial inoculation was conducted essentially as described (52, 55). Briefly, heavily anesthetized mice were immobilized in a stereotaxic chamber (Stoetling Co., Wood Dale, IL), and a small hole was drilled into the exposed skull above the right hemisphere at a distance of 2.5 mm to the right of bregma. Ten microliters of virus stock was injected by a micropump into the caudate putamen at a rate of 10 μL/20 min, and the wound was sutured (52). At the designated times, groups of animals were killed by asphyxiation with 100% CO2, and tissues were collected and processed for isolation of total cellular DNA and RNA, as described (49, 52, 53).
qPCR and Conventional PCR
The procedures for harvesting spleen cells, peritoneal macrophages (PMs), and brain tissues; the preparation of cellular DNA and RNA; and the detection of EcoHIV/NDK gag DNA or spliced vif message by qPCR were previously described (49, 52). Samples were run in duplicate in the AB7300 real-time thermal cycler (Life Technologies). Data analysis was performed with the 7300 System software according to the manufacturer’s instructions. For the detection of EcoHIV/NDK 2LTR circular DNA by qPCR, the reaction mixture contained custom-designed forward primer 2LTRF3 (5′-CTAGAGATCC CTCAGATCCGTTTAGT-3′), reverse primer 2LTRR1 (5′-TGGTGTGTAGTTCTGCCAATCG-3′), and probe 2LTRP (5′-(FAM)-TTTGGGTCTACAACACACAAGGCATCTTCC-(MGBNFQ)-3′). A standard curve for the quantitation of 2LTR copy number was constructed using graded numbers of plasmid TA-NDK2LTR containing the NDK U5 (3′-LTR)-U3 (5′-LTR) segment. Sample DNA content was normalized by murine β-globin DNA amplified by qPCR using forward primer MGBF (5′-CTGCCTCTGCTATCATGGGTAAT-3′), reverse primer MGBR (5′-TCACTGAGGCTGGCAAAGGT-3′), and probe MGBP (5′-FAM-TTAACGATGGCCTGAATC-MGBNFQ-3′). For the detection of double-spliced HIV/NDK nef transcript, RNA was isolated from brain tissue and reverse transcribed to cDNA as described above, and cDNA was amplified by conventional PCR using primers ND160 (5′-GAGTGAAAAATCTCTAGCAGTGGCGC-3′) and ND8002 (5′-GCTGAAGAGGCACAGGTTCCTCAGGTCG-3′). The PCR products were resolved on agarose gels and visualized by Southern blotting using a (32P)-labeled probe NDS3 (5′-GAAGAAGCGGAGACAGCGACGAAAACCTCC-3′) and standard methods. Murine β-actin was amplified in parallel to standardize RNA input. The predominant product of this PCR is a 342 bp nef transcript; minor products representing rev (364 bp) and tat (541 bp) can be seen upon film overexposure.
Cellular Gene Expression in Brain Tissues
The changes in the expression of genes from mouse brain samples were analyzed by qPCR using TaqMan chemistry and probes from the Universal Probe Library (Roche, Indianapolis, IN), as previously described (28). Data were normalized using the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Relative quantification used the comparative threshold cycle method (Applied Biosystems Technical Bulletin no. 2).
Histopathology and Immunohistochemistry
The procedures were conducted essentially as described (52, 53). Brains from dissected mice were fixed in 4% paraformaldehyde, then the right cerebral hemisphere was marked with ink, and the brain was sectioned in the coronal plane. Whole coronal sections approximately 3 mm in thickness were dehydrated overnight in a VIP Tissue-Tek tissue processor and embedded in paraffin in a Leica EG1160 embedding center. Six-micrometer-thick sections were cut on a Leica microtome and stained with hematoxylin and eosin for histopathological assessment. Selected paraffin blocks were cut at 6 μm for immunohistochemistry, which was performed using the Dako EnVision system (Dako, Carpinteria, CA), using diaminobenzidine as chromogen with light counterstaining with hematoxylin. The antibodies used were a rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP) (Dako) for labeling astrocytes, a rabbit polyclonal antibody to ionized calcium binding adaptor molecule-1 (Iba-1) (Wako, Osaka, Japan) for labeling macrophages/microglia, and a mouse monoclonal antibody 183-H12-5C (National Institutes of Health AIDS Reagent Program, Germantown, MD; catalog no. 3537) for the detection of HIV-1 p24 antigen. Photomicrographs were taken on a Zeiss Photomicroscope III equipped with a Nikon DN100 digital camera.
Quantification of immunostaining was performed using Volocity 5.5 image analysis software (PerkinElmer) using 10 to 20 images from multiple coronal brain sections (5–8 images/section from 2 to 3 mice per group). Each evaluated protein was quantified by determining the positive stain area as a percentage of the total stained area per microscope field or count of stained cells or lesion per field normalized by image area.
Confocal Microscopy
Mice were perfused first with phospate-buffered saline (PBS) followed by 4% paraformaldehyde, tissues were then dissected, flash-frozen, and stored at −80°C. Thirty-micrometer-thick coronal sections were cut on a Leica cryostat and stored in cryoprotectant at −20°C until staining. For immunofluorescence staining, sections were rinsed in PBS, treated with 10% fetal bovine serum (FBS) and 0.2% Triton X-100 in PBS for 1 hour, washed 3 times in PBS–0.2% Triton X-100, and incubated with chicken anti-GFP (1:500; Life Technologies) (for the detection of marker protein in EcoHIV/NL4-3-GFP) and with rabbit anti–Iba-1 (1:400; Wako) (to identify macrophages/microglia) in PBS–10% FBS + 0.2% Triton X-100, at 4°C overnight. Sections were then washed and incubated with goat anti-chicken Alexa 488 or goat anti-rabbit Alexa 555 conjugated antibodies (1:100; Life Technologies) in PBS–10% FBS + 0.2% Triton X-100, at 37°C for 2 hours, washed, and mounted onto slides with PROLONG GOLD ANTIFADE Reagent. Images including Z-stacking were captured with fully motorized Leica TCS FP5 confocal microscope and analyzed using Improvision Volocity software (PerkinElmer).
Statistical Analysis
Differences in HIV-1 burdens and other parameters between WT and IFNRKO mice were tested by paired Student t-test.Changes in cellular gene expression in brain tissues of infected mice were first normalized to respective uninfected controls then compared in t-test between WT and IFNRKO.
RESULTS
The studies used 2 IFNRKO mouse strains, based on strains 129Sv/Ev (22) and C57BL/6. The C57BL/6/IFNRKO strain was derived as described in Methods. Figure 1A shows an example of confirmatory screening of 7 C57BL/IFNRKO mice (lanes 1–7) versus WT control (lane 8). KO mice yielded a 689-bp product designed to span part of the neo gene transcript and IFNR flanking sequences; WT controls showed a smaller 571-bp PCR product representing part of uninterrupted IFNR transcript from the same region (Fig. 1A). Because each mouse sampled in lanes 1 to 7 showed a single 689-bp band, we conclude that these animals are homozygous for the IFNRKO allele. To determine the capacity of WT and IFNRKO mice for signaling through the IFN receptor, bone marrow–derived macrophages from mice on either background were activated by treatment with recombinant mouse IFN-α and tested for induction of 2 interferon-stimulated genes, i.e. IRF-7 and IFIT1, by qPCR 24 hours later. Both interferon-stimulated genes were induced in WT 129x1/Sv and C57BL/6 but not in their IFNRKO counterparts (Fig. 1B), confirming lack of Type I IFN responses in cells from KO mouse lines used in this study.
FIGURE 1.
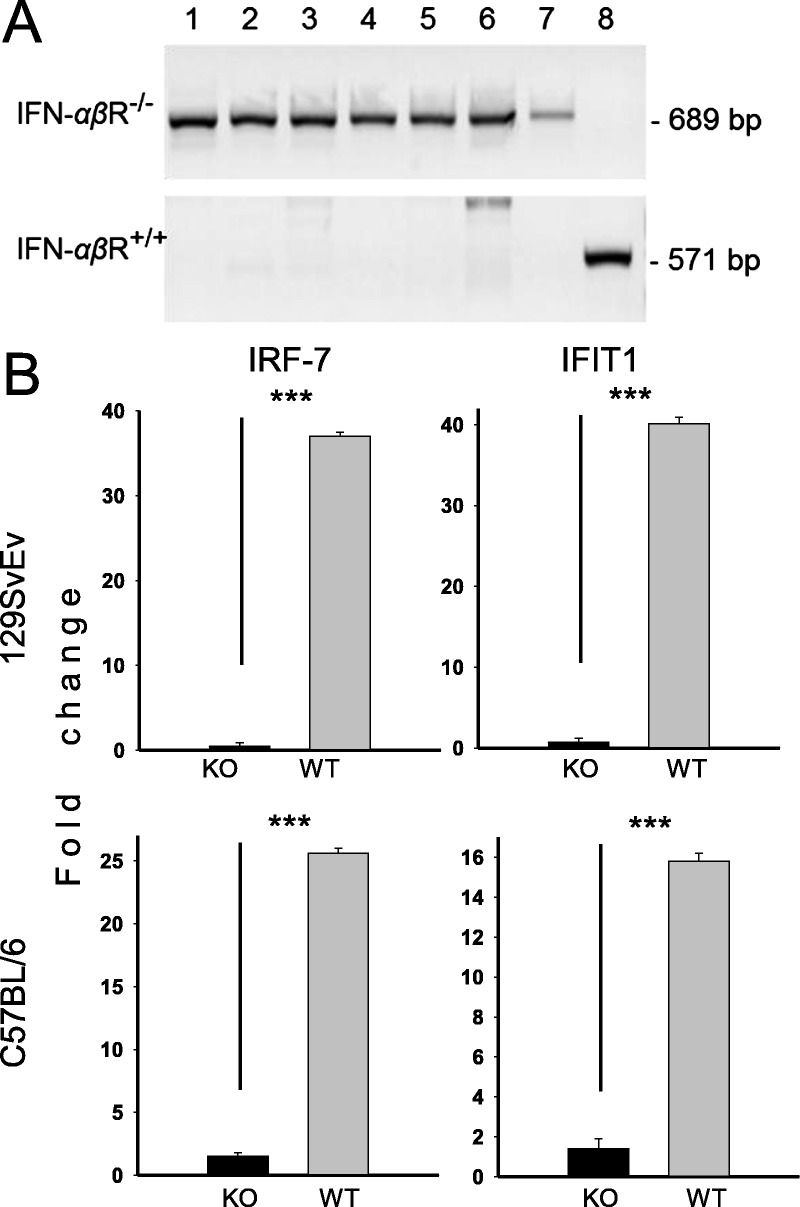
Confirmation of the interferon-α/β receptor–negative (IFN-αβR−/−) phenotype of mouse strains used. (A) Southern blot hybridization within the IFN-αβR gene showing the mobility change after insertion of the targeting vector. Data from 7 C57BL/IFNRKO mice are compared to a wild-type (WT) control. A 571-bp RNA product for intact (WT, lane 8) and 689-bp RNA products for mutant receptor gene alleles (lanes 1–7) are demonstrated. (B) Detection of responses to Type I interferon (IFN-α) in culture by cells from WT or IFN-αβR−/− mice. Bone marrow–derived macrophages were stimulated with recombinant mouse IFN-α and tested for induction of the IFN-stimulated genes IRF-7 and IFIT1 by qPCR after 24 hours. ***, p = 0.001. KO, knockout.
Next, we investigated the progress of HIV-1 infection and neuroinvasion in mice as a function of the integrity of IFN responses. 129 WT or IFNRKO mice were infected with EcoHIV by intraperitoneal injection and PMs were harvested at the times indicated after infection to measure spliced vif HIV-1 RNA, a form absent from the virus inoculum (Fig. 2A). Viral RNA was detected in both WT and IFNRKO mice up to 4 weeks after infection but the viral RNA burden was significantly greater in IFNRKO mice. To determine whether the inhibitory effect of IFN extends to HIV-1 replication in the brain, the experiment was repeated, and PMs and brain were harvested 4 weeks after infection and tested for vif RNA burden by qPCR (Fig. 2B). Results from PMs were similar to those shown in Figure 2A with viral RNA burdens significantly higher in PMs from IFNRKO mice compared to WT mice; however, we were unable to detect viral RNA by qPCR in brain tissue from either mouse strain (Fig. 2B). We then tested these brain tissues for HIV-1 DNA using the sensitive traditional PCR amplification in combination with Southern blotting (Fig. 2C), as previously described (53). HIV-1 DNA was detected with weak signals in 2 of 3 WT mice and with stronger signals in 3 of 3 IFNRKO mice (Fig. 2C). Similar results were obtained in IFNRKO mice on the C57BL/6 background infected by peripheral inoculation of EcoHIV (not shown). Overall, these results indicate that absence of functional Type I IFN responses increases HIV-1 expression in peripheral tissues and may result in increased virus burdens in the brain. Because traditional PCR is semiquantitative, the effect of higher HIV-1 expression in macrophages on HIV-1 neuroinvasion in peripherally infected IFNRKO animals could not be precisely determined.
FIGURE 2.
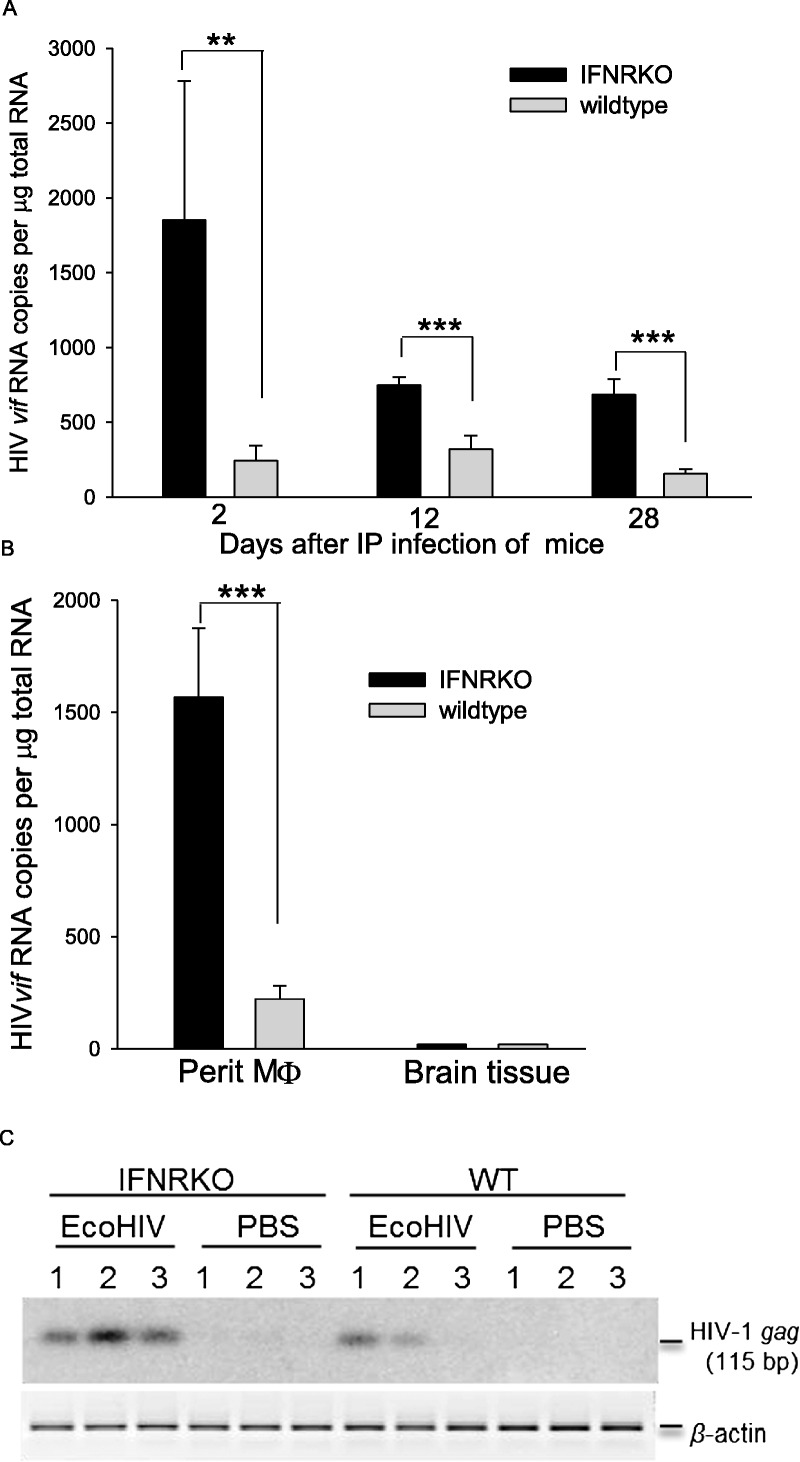
Absence of Type I interferon (IFN) receptors in mice permits increased systemic human immunodeficiency virus Type 1 (HIV-1) replication and viral neuroinvasion after intraperitoneal (IP) EcoHIV inoculation. (A) Interferon-α/β receptor–negative (IFNRKO) and wild-type (WT) 129x1 mice were infected by IP injection of 106 pg of p24 of EcoHIV/NDK, and at the indicated time points, peritoneal macrophages isolated from groups of infected mice (n = 3) were tested for expression of spliced vif mRNA by RT-qPCR. **, p = 0.01; ***, p = 0.001. (B, C) In a repeat experiment, mice infected as in (A) were tested 4 weeks after IP infection for vif mRNA expression by qPCR in peripheral macrophages (Perit M Φ) and brain tissue (B) and for HIV-1 gag DNA in brain tissue by Southern blot hybridization (C).
To increase the resolution of virological studies in mouse brain, we used our recently reported approach to initiate robust EcoHIV infection in mouse brain by stereotaxic intracerebral inoculation of virus into the basal ganglia region as described (52) (Fig. 3). 129x1 WT and IFNRKO mice were infected with EcoHIV by intracerebral injection, and brains were harvested at the indicated times after infection to measure HIV-1 RNA either by qPCR (Fig. 3A) or by conventional RT-PCR combined with Southern blot hybridization (Fig. 3B). To ensure that only de novo synthesized viral products are detected, we amplified spliced HIV-1 RNA species vif and nef, which are not packaged in virions. At 1 week after infection, vif RNA was clearly detected by qPCR in brain tissues of both WT and IFNRKO mice (Fig. 3A); although HIV-1 RNA burdens were lower in WT mice, the difference was not significant at this time point. By 2 and 4 weeks after infection, vif RNA was significantly lower in brains of WT mice than in IFNRKO brains (Fig. 3A), suggesting a marked effect of IFN signaling on HIV-1 replication. Similar results were obtained by a semiquantitative analysis of brain RNAs from this experiment by conventional RT-PCR amplifying spliced HIV-1 RNAs that share a splice acceptor site at the 3′ end of the viral genome (56), of which a 342-bp nef message is a predominant product (Fig. 3B). To determine whether Type I IFN acts at the level of HIV-1 DNA or RNA synthesis in the brain, we repeated the experiment in the same format twice, using either the 129x1 or the C57BL mouse strain. At 4 weeks after infection, we sampled viral RNA as well as DNA in brain tissues, amplifying 2-LTR circular DNA and vif RNA (Fig. 3C, D). We chose to amplify circular HIV-1 DNA because, unlike full-length DNA, this viral DNA form is absent from the HIV-1 inoculum and its amplification by qPCR provides a quantitative measure of HIV-1 infection de novo in mouse brain. As in the experiment shown in Figure 3A, IFNRKO mice in both backgrounds failed to control HIV-1 RNA expression in the brain as efficiently as did WT mice (Fig. 3C). It is striking, however, that there were no significant differences in viral DNA burden between IFNRKO and WT mice in either background (Fig. 3D). These findings extend the effects of the IFNRKO phenotype on HIV-1 expression from peripheral mononuclear cells to brain tissue, and they suggest that some IFN-responsive genes mitigate HIV-1 replication at the level of transcription in the brain.
FIGURE 3.
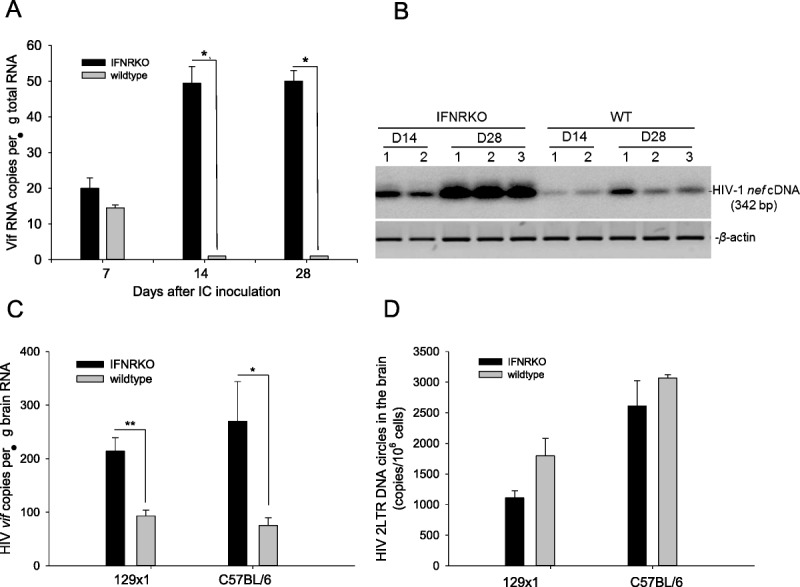
Human immunodeficiency virus Type 1 (HIV-1) expression is readily detectable after intracerebral (IC) inoculation of EcoHIV into mouse brain and virus expression is significantly greater in interferon-α/β receptor–negative (IFNRKO) mice versus wild-type (WT) mice. (A, B) IFNRKO and WT mice (129x1 strain) were inoculated IC with 106 pg of p24 of EcoHIV/NDK, and brain tissues from groups of mice (n = 3) were tested at the indicated time points for HIV-1 expression by QPCR amplification of the vif transcript (A) or RT-PCR and Northern blot hybridization for nef transcript (B). (C, D) The experiment was repeated using 2 IFNRKO mouse strains, 129x1/IFNRKO and C57BL/IFNRKO and their matching WT controls. Brain tissues were tested for vif expression (C) and HIV-1 2LTR circular DNA (D) by QPCR 30 days after EcoHIV IC inoculation. *, p = 0.05; **, p = 0.01.
In HIV-1–infected patients and in various animal models, HIV-1 neuropathogenesis has been associated with induction of a large number of cytokines, chemokines, and IFN-related genes (15, 29, 57–60). Here, we tested whether efficient EcoHIV infection of mouse brain also causes cellular gene dysregulation in the tissue and whether HIV-1 effects require Type I IFN signaling (Fig. 4). We chose 15 transcripts that have been described as being elevated either in HIV-1–infected patients (IL-1β, IRF-7, Stat-1, MyD88, and IL-6) (61, 62) or SIV-infected macaques (CD8 and CD163) (63, 64) or in brain cells stimulated with HIV-1 or Tat in tissue culture (MCP-1, C3, and IP-10) (65–67). WT or IFNRKO mice on a 129x1 background were injected intracerebrally (IC) with EcoHIV or PBS and brains were collected 4 weeks later for isolation of RNA. Cellular transcripts as well as GAPDH were amplified by qPCR, the expression of each transcript was normalized by GAPDH expression, and finally, the ratio of expression of each transcript in infected compared to PBS-injected brain tissue was calculated to determine its induction during HIV-1 infection (Fig. 4). Fourteen transcripts were significantly upregulated in the brain of infected WT and IFNRKO mice; induction ranged from roughly 1.2-fold for synaptic protein SNAP23 to more than 200-fold for CD8 in IFNRKO mice (Fig. 4). Seven transcripts were induced to significantly greater levels in IFNRKO than in WT brains, consistent with their enhanced expression of HIV-1 RNA shown in Figure 3. However, 7 other transcripts, including 3 encoded by genes linked to the Type I IFN pathway (IRF-7, STAT-1, and MyD88), were significantly induced by HIV-1 independent of the presence of a functional IFN receptor (Fig. 4). Expression of CD163, a marker of perivascular macrophages implicated in SIV encephalitis (64), was unchanged and serves as a specificity control in this panel. Thus, EcoHIV causes extensive dysregulation of cellular gene expression in mouse brain, which can be partially mitigated by functional IFN responses.
FIGURE 4.
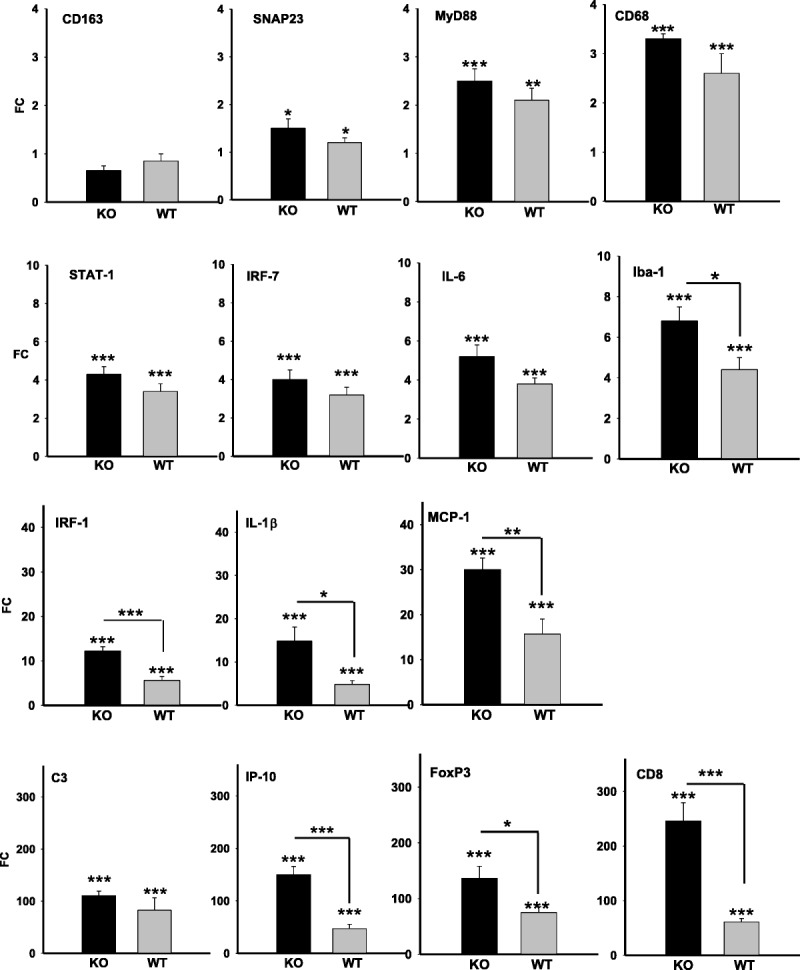
EcoHIV-infected interferon-α/β receptor–negative (IFNRKO) mice show greater dysregulation of some inflammatory and immune response genes in the brain versus wild-type (WT) mice. 129/IFNRKO (KO, black bars) and WT mice (gray bars) were infected by intracerebral injection with EcoHIV/NDK or with phosphate-buffered saline (PBS); 4 weeks later, the mice were killed and total cellular RNAs from brain tissues were compared by qPCR for expression of genes known to be involved in human immunodeficiency virus Type 1 (HIV-1) neuropathogenesis in patients. IFNRKO and WT mice inoculated with PBS served as respective controls for measurement of gene expression changes. FC, fold change.
Because productive HIV-1 infection in human brains generally correlates with overt pathological changes (68, 69), we next inquired about neuropathological changes in brain tissues from IC-infected mice, also as a function of the IFNR phenotype. C57BL/6 IFNRKO and WT mice received intracerebral injections of virus or PBS and evaluated 2 weeks later (Fig. 5). We counted perivascular lesions in hematoxylin and eosin–stained sections and used computerized image analysis to quantify Iba-1 staining as a measure of macrophage/microglial activation and GFAP staining as a measure of astrogliosis (Fig. 5). Representative fields from the right basal ganglia region in the vicinity of the injection site are shown in each category, and the frequencies of discrete pathological parameters and of their extent in this region are graphed. PBS injection was innocuous, indicating that IC inoculation alone did not lead to constitutive morphological changes in the brain, as previously reported (52). However, at 2 weeks after EcoHIV infection, brains from both WT and IFNRKO mice showed frequent perivascular infiltrates of mononuclear cells, as well as regions of excessive staining of lineage markers of both macrophage/microglia and astrocytes. The distinction between the maintenance and absence of IFN responses in the brains was quantitative, with each measure of neuropathogenesis significantly lower in WT mice, in which IFN responses were intact; when these responses were absent, there were significantly more perivascular lesions and greater Iba-1 and GFAP expression. As an internal control, we also evaluated pathological changes in the respective left (i.e. the uninjected) basal ganglia regions of affected brains and found only occasional low-level macrophage/microglial activation, astrogliosis, and no lesions (not shown). These observations are consistent with our previous results showing the highest expression of IC-injected EcoHIV near the inoculation site, with virus only slowly spreading to other brain regions (52). The differences in the extent of neuropathological changes between infected WT and IFNRKO mice were reproduced in 5 experiments, and they were independent of the EcoHIV backbone (NL4-3 or NDK) and mouse strain used (129x1 or C57BL). These findings support and extend those in Figure 4 showing that IFN responses reduce but do not eliminate cellular gene dysregulation during mouse brain infection.
FIGURE 5.
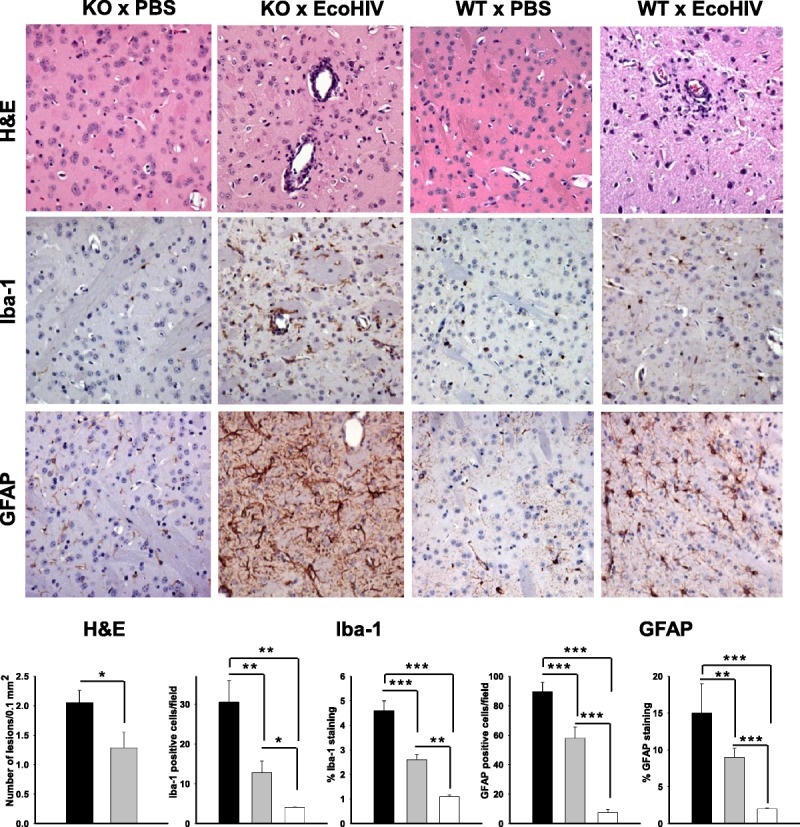
EcoHIV-infected interferon-α/β receptor–negative (KO) mice show more extensive brain abnormalities than infected wild-type (WT) mice. KO and WT mice received intracerebral infection with EcoHIV or phosphate-buffered saline (PBS). Two weeks later, the animals were killed, and brains were fixed, embedded in paraffin, and sectioned. Thin sections were stained with hematoxylin and eosin (H&E) or immunostained with an antibody to the macrophage/microglia marker Iba-1 or to the astrocyte marker glial fibrillary acidic protein (GFAP). The photomicrographs show representative fields at 10× from the right basal ganglia region in the vicinity of the injection site in each experimental system. There are perivascular mononuclear cells, extensive macrophage/microglial (MP/MG) activation, and astrocytosis in infected but not in control mice. Minimal pathology was found in the left (uninjected) hemispheres of infected mice (not shown). Graphs show means and SEM of quantitative analysis of perivascular lesion counts, MP/MG activation, and astrogliosis using 15 to 20 representative sections from 2 mice in each condition. Black bars, infected KO mice; gray bars, infected WT mice; open bars, uninfected WT mice. Asterisks indicate Student t-test p values for significance of differences among the conditions: *, p = 0.05; **, p = 0.01; ***, p = 0.001.
Finally, we compared brain tissues from EcoHIV-infected WT and IFNRKO mice at 2 weeks after IC inoculation for the presence of HIV-1 p24, a virion structural protein indicative of productive viral infection (Fig. 6A, B). In some experiments, we infected mice with EcoHIV/NL4-3–GFP, an EcoHIV clone modified to express GFP as a convenient surrogate marker of HIV-1–encoded proteins for cellular colocalization (Fig. 6C–K). Using immunohistochemistry and monoclonal anti-p24 antibody for p24 staining in paraffin-embedded brain sections, we found that IFNRKO mice expressed p24 more efficiently and had higher number of p24-positive cells than did WT mice (Fig. 6A, B), a result consistent with their more extensive expression of viral RNA (Fig. 4). The morphology of p24-positive cells in IFNRKO brains resembled that of macrophages/microglia (Fig. 6A), while p24 in WT brains resided mostly in round mononuclear cells (Fig. 6B). To identify the lineage of cells productively infected by HIV-1 in mouse brain, we used frozen brain sections and used fluorescence microscopy staining for Iba-1 to detect macrophages/microglia and GFP to detect HIV-1 (Fig. 6C–K). Brains of IFNRKO mice showed abundant GFP expression that frequently localized to Iba-1–positive cells with multiple cellular processes (Fig. 6C–H). Using Z-stacking and high-magnification confocal microscopy to visualize GFP-expressing HIV-1–infected cells in more detail, it is clear in the IFNRKO mice that they are Iba-1–positive (Fig. 6F–H), with highly ramified processes characteristic of activated microglia. Similarly, brains of infected WT mice contained HIV-1–expressing Iba-1–positive cells but at a lower frequency than IFNRKO mice, with many GFP-positive cells appearing as round cells without processes (Fig. 6I–K). Merged images in Figure 6H and K also show that some GFP-positive cells did not co-stain with a macrophage marker. A detailed study of EcoHIV tropism to mouse CNS cells is underway and will be reported separately. The present study shows that EcoHIV can establish productive infection in brain macrophages/microglia in mice in vivo, and this infection is more prominent in IFNRKO mice.
FIGURE 6.
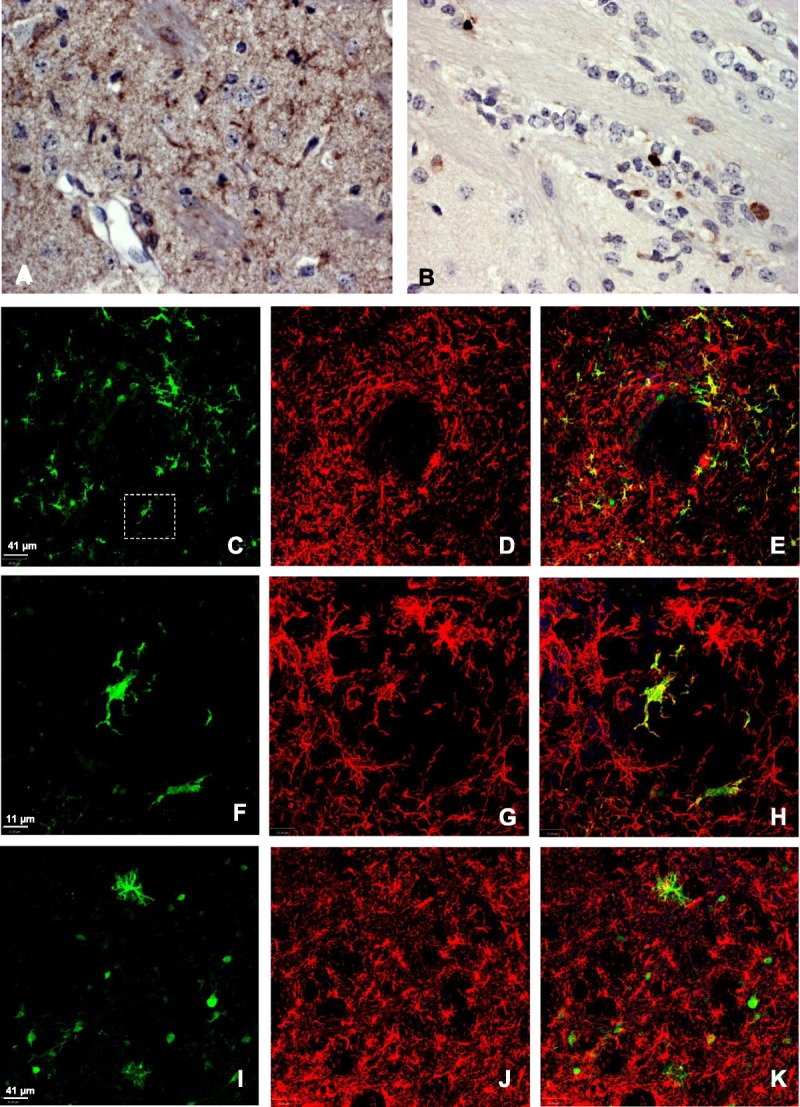
HIV-1 p24 and HIV-1–encoded marker green fluorescent protein (GFP) localize to microglia/macrophages in the brains of infected mice and are more efficiently expressed in interferon-α/β receptor–negative (IFNRKO) than wild-type (WT) animals. (A–K) At 2 weeks after intracerebral infection of mice with EcoHIV/NDK (A, B) or EcoHIV/NL4-GFP (C–K), the brains were removed, fixed, sectioned, and either stained for p24 by immunohistochemistry and evaluated by light microscopy (A, B) or co-stained for the macrophage marker Iba-1 and virally encoded GFP and evaluated by fluorescence microscopy (C–K). The figure compares virally coded protein expression in brain tissues of IFNRKO (A, C–H) to that of WT mice (B, I–K). Panels F to H show higher magnification of dashed area in Panel C enhanced by Z-stacking for better resolution of HIV-1–positive macrophages/microglia. Original magnification: (A, B) 25×. In B, p24-positive cells are mostly mononuclear, with Iba-1–negative, HIV-1–positive mononuclear cells also present in I to K (also WT). C, F, J: GFP (green); D, G, J: Iba-1 (red); E, H, K: merge.
DISCUSSION
Our results strongly suggest that innate immune responses mediated by Type I IFN can mitigate, albeit not arrest, systemic HIV-1 infection and neuropathogenesis in vivo. The closest parallel to our findings comes from studies on induction of viral encephalitis during infection of macaques with SIV (36). In this system, SIV entry into the brain early after virus exposure coincides with induction of IFN-β and imposition of SIV transcriptional latency that lasts through the asymptomatic period of disease, suggesting the role of IFN-β in delaying SIV neuropathogenesis (37, 38). Type 1 IFN has been described to control HIV-1 infection in tissue culture in a variety of cell types and at different stages in virus replication (23, 47, 61, 70). While studies in culture also indicate that HIV-1 has evolved mechanisms to counter many innate antiviral responses (44–48), the extent of HIV-1 escape from these constraints in vivo has not been determined. Our results suggest that functional IFN responses in vivo afford measurable advantage to the host in fighting HIV-1 infection, including that in the brain, at least in the animal model tested. In HIV-1–infected human beings, both monotherapy and adjunct therapy with recombinant IFN-α were found to reduce plasma virus burden (71–73), suggesting that augmentation of native IFN responses to HIV-1 can be clinically beneficial. A similar approach could be explored for the amelioration of HIV-1 brain disease.
The conclusions of the present study were made possible by the resource of mice lacking Type I IFN responses (22) and the ability of EcoHIV to infect a range of mouse strains (49, 51, 53, 74). Although the initial expression of IFN proteins themselves is not affected in IFNRKO mice (22), IFN responses are not transduced, and the mice succumb to cytopathic viruses (e.g. vesicular stomatitis virus and Semliki Forest virus) at doses 106-fold less than do WT mice (21, 22, 75). Consistent with these studies, we show that mice defective in IFNR signaling maintained significantly higher systemic and CNS HIV-1 burdens than did WT mice (Fig. 2), albeit lesser in extent than that seen with some other viruses (22, 75). Natural killer cells (76) or innate virus resistance factors (45) may also contribute to early HIV control in the mouse host. Because HIV-1 DNA levels were similar in infected WT and IFNRKO mouse brains (Fig. 3), the effect of Type I IFN signaling on EcoHIV infection in mouse brain seems to be mediated at the viral transcription level. This result is consistent with our finding that HIV-1 can infect macrophages/microglia in mouse brains (Fig. 6) and the demonstration by others that infection of cells of this lineage by HIV-1 in culture is blocked by Type I IFN at transcription (34, 35). Transcriptional silencing of HIV DNA in these cells could mitigate neuropathogenesis by reducing virus production and expression of viral neuropathogenic proteins, including Tat and Nef (77, 78). Further studies are needed to clarify the mechanism of HIV-1 inhibition by IFN in mice and, in particular, why this block seems to be incomplete.
Intracerebral inoculation of EcoHIV into both WT and IFNRKO mice caused neuropathological and molecular changes with characteristics similar to those in human HAND (28, 29, 68, 69, 79, 80). However, compared to infected WT mice, higher HIV-1 RNA burdens in brain tissues of infected IFNRKO mice correlated with increased brain pathology in these animals, including significantly greater numbers of perivascular inflammatory lesions, increased microglial and astrocyte activation, and increased induction of inflammatory and other cellular genes previously linked to neuropathogenesis in HIV-1–infected humans (Figs. 4–6). Conversely, because infected IFNRKO and WT mice differ singularly in their ability to signal through IFN receptors (22), our results strongly suggest that functional IFN responses mitigate HIV-1 neuropathogenesis through suppression of HIV-1 expression in the brain. This was most evident in the clear difference between infected IFNKO and WT mice in the presence of HIV-1–positive macrophages/microglia, the main cellular agents of HIV-1–mediated neuropathogenesis (Fig. 6) (15, 16). In fact, the detection of p24-positive (and marker protein GFP-positive) macrophages/microglia in infected IFNRKO mice bears similarity to HIV encephalitis in humans (68, 69) and to experimental HIV encephalitis in other animal models (55, 81–83). These results also support a causal relationship between the extent of HIV-1 expression in the brain and the severity of brain pathology (28, 29, 84). However, IFNRKO mice infected with EcoHIV by intracerebral inoculation do not display the high virus burdens, multinucleated giant cells, and gross morphological changes that are typical of HIV or SIV encephalitis (68, 69, 81).
Recent studies indicate that antiviral inflammatory responses are intact in IFNRKO mice despite the absence of IFN signaling (85), indicating that this strain offers the opportunity to distinguish between these potential pathogenic pathways. With differences in HIV-1 expression in the brain linked to IFN responses, we were able to investigate viral neuropathogenesis in this model as a function of cellular products that are induced by virus expression (Fig. 4). HIV-1 and its proteins have been shown to be directly neuropathogenic (77, 86–88) and induce large number of brain cell gene products implicated in neuroinflammation and brain disease (62, 65, 89, 90). Here, 14 of the 15 cellular transcripts shown were induced by EcoHIV infection in brains of WT and IFNRKO mice, and of these, 7 were more significantly induced in the IFNRKO strain. The latter observation indicates that extracellular Type I IFN is not required for their induction in this system and supports the contention that HIV-1 products and the induction of intermediate cellular products are responsible for the enhanced gene expression. Indeed, if induction of the 7 inflammatory and immune response–linked genes overexpressed in IFNRKO brain is considered pathogenic, it can be concluded that, in this case, Type I IFN protects against HIV-1–associated neuropathogenesis. It should also be noted that EcoHIV expresses all HIV-1 genes except gp120 (53), and therefore, the HIV-1–associated neuropathogenesis observed in this work occurs independently of gp120. Because EcoHIV transcribes well in mouse brain (Fig. 3) and because Tat is expressed systemically in EcoHIV–infected mice (53), the virus likely expresses functional Tat in the brain, which can contribute to the observed pathological findings alone (91) or together with other viral proteins such as Nef or Vpr (88, 92).
We interpret extensive overexpression of lineage-restricted markers like CD8 and FoxP3 as evidence for the migration of CD8-positive and regulatory T cells, respectively, into the brain, as previously observed during HIV-1 or SIV infection (63, 93). IRF-1 is induced very early in HIV-1 infection, and IP-10 can be induced by HIV-1 Nef, providing a potential link between greater induction of these cytokines in IFNRKO mice and increased viral expression (92, 94). However, there are multiple routes to cellular gene induction during HIV-1 infection. For example, interleukin (IL)-1β produced by microglia in HIV-1–infected brain (62), and as shown here, may itself drive the expression of MCP-1 in astrocytes (95). The degree of induction of cellular transcripts during EcoHIV infection is consistent with the neuropathology observed in WT mice and the more extensive neuropathology observed in IFNRKO mice (Fig. 5).
In summary, our studies support a protective role of Type I IFN in HIV-1 expression in the periphery and in the brain. Our work demonstrates that this central, innate immune pathway reduces but does not eliminate HIV-1 neuropathogenesis through its induction of posttranscriptional latency in the brain. Therefore, Type I IFN may supplement existing antiretroviral therapies to minimize HIV-1 pathogenesis.
ACKNOWLEDGMENTS
The authors would like to thank Dr Joan Durbin, currently at Rutgers New Jersey Medical School, for donating IFNRKO mice on the 129Sv/Ev background, Dr Galina Bentsman for help in EcoHIV propagation, and Ms Ilene M. Totillo for manuscript preparation.
Footnotes
Send correspondence and reprint requests to: David J. Volsky, PhD, Molecular Virology Division, St. Luke’s-Roosevelt Hospital Center, 432 W 58th St, Antenucci Research Building, Room 709, New York, NY; E-mail: djv4@columbia.edu
Dr Ichiyama is currently with the Division of Virology, Department of Infection and Immunity, Jichi Medical University School of Medicine, Shimotsuke-Shi, Tochigi-Ken, Japan.
Work in this article was supported by grants DA 017618, MH 083627, and NS 061646 from the National Institutes of Health, Public Health Service.
The authors declare they have no competing interests.
REFERENCES
- 1. Antinori A, Arendt G, Becker JT, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007: 69; 1789– 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mateen FJ, Shinohara RT, Carone M, et al. Neurologic disorders incidence in HIV+ vs HIV- men: Multicenter AIDS Cohort Study, 1996–2011. Neurology 2012: 79; 1873– 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harezlak J, Buchthal S, Taylor M, et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS 2011: 25; 625– 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robertson KR, Smurzynski M, Parsons TD, et al. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS 2007: 21; 1915– 21 [DOI] [PubMed] [Google Scholar]
- 5. Sacktor N. The epidemiology of human immunodeficiency virus–associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol 2002: 8; 115– 21 [DOI] [PubMed] [Google Scholar]
- 6. Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010: 24; 1243– 50 [DOI] [PubMed] [Google Scholar]
- 7. Heaton RK, Franklin DR, Ellis RJ, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J Neurovirol 2011: 17; 3– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ellis RJ, Badiee J, Vaida F, et al. CD4 nadir is a predictor of HIV neurocognitive impairment in the era of combination antiretroviral therapy. AIDS 2011: 25; 1747– 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heaton RK, Clifford DB, Franklin DR, Jr, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010: 75; 2087– 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCombe JA, Vivithanaporn P, Gill MJ, et al. Predictors of symptomatic HIV-associated neurocognitive disorders in universal health care. HIV Med 2013: 14; 99– 107 [DOI] [PubMed] [Google Scholar]
- 11. Davis LE, Hjelle BL, Miller VE, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 1992: 42; 1736– 39 [DOI] [PubMed] [Google Scholar]
- 12. Enting RH, Prins JM, Jurriaans S, et al. Concentrations of human immunodeficiency virus Type 1 (HIV-1) RNA in cerebrospinal fluid after antiretroviral treatment initiated during primary HIV-1 infection. Clin Infect Dis 2001: 32; 1095– 99 [DOI] [PubMed] [Google Scholar]
- 13. Mogensen TH, Marinovskij E, Larsen CS. Acute demyelinizating encephalomyelitis (ADEM) as initial presentation of primary HIV infection. Scand J Infect Dis 2007: 39; 630– 34 [DOI] [PubMed] [Google Scholar]
- 14. Valcour V, Chalermchai T, Sailasuta N, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis 2012: 206; 275– 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nat Rev Immunol 2005: 5; 69– 81 [DOI] [PubMed] [Google Scholar]
- 16. Lipton S, Gendelman HE. Dementia associated with the acquired immunodeficiency syndrome. N Engl J Med 1995: 233; 934– 40 [DOI] [PubMed] [Google Scholar]
- 17. Chang L, Ernst T, Witt MD, et al. Persistent brain abnormalities in antiretroviral-naive HIV patients 3 months after HAART. Antivir Ther 2003: 8; 17– 26 [PubMed] [Google Scholar]
- 18. Lentz MR, Kim WK, Lee V, et al. Changes in MRS neuronal markers and T cell phenotypes observed during early HIV infection. Neurology 2009: 72; 1465– 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sailasuta N, Ross W, Ananworanich J, et al. Change in brain magnetic resonance spectroscopy after treatment during acute HIV infection. PLoS One 2012: 7; e49272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spudich S, Gisslen M, Hagberg L, et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J Infect Dis 2011: 204; 753– 60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hwang SY, Hertzog PJ, Holland KA, et al. A null mutation in the gene encoding a Type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci U S A 1995: 92; 11284– 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Müller U, Steinhoff U, Reis LF, et al. Functional role of Type I and Type II interferons in antiviral defense. Science 1994: 264; 1918– 21 [DOI] [PubMed] [Google Scholar]
- 23. Pitha PM. Innate antiviral response: Role in HIV-1 infection. Viruses 2011: 3; 1179– 203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang BX, Fish EN. The yin and yang of viruses and interferons. Trends Immunol 2012: 33; 190– 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lutfalla G, Gardiner K, Proudhon D, et al. The structure of the human interferon alpha/beta receptor gene. J Biol Chem 1992: 267; 2802– 9 [PubMed] [Google Scholar]
- 26. Uzé G, Lutfalla G, Bandu M-T, et al. Behavior of a cloned murine interferon α/β receptor expressed in homospecific or heterospecific background. Proc Natl Acad Sci U S A 1992: 89; 4774– 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 2011: 1; 519– 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borjabad A, Morgello S, Chao W, et al. Significant effects of antiretroviral therapy on global gene expression in brain tissues of patients with HIV-1–associated neurocognitive disorders. PLoS Pathog 2011: 7; e1002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gelman BB, Chen T, Lisinicchia JG, et al. The National NeuroAIDS Tissue Consortium brain gene array: Two types of HIV-associated neurocognitive impairment. PLoS One 2012: 7; e46178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Q, Schacker T, Carlis J, et al. Functional genomic analysis of the response of HIV-1–infected lymphatic tissue to antiretroviral therapy. J Infect Dis 2004: 189; 572– 82 [DOI] [PubMed] [Google Scholar]
- 31. Masliah E, Roberts ES, Langford D, et al. Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis. J Neuroimmunol 2004: 157; 163– 75 [DOI] [PubMed] [Google Scholar]
- 32. Roberts ES, Burudi EME, Flynn C, et al. Acute SIV infection of the brain leads to upregulation of IL6 and interferon-regulated genes: Expression patterns throughout disease progression and impact on neuroAIDS. J Neuroimmunol 2004: 157; 81– 92 [DOI] [PubMed] [Google Scholar]
- 33. Van den Bergh R, Florence E, Vlieghe E, et al. Transcriptome analysis of monocyte-HIV interactions. Retrovirology 2010: 7; 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheney KM, McKnight A. Interferon-alpha mediates restriction of human immunodeficiency virus Type-1 replication in primary human macrophages at an early stage of replication. PLoS One 2010: 5; e13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goujon C, Malim MH. Characterization of the alpha interferon–induced postentry block to HIV-1 infection in primary human macrophages and T cells. J Virol 2010: 84; 9254– 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clements JE, Babas T, Mankowski JL, et al. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): Steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis 2002: 186; 905– 13 [DOI] [PubMed] [Google Scholar]
- 37. Alammar L, Gama L, Clements JE. Simian immunodeficiency virus infection in the brain and lung leads to differential Type I IFN signaling during acute infection. J Immunol 2011: 186; 4008– 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barber SA, Gama L, Dudaronek JM, et al. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus–macaque model. J Infect Dis 2006: 193; 963– 70 [DOI] [PubMed] [Google Scholar]
- 39. Thompson KA, Cherry CL, Bell JE, et al. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol 2011: 179; 1623– 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fauci AS. Multifactorial nature of human immunodeficiency virus disease: Implications for therapy. Science 1993: 262; 1011– 18 [DOI] [PubMed] [Google Scholar]
- 41. Mandl JN, Barry AP, Vanderford TH, et al. Divergent TLR7 and TLR9 signaling and Type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med 2008: 14; 1077– 87 [DOI] [PubMed] [Google Scholar]
- 42. Sas AR, Bimonte-Nelson H, Smothers CT, et al. Interferon-α causes neuronal dysfunction in encephalitis. J Neurosci 2009: 29; 3948– 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bosinger SE, Johnson ZP, Folkner KA, et al. Intact Type I interferon production and IRF7 function in sooty mangabeys. PLoS Pathog 2013: 9; e1003597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doehle BP, Chang K, Fleming L, et al. Vpu-deficient HIV strains stimulate innate immune signaling responses in target cells. J Virol 2012: 86; 8499– 506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008: 451; 425– 30 [DOI] [PubMed] [Google Scholar]
- 46. Peng G, Lei KJ, Jin W, et al. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti–HIV-1 activity. J Exp Med 2006: 203; 41– 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pillai SK, Abdel-Mohsen M, Guatelli J, et al. Role of retroviral restriction factors in the interferon-α–mediated suppression of HIV-1 in vivo. Proc Natl Acad Sci U S A 2012: 109; 3035– 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sheehy AM, Gaddis NC, Choi JD, et al. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002: 418; 646– 50 [DOI] [PubMed] [Google Scholar]
- 49. Hadas E, Borjabad A, Chao W, et al. Testing antiretroviral drug efficacy in conventional mice infected with chimeric HIV-1. AIDS 2007: 21; 905– 9 [DOI] [PubMed] [Google Scholar]
- 50. Hadas E, Chao W, He H, et al. Transmission of chimeric HIV by mating in conventional mice: Prevention by pre-exposure antiretroviral therapy and reduced susceptibility during estrus. Dis Model Mech 2013; 1292– 98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Im E-J, Hong JP, Roshorm Y, Bridgeman A, et al. Protective efficacy of serially up-ranked subdominant CD8 T cell epitopes against virus challenges. PLoS Pathog 2011: 7; e1002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kelschenbach JL, Saini M, Hadas E, et al. Mice chronically infected with chimeric HIV resist peripheral and brain superinfection: A model of protective immunity to HIV. J Neuroimmune Pharmacol 2012: 7; 380– 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Potash MJ, Chao W, Bentsman G, et al. A mouse model for study of systemic HIV-1 infection, antiviral immune responses, and neuroinvasiveness. Proc Natl Acad Sci U S A 2005: 102; 3760– 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dou H, Destache CJ, Morehead JR, et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood 2006: 108; 2827– 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Persidsky Y, Limoges J, McComb R, et al. Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol 1996: 149; 1027– 53 [PMC free article] [PubMed] [Google Scholar]
- 56. Schwartz S, Felber BK, Benko DM, et al. Cloning and functional analysis of multiply spliced mRNA species of human immunodeficiency virus Type 1. J Virol 1990: 64; 2519– 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Borjabad A, Brooks AI, Volsky DJ. Gene expression profiles of HIV-1–infected glia and brain: Toward better understanding of the role of astrocytes in HIV-1–associated neurocognitive disorders. J Neuroimmune Pharmacol 2010: 5; 44– 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lane TE, Buchmeier MJ, Watry DD, et al. Expression of inflammatory cytokines and inducible nitric oxide synthase in brains of SIV-infected rhesus monkeys: Application to HIV-induced central nervous system disease. Mol Med 1996: 2; 27– 37 [PMC free article] [PubMed] [Google Scholar]
- 59. Speth C, Dierich MP, Sopper S. HIV-infection of the central nervous system: The tightrope walk of innate immunity. Mol Immunol 2005: 42; 213– 28 [DOI] [PubMed] [Google Scholar]
- 60. Zhu Y, Jones G, Tsutsui S, et al. Lentivirus infection causes neuroinflammation and neuronal injury in dorsal root ganglia: Pathogenic effects of STAT-1 and inducible nitric oxide synthase. J Immunol 2005: 175; 1118– 26 [DOI] [PubMed] [Google Scholar]
- 61. Baca-Regen L, Heinzinger N, Stevenson M, et al. alpha Interferon–induced antiretroviral activities: Restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus Type 1–infected monocytes. J Virol 1994: 68; 7559– 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xing HQ, Hayakawa H, Gelpi E, et al. Reduced expression of excitatory amino acid transporter 2 and diffuse microglial activation in the cerebral cortex in AIDS cases with or without HIV encephalitis. J Neuropathol Exp Neurol 2009: 68; 199– 209 [DOI] [PubMed] [Google Scholar]
- 63. Marcondes MC, Burudi EM, Huitron-Resendiz S, et al. Highly activated CD8(+) T cells in the brain correlate with early central nervous system dysfunction in simian immunodeficiency virus infection. J Immunol 2001: 167; 5429– 38 [DOI] [PubMed] [Google Scholar]
- 64. Kim W-K, Alvarez X, Fisher J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol 2006: 168; 822– 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Conant K, Major EO. Astrocytes as mediators of CNS injury in AIDS. In: Gendelman HE, Lipton AS, Epstein L, Swindells S, eds. The Neurology of AIDS. New York, NY: Chapman & Hall, 1998: 147– 55 [Google Scholar]
- 66. Kutsch O, Oh J-W, Nath A, et al. Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus Type 1 Tat in astrocytes. J Virol 2000: 74; 9214– 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Speth C, Stöckl G, Mohsenipour I, et al. Human immunodeficiency virus Type 1 induces expression of complement factors in human astrocytes. J Virol 2001: 75; 2604– 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Budka H. The neuropathology of HIV-associated brain disease. In: Gendelman HE, Grant I, Everall IP, Lipton SA, Swindells S, eds. The Neurology of AIDS. New York, NY: Oxford University Press, 2005: 375– 91 [Google Scholar]
- 69. Sharer LR. Neuropathologic aspects of HIV-1 infection in children. In: Gendelman HE, Lipton SA, Epstein L, Swindells S, eds. The Neurology of AIDS. New York, NY: Chapman & Hall, 1998: 408– 18 [Google Scholar]
- 70. Sirois M, Robitaille L, Allary R, et al. TRAF6 and IRF7 control HIV replication in macrophages. PLoS One 2011: 6; e28125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Asmuth DM, Murphy RL, Rosenkranz SL, et al. Safety, tolerability, and mechanisms of antiretroviral activity of pegylated interferon alfa-2a in HIV-1–monoinfected participants: A Phase II clinical trial. J Infect Dis 2010: 201; 1686– 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hatzakis A, Gargalianos P, Kiosses V, et al. Low-dose IFN-alpha monotherapy in treatment-naive individuals with HIV-1 infection: Evidence of potent suppression of viral replication. J Interferon Cytokine Res 2001: 21; 861– 69 [DOI] [PubMed] [Google Scholar]
- 73. Tavel JA, Huang CY, Shen J, et al. Interferon-alpha produces significant decreases in HIV load. J Interferon Cytokine Res 2010: 30; 461– 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saini M, Hadas E, Volsky DJ, et al. Vaccine-induced protection from infection of mice by chimeric human immunodeficiency virus Type 1, EcoHIV/NL4-3. Vaccine 2007: 25; 8660– 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Johnson TR, Mertz SE, Gitiban N, et al. Role for innate IFNs in determining respiratory syncytial virus immunopathology. J Immunol 2005: 174; 7234– 41 [DOI] [PubMed] [Google Scholar]
- 76. Alter G, Heckerman D, Schneidewind A, et al. HIV-1 adaptation to NK-cell–mediated immune pressure. Nature 2011: 476; 96– 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis 2002: 186 (Suppl. 2); S193– 98 [DOI] [PubMed] [Google Scholar]
- 78. Ranki A, Nyberg M, Ovod V, et al. Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS 1995: 9; 1001– 8 [DOI] [PubMed] [Google Scholar]
- 79. Gray F, Adle-Biassette H, Chretien F, et al. Neuropathology and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol 2001: 20; 146– 55 [PubMed] [Google Scholar]
- 80. Masliah E, DeTeresa RM, Mallory ME, et al. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS 2000: 14; 69– 74 [DOI] [PubMed] [Google Scholar]
- 81. Clements JE, Mankowski JL, Gama L, et al. The accelerated simian immunodeficiency virus macaque model of human immunodeficiency virus–associated neurological disease: From mechanism to treatment. J Neurovirol 2008: 14; 309– 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gorantla S, Makarov E, Finke-Dwyer J, et al. Links between progressive HIV-1 infection of humanized mice and viral neuropathogenesis. Am J Pathol 2010: 177; 2938– 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zink MC, Laast VA, Helke KL, et al. From mice to macaques—animal models of HIV nervous system disease. Curr HIV Res 2006: 4; 293– 305 [DOI] [PubMed] [Google Scholar]
- 84. Glass JD, Fedor H, Wesselingh SL, et al. Immunocytochemical quantitation of human immunodeficiency virus in the brain: Correlations with dementia. Ann Neurol 1995: 38; 755– 62 [DOI] [PubMed] [Google Scholar]
- 85. Goodman AG, Zeng H, Proll SC, et al. The alpha/beta interferon receptor provides protection against influenza virus replication but is dispensable for inflammatory response signaling. J Virol 2010: 84; 2027– 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim BO, Liu Y, Ruan Y, et al. Neuropathologies in transgenic mice expressing human immunodeficiency virus Type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol 2003: 162; 1693– 707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Magnuson DS, Knudsen BE, Geiger JD, et al. Human immunodeficiency virus Type 1 tat activates non-N-methyl-d-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol 1995: 37; 373– 80 [DOI] [PubMed] [Google Scholar]
- 88. Power C, Hui E, Vivithanaporn P, et al. Delineating HIV-associated neurocognitive disorders using transgenic models: The neuropathogenic actions of Vpr. J Neuroimmune Pharmacol 2012: 7; 319– 31 [DOI] [PubMed] [Google Scholar]
- 89. Kim S-Y, Li J, Bentsman G, et al. Microarray analysis of changes in cellular gene expression induced by productive infection of primary human astrocytes: Implications for HAD. J Neuroimmunol 2004: 157; 17– 26 [DOI] [PubMed] [Google Scholar]
- 90. Toggas SM, Masliah E, Rockenstein EM, et al. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 1994: 367; 188– 93 [DOI] [PubMed] [Google Scholar]
- 91. Zou W, Kim BO, Zhou BY, et al. Protection against human immunodeficiency virus Type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving glial fibrillary acidic protein. Am J Pathol 2007: 171; 1923– 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. van Marle G, Henry S, Todoruk T, et al. Human immunodeficiency virus Type 1 Nef protein mediates neural cell death: A neurotoxic role for IP-10. Virology 2004: 329; 302– 18 [DOI] [PubMed] [Google Scholar]
- 93. Liu J, Gong N, Huang X, et al. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1–associated neurodegeneration. J Immunol 2009: 182; 3855– 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sgarbanti M, Borsetti A, Moscufo N, et al. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J Exp Med 2002: 195; 1359– 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Burkert K, Moodley K, Angel CE, et al. Detailed analysis of inflammatory and neuromodulatory cytokine secretion from human NT2 astrocytes using multiplex bead array. Neurochem Int 2012: 60; 573– 80 [DOI] [PubMed] [Google Scholar]