

Abstract
Even though congenital heart disease is the most prevalent malformation, little is known about how mutations affect cardiovascular function during development. Akt1 is a crucial intracellular signaling molecule, affecting cell survival, proliferation, and metabolism. The aim of this study was to determine the role of Akt1 on prenatal cardiac development. In utero echocardiography was performed in fetal wild-type, heterozygous, and Akt1-deficient mice. The same fetal hearts were imaged using ex vivo micro-computed tomography (μCT) and histology. Neonatal hearts were imaged by in vivo magnetic resonance imaging. Additional ex vivo neonatal hearts were analyzed using histology and real-time PCR of all three groups. In utero echocardiography revealed abnormal blood flow patterns at the mitral valve and reduced contractile function of Akt1 null fetuses, while ex vivo μCT and histology unraveled structural alterations such as dilated cardiomyopathy and ventricular septum defects in these fetuses. Further histological analysis showed reduced myocardial capillaries and coronary vessels in Akt1 null fetuses. At neonatal age, Akt1-deficient mice exhibited reduced survival with reduced endothelial cell density in the myocardium and attenuated cardiac expression of vascular endothelial growth factor A and collagen Iα1. To conclude, this study revealed a central role of Akt1 in fetal cardiac function and myocardial angiogenesis inducing fetal cardiomyopathy and reduced neonatal survival. This study links a specific physiological phenotype with a defined genotype, namely Akt1 deficiency, in an attempt to pinpoint intrinsic causes of fetal cardiomyopathies.
Keywords: Akt1, echocardiography, fetus, heart, mice
Introduction
Congenital heart disease affects almost 1% of human births and is the most prevalent malformation in the fetal and neonatal period (Hoffman et al. 2004). Significant progress has been made over the past decade in elucidating genetic and environmental factors causing these malformations in animal models and clinical studies (Andelfinger 2008). Functional analysis of fetal hearts in utero would provide a powerful noninvasive tool to associate a given genotype not only with morphological changes, but also with functional alterations. Characterizing fetal circulatory physiology using ultrasound biomicroscopy has been initiated in the past decade (Aristizabal et al. 1998; Pedra et al. 2002; Phoon and Turnbull 2003), allowing mechanistic understanding of cardiac malformations. The use of magnetic resonance imaging (MRI) for in utero cardiac imaging has been demonstrated (Nieman et al. 2009), yet reconstruction of the MR images remains challenging due to embryonic motion.
Akt, a protein kinase, plays a crucial role in processes such as cell survival, growth and proliferation, metabolism, and organogenesis (Yang et al. 2003; Plaks et al. 2010; Vandoorne et al. 2010). Of the three Akt isoforms, Akt1 plays an essential role in the induction of angiogenesis as a downstream effector of vascular endothelial growth factor (VEGF-A) through the phosphoinositide 3-kinase -Akt signaling pathway (Chen et al. 2005). Previous studies including those from our lab have described reduced angiogenesis in Akt1 deficient placenta and bone (Yang et al. 2003; Plaks et al. 2010; Vandoorne et al. 2010). Expression and phosphorylation levels of Akt isoforms in the heart of lacking the Akt1 gene (Akt1−/−) showed that Akt1 is the main isoform in the heart (Chang et al. 2010). Furthermore, Akt1−/− fetuses exhibit ventricular dilatation with ventricular septum defects (VSDs) on histological sections, yet apoptosis in Akt1−/− fetuses has not been found different than wild-type embryos (Chang et al. 2010). These defects in Akt1−/− fetuses presumably lead to impaired neonatal cardiac function as detected by echocardiographic examination due to enhanced p38MAPK activation (Chang et al. 2010). Consequently, it has been assumed that Akt1−/− mice exhibit enhanced postnatal mortality (Cho et al. 2001; Yang et al. 2003; Plaks et al. 2010) due to progressive contractile failure of neonatal Akt1−/− hearts (Chang et al. 2010).
We hypothesized that mouse fetuses systemically lacking Akt1 might display early cardiac failure, not detected on postmortem histology. We sought to characterize blood flow velocities, to achieve a better mechanistic appreciation of fetal cardiac failure, by applying multi-parameter analyses of in utero fetuses with echocardiography. The complex interaction between VEGF-A and Akt (Chen et al. 2005), led us to also assess myocardial angiogenesis in Akt1−/− mice during development.
Material and Methods
Animals
Animal experiments were approved by the Weizmann Institutional Animal Care and Use Committee following US National Institutes of Health, European Commission and Israeli guidelines. For in utero echocardiography, 14 heterozygous (Akt1+/−) females were mated with 3 Akt1+/− males, yielding in total 24 wild-type (Akt1+/+), 31 Akt1+/−, 18 Akt1−/− mice on a C57bl/6 background (Yang et al. 2003) at two fetal ages, namely embryonic day (E) 16.5 and E18.5. After echocardiography imaging sessions, dams and fetuses were sacrificed using an overdose of intraperitoneal (IP) pentobarbital, a sample was taken from the tail of each fetus for genotyping, and the fetal hearts were prepared for either ex vivo μCT imaging or histological analysis. Neonatal Akt1+/+, Akt1+/− and Akt1−/− mice were generated through heterozygote breeding pairs and age-matched littermates were used. Furthermore, 9 Akt1+/+, 8 Akt1+/−, 7 Akt1−/− neonates, of which 2–3 per genotype were in vivo imaged with cardiac MRI, were sacrificed at postnatal day 3 (P3) for histology, or for quantitative real-time polymerase chain reaction (qRT-PCR). Mice were housed at 21 ± 1°C, 40–50% humidity, on a 12 h light–dark cycle, with ad libitum access to water and standard rodent food.
Genotyping
Tail samples were lysed overnight at 55°C in the presence of proteinase-K and DNA lysis buffer (Sigma-Aldrich, Saint-Louis, MO), and then denatured by heating at 85°C for 45 min. The resultant supernatant was used in a PCR reaction to identify the genotype of the mice. The following primers were used in the PCR reaction: (1) Akt1 forward- 5′-TTGTCTCACGTGCTTTCTCG-3′, (2) Akt1 reverse – 5′-5′-CCTGCTGGGTCAGTAAAGA-3′, (3) LacZ-cassette – 5′-GCGGATTGACCGTAATGG-3′; The amplified PCR product was subjected to agarose-gel electrophoresis and visualized using an infrared detection system.
In utero echocardiography
For in utero echocardiography 14 Akt1+/− females were mated with Akt1+/− males. Pregnant Akt1+/− dams were imaged at embryonic days E16.5 (n = 8) and E18.5 (n = 6), yielding, respectively, 17 and 8 Akt1−/−, 15 and 16 Akt1+/−, and 9 and 9 Akt1+/+ fetuses measured for, respectively, E16.5 and E18.5. These ages were chosen to avoid nucleated red blood cells in the blood of younger fetuses which contain hyperechogenic blood that hinders delineation of myocardial borders at echocardiography. In utero fetal echocardiography was performed using a Vevo 770 high-resolution ultrasound system with a RMV770B scanhead (Visualsonics, Toronto, Canada).
Animal handling
Dams were anesthetized (isoflurane; induction 3.5%, maintenance 1.25–2%, in oxygen). Fur of the mother's abdomen was removed and prewarmed ultrasound transmission gel was applied over the entire abdomen. Body temperature was kept at 36–37°C using a preheated platform, and heart rate (HR) at 400–550 beats per minute (bpm) measured with ECG leads, by adjusting isoflurane level. Following the echocardiography imaging session, dams and fetuses were sacrificed using an overdose of IP pentobarbital, a sample was taken from the tail of each fetus for genotyping, and the fetal hearts were prepared for ex vivo μCT imaging or histological analysis. Care was taken during imaging and sacrifice to accurately record the position of each fetus such that the genetic, echocardiographic and histological data would be paired correctly.
Imaging protocol
In order to image fetal cardiac structure, B-mode (lateral resolution: 115 μm) was performed through the mother's abdominal wall. For each measurement, at least three consecutive cardiac cycles were measured and averaged. To decrease experimental bias, all of the echocardiographic measurements were performed without knowing the genotype. Echocardiographic measurements are sensitive to the imaging planes used for ultrasound data acquisition. As echocardiographic measurements depend on fetal position, not all echocardiographic parameters could be acquired for each fetus. The left side of the fetuses was determined using B-mode imaging to determine the left ventricle (LV). Measurements were done using M-mode and Pulsed-wave (PW) Doppler mode. Inflow and outflow blood velocities at the mitral valve were registered using the PW Doppler mode. Echocardiography was performed as described before in C57bl/6 fetal hearts (Yu et al. 2008).
Image analysis
From inflow waveforms the E peak (early) and A peak (active) were measured to calculate the E/A ratio. For outflow of the LV, isovolumetric relaxation time (IRT), isovolumetric contraction time (ICT), and time (ET) were measured. The myocardial performance index (MPI), clinically referred to as Tei index, was calculated as:
Fractional shortening (FS) was calculated from M-mode images (axial resolution = 55 μm) by measuring left ventricular internal diameter (LVID) during systole (LVIDs) and diastole (LVIDd) as:
As these ultrasound measurements were two-dimensional, we did not derive three-dimensional (3D) parameters.
Ex vivo μCT of fetal hearts
Ex vivo μCT was performed at E16.5 and E18.5 hearts (n = 4 for each genotype, each age). Samples were fixed, treated with Lugol's solution (Degenhardt et al. 2010), and examined using a Micro-XCT 400 system (Xradia, Pleasanton, CA). A total of 300 projection images were taken with magnification 4×, exposure time of 5 sec and voxel size 4.6 μm. Image analyses, including 3D segmentation, 3D volume rendering, and volume measurements, were performed using Avizo software (VSG International, Burlington, MA).
Neonate cardiac magnetic resonance imaging
Neonatal mice at postnatal day 3 (P3) (Akt1+/+: n=2; Akt1+/-: n=3; Akt1-/-: n=1) were imaged in vivo before euthanasia for ex vivo histology. Cardiac MRI was performed on a 9.4T using a linear resonator for excitation and an actively decoupled 2-cm surface coil for detection (Bruker, Rheinstetten, Germany). Mice were anesthetized (isoflurane; induction 3.5% in oxygen in a box, maintenance 1.25% in oxygen inhaled through a nose cone. Body temperature of the animals was kept between 36 and 37°C using a warming water blanket (Bruker, Germany) and HR was monitored using a fiber-optic system (Small animal Inc., Stony Brook, NY), and kept at 300–400 bpm. Gradient echo FLASH sequence was used with retrospective gating, acquiring 10 frames per cardiac cycle for both short-axis and (two-chamber and four-chamber) long-axis images (Intragate, Bruker, Germany). Imaging parameters for short axis: four to six slices from base to apex; Slice thickness: 0.8; Interslice distance: 1 mm; flip angle 10°; TR/TE 6.2716/2.9106 msec; number of repetitions 80; matrix 256 × 256; field of view (FOV) 20 × 20 mm; acquisition time 7 min 8.1 sec. Imaging parameters for long axis: Slice thickness: 1 mm; flip angle 10°; TR/TE 5.97/2.878 msec; number of repetitions 300; matrix 256 × 256; FOV 20 × 20 mm; acquisition time 4 min 2.829 sec.
Histology and Fluorescence Microscopy
Hearts were fixed, embedded in paraffin, and sectioned serially at 4 μm thickness (E16.5 hearts [n = 4 for each group] and P3 hearts [n = 3–4 for each group]). Tissue sections were stained with hematoxylin and eosin (H&E). Light microscopic images were taken with a Nikon E800 camera (Nikon, Tokyo, Japan) and quantitative histological analysis was performed using ImageJ. Coronal H&E cardiac sections (three sections/animal) were prepared from the middle of the heart. For coronary blood vessels, the number of vessels larger than 90 μm was calculated in the fetal heart close to the epicardium. For myocardial capillaries, the number of vessels within one high-power field (HPF) on myocardial sections was calculated. Endothelial cells in the myocardium at P3 were visualized by using Bandeiraea simplicifolia isolectin B4 (lectin-fluorescein isothiocyanate [FITC]) staining (1:100; Sigma-Aldrich L2895) (May et al. 2008). Fluorescent images were acquired on a Zeiss Axio observer microscope (Zeiss, Jena, Germany) equipped with a fluorescence illuminator and an Olympus DP72 camera using the Cell∧A camera-controlling software (Olympus, Center Valley, PA). The percent area green (lectin-FITC) fluorescent staining in each field of view was calculated using ImageJ software; the same threshold was applied for all samples (three to five sections/animal).
RNA extraction and QRT-PCR
Quantitative real-time polymerase chain reaction was performed on hearts excised from P3 mice. After euthanasia (as previously described), hearts were rapidly excised, frozen, and RNA was isolated using the NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany). Total RNA (200 ng) was used for reverse transcription using SuperScript II RNase H-reverse (Invitrogen, Carlsbad, CA). QRT-PCR was performed using the StepOnePlus QRT-PCR system (Applied Biosystems, Carlsbad, CA) with the following primers: mouse VEGF-A (Forward primer: 5′-CATTCCTGGCCCTGAGTCAA and Reverse primer: 5′-GGTTGGAACCGGCATCTTTA); mouse collagen Iα1 (COL1A1; Forward primer: 5′-GAAACCCGAGGTATGCTTGA and Reverse primer: 5′- GACCAGGAGGACCAGGAAGT). The transcription level was normalized to the expression of Hypoxanthine Phosphoribosyltransferase (HPRT, Forward primer: 5′-GGTCCTTTTCACCAGCAA and Reverse primer: 5′-GCAGTACAGCCCCAAAATGG). Each sample ran at least in duplicates.
Statistical analysis
All data are presented as mean ± SEM. Statistical analysis was performed with JMP (JMP 6.0.3, SAS Institute, Cary, NC). All data were analyzed using one-way analyses of variance (ANOVA) followed by Tukey HSD All-Pairwise Comparisons test. Differences were considered significant at P < 0.05.
Results
Abnormal blood flow pattern at the mitral valve and reduced contractile function of Akt1-deficient fetuses
In utero fetal ultrasound detected similar fetal HRs for all genotypes. Blood PW waveforms at the mitral valve for fetal E16.5 and E18.5 hearts, revealed a reduced early (E) ventricular filling velocity in Akt1+/− and Akt1−/− fetuses compared to Akt1+/+ fetuses (Fig. 1A and B). Consequently, the E/A ratio, which assesses compliancy of the heart as well as diastolic function, was reduced in Akt1+/− fetuses at E16.5 and in Akt1−/− fetuses at E16.5 and E18.5 compared to Akt1+/+ fetuses. This is consistent with a restrictive filling pattern in Akt1−/− fetuses (Table 1). E/A ratio increased from E16.5 to E18.5 in all genotypes, as described before (Yu et al.2008). MPI values, combining both systolic and diastolic cardiac performance, were significantly increased in Akt1+/− and Akt1−/− at E16.5 and E18.5 compared to Akt1+/+, suggesting reduced cardiac function. FS decreased significantly in Akt1−/− fetuses at E16.5 and E18.5 and in Akt1+/− fetuses at E16.5 (Table 1). Moderate pericardial effusion, previously described for fetal heart failure (Ranger et al. 1998), was seen in 3 of 17 E16.5 Akt1−/− fetuses (Fig. 1C) and 1 of 8 E18.5 Akt1−/− fetuses. We could not observe any dead fetuses. Thus, Akt1−/− fetuses exhibited abnormal blood flow velocities and reduced cardiac function during systole and diastole.
Figure 1.
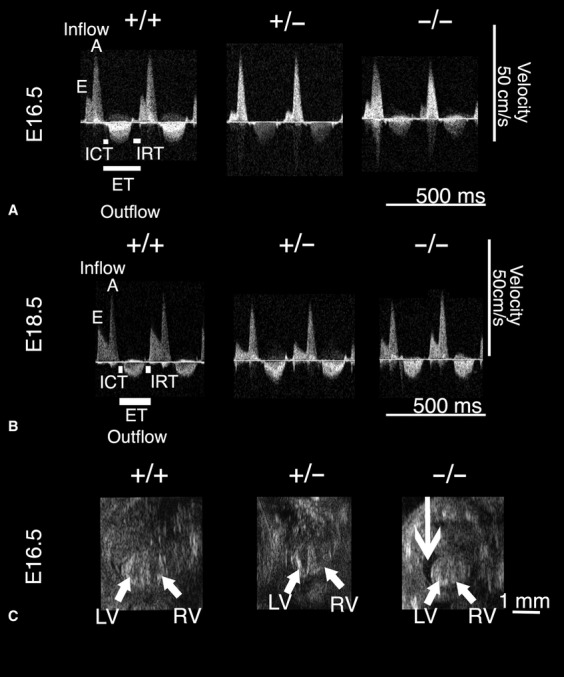
Akt1 disruption affected fetal cardiac function at embryonic day (E) 16.5 and E18.5. (A and B) In utero PW Doppler-positive blood velocities represent the inflow with early filling resulting in the E wave, and active filling (A wave), whereas negative peaks stand for outflow velocities from the LV encompassing ICT, IRT and ET. Representative E16.5 (A) and E18.5 (B) Doppler waveforms of Akt1+/− and Akt1−/− fetuses showed decreased E peak, resulting in a reduced E/A ratio compared to Akt1+/+. (C) B-mode ultrasound biomicroscopy (UBM) images depicted the fetal left ventricle (LV) and right ventricle (RV). Mild pericardial effusion (arrow), described for fetal heart failure, was found in some Akt1−/− fetuses. +/+, Akt1+/+; +/−, Akt1+/−; −/−, Akt1−/−.
Table 1.
Echocardiographic measurements
| Fetal age | E16.5 | E18.5 | ||||
|---|---|---|---|---|---|---|
| Genotype Akt1 | +/+ | +/− | −/− | +/+ | +/− | −/− |
| n fetuses PW doppler | 17 | 15 | 9 | 8 | 16 | 9 |
| Heart rate (bpm) | 246 ± 8 | 217 ± 8 | 235 ± 18 | 231 ± 19 | 252 ± 11 | 224 ± 19 |
| E/A ratio | 0.39 ± 0.01 | 0.34 ± 0.01* | 0.29 ± 0.02* | 0.45 ± 0.03 | 0.39 ± 0.02 | 0.36 ± 0.02* |
| Isovolumetric contraction time (msec) | 18.7 ± 0.8 | 27.9 ± 1.4* | 25.2 ± 2.5* | 16.2 ± 1.2 | 21.5 ± 1.3 | 24.7 ± 2.9* |
| Isovolumetric relaxation time (msec) | 29.0 ± 1.4 | 32.5 ± 1.0 | 35.4 ± 1.3* | 28.0 ± 1.4 | 28.0 ± 1.2 | 30.0 ± 1.8 |
| Ejection time (msec) | 103.5 ± 4.0 | 112 ± 2.6 | 106.4 ± 6.7 | 115.8 ± 7.7 | 99 ± 3.2 | 102.6 ± 8.9 |
| Myocardial performance index | 0.46 ± 0.01 | 0.54 ± 0.02** | 0.52 ± 0.03** | 0.40 ± 0.02 | 0.50 ± 0.02** | 0.53 ± 0.02** |
| n fetuses M-mode | 12 | 15 | 7 | 8 | 13 | 6 |
| Fractional Shortening (%) | 42.7 ± 2.7 | 33.6 ± 2.5* | 29.8 ± 3.7* | 46.1 ± 2.8 | 43.0 ± 1.9 | 35.7 ± 2.4* |
A +/+, Akt1+/+; +/−, Akt1+/−; −/−, Akt1−/−.
P < 0.05;
P < 0.01 vs. Akt1+/+.
Left ventricular volume and fetal weight are reduced in Akt1-deficient fetuses
In order to examine myocardial morphology in high-resolution and 3D, ex vivo μCT was used to validate in utero echocardiography. While the ventricular septum in E16.5 and E18.5 Akt1−/− fetal hearts appeared thinned, CT examination of isolated hearts failed to expose any VSD's in either Akt1−/− or Akt1+/− hearts (Fig. 2A and B). The myocardium of Akt1−/− fetal hearts appeared thinned and had a dilated morphology, as compared to Akt1+/+ fetal hearts. LV myocardial mass was significantly lower at E16.5 in Akt1+/−, and at E18.5 in Akt1+/− and Akt1−/− compared to Akt1+/+ fetal hearts. Total myocardial mass was also reduced at E16.5 and E18.5 Akt1−/− fetuses compared to Akt1+/+ fetuses (Fig. 2C and D). Fetal Akt1−/− body weight was significantly reduced, leaving LV mass index similar for all groups. Intrauterine growth retardation with reduced body weight has been previously described in Akt1−/− mice (Cho et al. 2001; Yang et al. 2003; Plaks et al. 2010).
Figure 2.
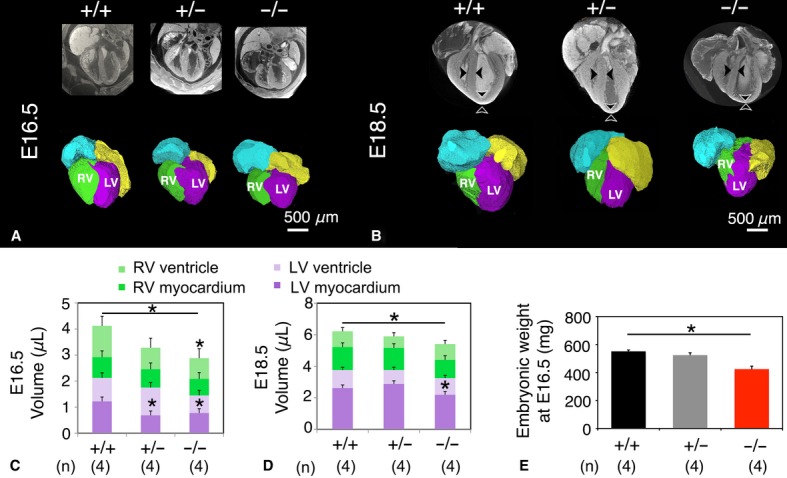
Akt1−/− fetuses displayed smaller hearts and reduced fetal weight. (A and B) Ex vivo μCT images and volume-rendered hearts revealing reduced size with a thinner myocardium and ventricular septum with a dilated morphology in Akt1−/− mice (B, arrowheads). (C and D) Quantification of volumes of fetal heart at E16.5 (C) and E18.5 (D), illustrating reduced Akt1−/− myocardial volume. (E) Akt1−/− fetuses displayed significantly lower weight at E16.5 compared to Akt1+/+ fetuses. RV, right ventricle; LV, left ventricle; +/+, Akt1+/+; +/−, Akt1+/−; −/−, Akt1−/−; *P < 0.05.
Altered cardiac morphology, reduced amount of coronary blood vessels, and decreased number of myocardial capillaries observed in Akt1-deficient fetuses
Histological analysis of fetal hearts at E16.5 revealed in one of four Akt1−/− fetuses a large VSD. All Akt1−/− fetuses displayed a thin myocardium and a dilated morphology (Fig. 3A), consistent with prior histological data (Chang et al. 2010). Interestingly, we observed a reduced presence of coronary blood vessels larger than 90 μm per cardiac section (Fig. 3A and D). Close-ups of the myocardial veins showed thinning of the perivascular wall in Akt1−/− fetal myocardium (Fig. 3B). When looking at the highest magnification we could detect a decreased amount of myocardial capillaries per HPF in E16.5 Akt1−/− fetuses compared to Akt1+/+ fetuses (Fig. 3C and E). The total amount of coronary blood vessels and myocardial capillaries in Akt1+/− fetuses was not different from the amount in Akt1+/+ fetuses (Fig. 3).
Figure 3.
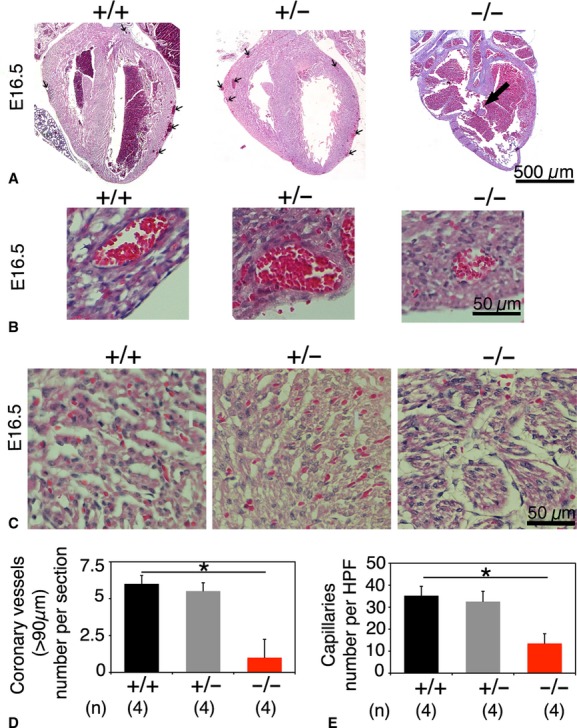
Histological analysis of E16.5 fetal Akt1+/+, Akt1+/−, and Akt1−/− hearts, coronary blood vessels and myocardial capillaries. (A) Some Akt1−/− fetal hearts displayed ventricular septum defects (VSDs; large arrow). Coronary vessels (>90 μm) located beneath the epicardium were rarely present in Akt1−/− fetal hearts (small arrows). (B) Akt1−/− myocardial veins are smaller and have a thinner perivascular wall than Akt1+/+ veins (C) Akt1−/− myocardium exhibited less myocardial capillaries than Akt1+/+ myocardium. (D) Quantification of the amount of coronary vessels (>90 μm) per section. (E) Quantification of the amount of myocardial capillaries per high power field (HPF). (*P < 0.05).
Neonatal Akt1−/− mice displayed reduced survival and impaired myocardial angiogenesis
Of 200 mice genotyped at 4 weeks old, 54 were Akt1+/+ (27% instead of the expected 25%), 115 were Akt1+/− (57.5% instead of the expected 50%), and only 31 were Akt1−/− (15.5%; fewer than the expected 25%; Fig. 4D). Although Akt1−/− mice exhibited dilated LV morphology and thin myocardium throughout development, 62% of these Akt1−/− mice survive to adulthood. Akt1−/− mice exhibited enhanced peri-natal mortality as shown before (Cho et al. 2001; Chang et al. 2010; Plaks et al. 2010). Cine MRI of neonatal Akt1−/− mice showed a trend of reduced cardiac function and reduced LV mass compared to Akt1+/+ littermates (Fig. 4A), consistent with previous echocardiographic measurements at P2 (Chang et al. 2010). Reduction in LV mass in neonatal Akt1−/− mice is also visible on coronal H&E sections (Fig. 4B). To assess the level of angiogenesis in neonatal Akt1−/− hearts, we measured the density of endothelial cells and their precursors in the myocardium. Lectin staining indicated a significantly lower quantity of endothelial cells in P3 Akt1−/− myocardium compared to Akt1+/− and Akt1+/+ (Fig. 4C and D). RT-PCR of neonatal hearts revealed reduced expression of VEGF-A in Akt1−/− mice compared to Akt1+/+ and Akt1+/− mice, as an indication for reduced angiogenesis (Fig. 4E). Moreover, reduced expression of collagen Iα1, the most abundant collagen in connective tissue, was found in Akt1−/− mice compared to Akt1+/+ and Akt1+/− mice (Fig. 4F).
Figure 4.
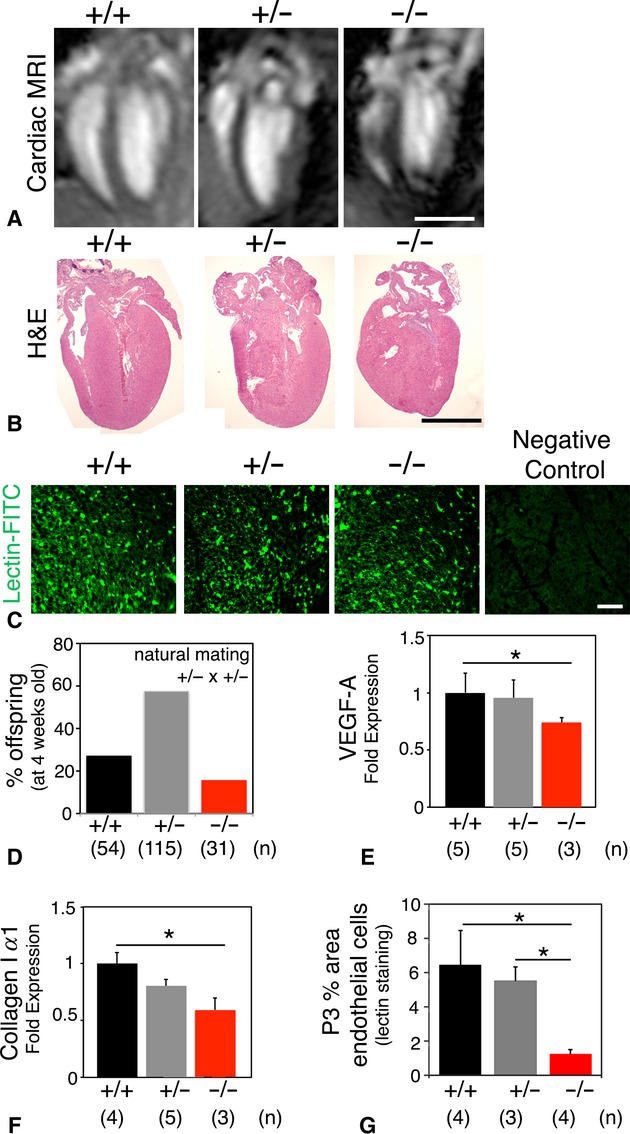
Akt1 deficiency at postnatal day 3 (P3) results in detectable changes in neonatal cardiac function, morphology and myocardial angiogenesis. (A) Neonatal cardiac MRI showing reduced cardiac size and altered morphology in Akt1−/− mice (scale bar 1 mm). (B) H&E staining of neonatal hearts confirming reduced cardiac size and altered structure in Akt1−/− mice (scale bar 1 mm). (C) Immunostaining using endothelial-cell specific lectin (green fluorescent) and negative control without lectin-FITC performed on myocardial sections of P3 mice; staining represent endothelial cells and endothelial cell precursors (scale bar 50 μm). (D) Of 200 mice genotyped at 4 weeks old, 54 were Akt1+/+, 115 were Akt1+/−, and only 31 were Akt1−/− (of which 62% survived to 4 weeks). (E) QRT-PCR of VEGF-A expression in P3 hearts, indicating reduced VEGF-A expression in Akt1−/−. (F) QRT-PCR of Collagen Iα1 expression in P3 hearts, indicating reduced Collagen type Iα1 expression in Akt1−/−. (G) Quantification of endothelial-cell specific lectin staining at the myocardium of P3; Akt1−/− is characterized by a reduced amount of green fluorescent lectin-stained endothelial cells compared to Akt1+/+ and Akt1+/−. +/+, Akt1+/+; +/−, Akt1+/−; −/−, Akt1−/−; *P < 0.05).
Discussion
This in utero echocardiographic study showed morphological cardiac changes in Akt1−/− fetuses resulting in abnormal mitral flow patterns and diastolic as well as systolic cardiac dysfunction. Ex vivo 3D μCT analysis revealed a reduced left ventricular volume, a dilated morphology with thinner fetal myocardium. Ex vivo histology confirmed altered cardiac morphology, and illustrated a reduced number of coronary blood vessels and myocardial capillaries in Akt1−/− fetuses. In neonatal Akt1−/− mice in vivo cardiac MRI and histology confirmed altered cardiac function and structure. Impaired myocardial angiogenesis with concomitant aberrant fibrinogenesis proceeded after birth in neonatal Akt1−/− mice displaying reduced survival.
In this study we used multiple imaging modalities leading to unique and powerful datasets to generate simultaneous high-resolution anatomical and functional information. Multiparameter in utero echocardiographic analyses could associate a functional phenotype with the genotype of Akt1−/− fetuses. PW Doppler analysis illustrated continuous reduced cardiac performance at late-stage fetuses. Altered fetal cardiac morphology appeared to contribute to fetal systolic and diastolic dysfunction. Only few fetuses displayed pericardial perfusions on echocardiography. These results are consistent with the cardioprotective properties previously reported for Akt (Matsui et al. 2001; Mangi et al. 2003). For fetal Akt1+/− hearts, we demonstrated early-impaired cardiac function, an intermediate phenotype between Akt1+/+ and Akt1−/− heart, underlining the intermediate phenotype reported before (Plaks et al. 2010; Vandoorne et al. 2010). Echocardiographic measures of wild-type Akt1+/+ fetuses from Akt1+/− dams were consistent with a previous publication on wild-type C57bl/6 fetal hearts (Yu et al. 2008).
Post mortem histology and μCT analysis of fetuses previously imaged by in utero echocardiography further linked the functional phenotype with a structural phenotype in Akt1−/− fetuses. Dilated morphology, thinning of the myocardium, and an occasional VSD were observed in late-stage Akt1−/− fetuses as previously also described in E14.5 fetuses (Chang et al. 2010). Although some major morphological changes were detected, in utero mortality was not identified at echocardiography. Given the reduced number of coronary vessels and myocardial capillaries, and thinning of the perivascular wall detected in Akt1−/− fetuses on histological analyses, we speculate that impaired myocardial angiogenesis and coronary vasculogenesis may be causing Akt1−/− fetal cardiac dysfunction. Residual and low levels of myocardial ischemia resulting from impaired microcirculatory formation may contribute to a morphology of ventricular wall thinning and LV dilation in Akt1−/− fetuses similar to what is observed in chronic myocardial ischemia (Reese et al. 2002). Impaired development of the coronary vascular system can thus directly lead to impaired cardiac function (Reese et al. 2002).
At neonatal age, we reported reduced angiogenesis in Akt1−/− hearts, with a smaller capillary network and decreased levels of VEGF-A. Remodeling of the extracellular matrix is essential for proper angiogenesis (Ivkovic et al. 2003). Expression of collagen Iα1, a marker for fibrinogenesis, was found reduced in Akt1−/− neonates compared to Akt1+/+ and Akt1+/− neonatal mice, indicating disturbed remodeling of the extracellular matrix. Similar matrix abnormalities have been previously observed in Akt1−/− mice (Somanath et al. 2007). Reduced angiogenesis and fibrinogenesis could further contribute to decreased survival in Akt1−/− mice. In neonatal Akt1−/− mice impaired cardiac function has previously been revealed using echocardiography, along with a thin myocardium and VSDs in the hearts of these mice (Chang et al. 2010). We underlined the same trend with a limited amount of animals using cardiac MRI at P3. Postnatal mortality and growth retardation in Akt1−/− mice has been associated with placental insufficiency (Yang et al. 2003). However, when placental Akt1 was rescued by tetraploid complementation, growth retardation and phenotypical changes in the fetus proper remained (Plaks et al. 2010). Smaller neonatal hearts and growth retardation have also been shown in studies downregulating VEGF (Gerber et al. 1999). During fetal development of the heart, VEGF is expressed at the atrioventricular field forming the heart septa (Miquerol et al. 2000). Reduced VEGF levels might ascribe to VSDs detected in Akt1−/− mice during early development. Yet also the disruption of other downstream targets of Akt1 such as GATA4, NFAT or MAPK (Sussman et al. 2011) during cardiac development has been assigned to cause VSDs (Ransom and Srivastava 2007). In embryonic Akt1−/− hearts p38MAPK is substantially activated (Chang et al. 2010).
In human fetuses, VSD is the most common congenital heart disease, seen in 1.5–3.5 per 1000 live births, and accounting for 30% of all cardiac anomalies (Barboza et al. 2002). Interestingly, dilated cardiomyopathies are rare, accounting for 8–11% of fetal cardiovascular abnormalities (Pedra et al.2002). Dilated cardiomyopathy is the end result of various cardiac disease processes and is characterized by the dilation of cardiac chambers and the reduction in systolic function (Pedra et al.2002). It remains unclear whether mutations in Akt1 gene play a role in human cardiac disease. The Akt1−/− model might present an interesting model for studying growth retardation, VSDs, and cardiomyopathy in utero.
The primary limitation of this study is our use of systemically Akt1 ablated mice, and thus the observed phenotype could be attributed to the contribution of multiple cell types. Additionally, we could not perform serial imaging of the mice during early development, as genotyping was required and there are very limited noninvasive methods described to landmark fetuses and neonatal mice (Kulandavelu et al. 2006; Avni et al. 2012). In addition, the lateral spatial resolution of the ultrasound system was limited to 115 μm, which given the small fetal heart size in mice (1–2 mm), may have limited in utero detection of VSDs. Finally, we did not assess umbilical and peripheral flows. Akt1−/− fetuses have a smaller and less perfused placenta and have phenotypical changes in the vascular bed of several organs (Yang et al. 2003; Plaks et al. 2010; Vandoorne et al. 2010). It is possible that the observed phenotype is complicated by differences in placental and peripheral perfusion in the Akt1−/− fetuses.
In conclusion, in utero echocardiography appeared to be a powerful tool to characterize cardiac function in late-stage fetal mice with loss of Akt1. The hearts of growth-retarded Akt1−/− fetuses exhibited abnormal blood flow patterns at the mitral valve and reduced contractile function. We were not only able to link these functional changes to structural alterations like dilated cardiomyopathy and VSDs using ex vivo methods, but we have also shown reduced amount of myocardial capillaries and coronary vessels in Akt1−/− fetal hearts. These changes continued after birth and may account for reduced survival of Akt1−/− neonates. Researching fetal cardiovascular function as well as delineation of the underlying pathogenesis might improve the prenatal and perinatal management providing prognostic information and improving outcomes of affected pregnancies.
Acknowledgments
We thank Jonathan Leor for scientific input, Yonathan Kubani for genotyping, Or David for animal care and Gila Meir for experimental support.
Conflict of Interest
None declared.
References
- Andelfinger G. Genetic factors in congenital heart malformation. Clin. Genet. 2008;73:516–527. doi: 10.1111/j.1399-0004.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- Aristizabal O, Christopher DA, Foster FS, Turnbull DH. 40-MHZ echocardiography scanner for cardiovascular assessment of mouse embryos. Ultrasound Med. Biol. 1998;24:1407–1417. doi: 10.1016/s0301-5629(98)00132-x. [DOI] [PubMed] [Google Scholar]
- Avni R, Raz T, Biton IE, Kalchenko V, Garbow JR, Neeman M. Unique in utero identification of fetuses in multifetal mouse pregnancies by placental bidirectional arterial spin labeling MRI. Magn. Reson. Med. 2012;68:560–570. doi: 10.1002/mrm.23246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboza JM, Dajani NK, Glenn LG, Angtuaco TL. Prenatal diagnosis of congenital cardiac anomalies: a practical approach using two basic views. Radiographics. 2002;22:1125–1137. doi: 10.1148/radiographics.22.5.g02se171125. discussion 1137–1128. [DOI] [PubMed] [Google Scholar]
- Chang Z, Zhang Q, Feng Q, Xu J, Teng T, Luan Q, et al. Deletion of Akt1 causes heart defects and abnormal cardiomyocyte proliferation. Dev. Biol. 2010;347:384–391. doi: 10.1016/j.ydbio.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Wright AC, Horng D, Padmanabhan A, Epstein JA. Rapid 3D phenotyping of cardiovascular development in mouse embryos by micro-CT with iodine staining. Circ. Cardiovasc. Imaging. 2010;3:314–322. doi: 10.1161/CIRCIMAGING.109.918482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, et al. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- Hoffman JI, Kaplan S, Liberthson RR. Prevalence of congenital heart disease. Am. Heart J. 2004;147:425–439. doi: 10.1016/j.ahj.2003.05.003. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, et al. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulandavelu S, Qu D, Sunn N, Mu J, Rennie MY, Whiteley KJ, et al. Embryonic and neonatal phenotyping of genetically engineered mice. ILAR J. 2006;47:103–117. doi: 10.1093/ilar.47.2.103. [DOI] [PubMed] [Google Scholar]
- Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS, et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat. Med. 2003;9:1195–1201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- Matsui T, Tao J, Lee F, del Monte KH, Li L, Picard M, et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- May D, Gilon D, Djonov V, Itin A, Lazarus A, Gordon O, et al. Transgenic system for conditional induction and rescue of chronic myocardial hibernation provides insights into genomic programs of hibernation. Proc. Natl. Acad. Sci. USA. 2008;105:282–287. doi: 10.1073/pnas.0707778105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- Nieman BJ, Szulc KU, Turnbull DH. Three-dimensional, in vivo MRI with self-gating and image coregistration in the mouse. Magn. Reson. Med. 2009;61:1148–1157. doi: 10.1002/mrm.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedra SR, Smallhorn JF, Ryan G, Chitayat D, Taylor GP, Khan R, et al. Fetal cardiomyopathies: pathogenic mechanisms, hemodynamic findings, and clinical outcome. Circulation. 2002;106:585–591. doi: 10.1161/01.cir.0000023900.58293.fe. [DOI] [PubMed] [Google Scholar]
- Phoon CK, Turnbull DH. Ultrasound biomicroscopy-Doppler in mouse cardiovascular development. Physiol. Genomics. 2003;14:3–15. doi: 10.1152/physiolgenomics.00008.2003. [DOI] [PubMed] [Google Scholar]
- Plaks V, Berkovitz E, Vandoorne K, Berkutzki T, Damari GM, Haffner R, et al. Survival and size are differentially regulated by placental and fetal PKBalpha/AKT1 in mice. Biol. Reprod. 2010;84:537–545. doi: 10.1095/biolreprod.110.085951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, Hoey FC, de la Brousse T, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Ransom J, Srivastava D. The genetics of cardiac birth defects. Semin. Cell Dev. Biol. 2007;18:132–139. doi: 10.1016/j.semcdb.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Reese DE, Mikawa T, Bader DM. Development of the coronary vessel system. Circ. Res. 2002;91:761–768. doi: 10.1161/01.res.0000038961.53759.3c. [DOI] [PubMed] [Google Scholar]
- Somanath PR, Kandel ES, Hay N, Byzova TV. Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J. Biol. Chem. 2007;282:22964–22976. doi: 10.1074/jbc.M700241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman MA, Volkers M, Fischer K, Bailey B, Cottage CT, Din S, et al. Myocardial AKT: the omnipresent nexus. Physiol. Rev. 2011;91:1023–1070. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandoorne K, Magland J, Plaks V, Sharir A, Zelzer E, Wehrli F, et al. Bone vascularization and trabecular bone formation are mediated by PKB alpha/Akt1 in a gene-dosage-dependent manner: in vivo and ex vivo MRI. Magn. Reson. Med. 2010;64:54–64. doi: 10.1002/mrm.22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, et al. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J. Biol. Chem. 2003;278:32124–32131. doi: 10.1074/jbc.M302847200. [DOI] [PubMed] [Google Scholar]
- Yu Q, Leatherbury L, Tian X, Lo CW. Cardiovascular assessment of fetal mice by in utero echocardiography. Ultrasound Med. Biol. 2008;34:741–752. doi: 10.1016/j.ultrasmedbio.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]