

Abstract
Metabolic Flux Analysis is now viewed as essential to elucidate the metabolic pattern of cells and to design appropriate genetic engineering strategies to improve strain performance and production processes. Here, we investigated carbon flux distribution in two Streptomyces coelicolor A3 (2) strains: the wild type M145 and its derivative mutant M1146, in which gene clusters encoding the four main antibiotic biosynthetic pathways were deleted. Metabolic Flux Analysis and 13C-labeling allowed us to reconstruct a flux map under steady-state conditions for both strains. The mutant strain M1146 showed a higher growth rate, a higher flux through the pentose phosphate pathway and a higher flux through the anaplerotic phosphoenolpyruvate carboxylase. In that strain, glucose uptake and the flux through the Krebs cycle were lower than in M145. The enhanced flux through the pentose phosphate pathway in M1146 is thought to generate NADPH enough to face higher needs for biomass biosynthesis and other processes. In both strains, the production of NADPH was higher than NADPH needs, suggesting a key role for nicotinamide nucleotide transhydrogenase for redox homeostasis. ATP production is also likely to exceed metabolic ATP needs, indicating that ATP consumption for maintenance is substantial.Our results further suggest a possible competition between actinorhodin and triacylglycerol biosynthetic pathways for their common precursor, acetyl-CoA. These findings may be instrumental in developing new strategies exploiting S. coelicolor as a platform for the production of bio-based products of industrial interest.
Introduction
Since the late 1960s, considerable effort has been devoted to discover new antibiotics. However, the discovery of biologically active molecules has proved progressively more difficult due to biochemically and technically long and costly processes. Furthermore, the prevalence of multi-resistant bacteria has now increased quite substantially [1]. Therefore, discovering novel antibiotics is now seen as critical for public health and medical research programs.
In this regard, the soil-inhabiting, Gram-positive bacteria, belonging to the genus Streptomyces, are significant sources of new bio-active molecules [2]. While these bacteria produce up to 70% of antibiotics currently used in medical prescriptions as well as many valuable compounds (such as immune-suppressors or anti-carcinogenic agents), recent publications of the genome sequence from several Streptomyces species [3]–[5] highlighted that their potential for the production of new secondary metabolites remains enormous. In fact, genome mining revealed that for a given Streptomyces species, the genome comprises 20 to 40 gene clusters presumably involved in the production of many secondary metabolites, while three to five only are currently produced under ordinary laboratory culture conditions.
The genetic organization of genes encoding proteins involved in secondary metabolite production has been studied extensively. Several gene clusters encoding antibiotic biosynthetic pathways, as well as specific or pleiotropic regulators, have now been described [6].
However, although primary metabolism provides precursors, reductive power and energy used for biomass and secondary metabolites biosyntheses, an integrated picture of relationships between primary and secondary metabolism is still missing. A metabolic flux analysis describing carbon flux distribution within the metabolic network, that would complete previous transcriptomic [7] and proteomic studies [8], would clarify the mechanisms by which primary metabolism sustains antibiotic production. This would in turn be of crucial importance for microbiological engineering of Streptomyces strains in order to define conditions under which the accumulation of precursors, preferential expression of the above-mentioned gene clusters, or antibiotic synthesis are optimal.
Streptomyces coelicolor A3 (2) M145 is a model strain often used to investigate the regulation of antibiotic production and its genome was the first one to be sequenced and annotated amongst Streptomyces species [3]. Depending on growth conditions, this strain may produce up to four antibiotics, but generally synthesizes mainly the blue pigmented, polyketide antibiotic actinorhodin as the major product [9].
Considering the production yield of S. coelicolor M145, which presumably indicates an efficient synthesis of the polyketide precursor acetyl-CoA, S. coelicolor M145 has been chosen as a ‘genetic platform’ to create a super-host for the heterologous expression of gene clusters associated with secondary metabolism from other Streptomyces species as well as organisms from other genera [10]–[12]. In order to avoid competition between native and heterologous biosynthetic pathways for a limited pool of metabolic precursors, the four major antibiotic clusters of S. coelicolor M145 were deleted, yielding strain M1146 [11].
In S. coelicolor M145, production of the Calcium-Dependent Antibiotic (CDA) coincides with growth [13], whereas production of actinorhodin, undecylprodigiosin and methylenomycin usually take place at the onset of the stationary phase when growth rate has slowed down [14], [15]. In the case of actinorhodin, the shift between the growth and the production phases is accompanied by a series of transcriptomic and proteomic switches leading to carbon redirection to actinorhodin synthesis [7], [8], [16].
In the present paper, we developed a N-limited minimal medium to study S. coelicolor metabolic fluxes under steady-state conditions. N-limitation was chosen as a typical condition promoting actinorhodin production by S. coelicolor M145, as previously described [17]–[19]. Under such conditions, both M145 and M1146 strains grow exponentially at a very low growth rate (μ∼0.04 h−1), M145 producing only actinorhodin during its growth phase.
We took advantage of metabolic steady conditions to compute a carbon flux distribution in S. coelicolor using a combination of stoichiometric inventory described by Holms [20], enzyme activity assays, mass-balance (Metabolic Flux Analysis) and 13C isotopic methods (13C Metabolic Flux Analysis). Data from genome [3], transcriptome [7] and proteome [8] analyses were used to constraint and define operating metabolic pathways.
We elucidate the flux distribution in primary carbon metabolism in both S. coelicolor M145 and M1146 strains and compare them so as to gain insights on effects of the impairment of secondary metabolism on metabolic homeostasis.
Materials and Methods
Biological material
The present study was carried out with S. coelicolor A3 (2) strain M145 and S. coelicolor A3 (2) strain M1146 (act, red, cpk, cda deletions). S. coelicolor strain A3 (2) M1146 cannot produce actinorhodin (ACT), undecylprodigiosin (RED) and calcium dependent antibiotics (CDA) and no cryptic type I polyketide synthase (Cpk) was expressed. Both strains were a generous gift from M.J. Bibb and J.P. Gomez-Escribano from the John Innes Centre (Norwich, UK).
Media and growth conditions
Batch cultures were grown in a bioreactor (Infors Labfors 3) with a 2 litre working volume, 2 Rushton-type impellers and 3 baffles. The bioreactor was equipped with a cooled condenser to avoid medium evaporation. Temperature and pH were controlled at 28°C and 7.0, respectively, with automatic addition of NaOH (1 mol L−1) or HCl (0.5 mol L−1). CO2 and O2 concentrations were monitored with a Bluesens BCpreFerm gas analyzer. Aeration was adjusted to 0.4 volume air (volume culture)−1 min−1 (vvm) and agitation was 800 rpm so as to maintain dissolved oxygen near 70% saturation (aerobic conditions).
A defined minimal medium (File S1) was developed according to Egli and Fiechter [21]. Based on theoretical excess factors for mineral elements and vitamins with respect to carbon [21] the medium, although N-limited, allowed S. coelicolor strains to reach a bacterial density of 9.6 g (cell dry weight) L−1 and to grow for more than 5 generations in the exponential growth phase. Note that exponential growth during at least 5 generations was a prerequisite for steady-state isotope labeling in metabolites [22]. Isotopic labeling was conducted with a mixture consisting of 80% unlabelled glucose and 20% [1-13C] glucose (CortecNet). For fermentation experiments, 100 mL of medium no. 1 (15.0 g L−1 glucose, 15.0 g L−1 glycerol, 15.0 g L−1 bacto peptone (Difco 0118-07), 3.0 g L−1 NaCl, 1.0 g L−1 CaCO3, pH 7.2) in a baffled polycarbonate 500-mL Erlenmeyer flask were inoculated with 109 spores from a 30% glycerol stock. The culture was incubated at 28°C and agitated with a magnet (NuovaII agitator, Bioblock Scientific/Sybron thermolyne) for 48 h. Eight mL were then used to inoculate 100 mL of medium no. 2 (33.0 g L−1 glucose, 15.0 g L−1 yeast extract (BD 212720), pH 7.2). The second culture was incubated for 24 h. Cells from 40 mL were collected by centrifugation (3,200 g, 5 min, room temperature), suspended in 20 mL of TES solution (270.5 mmol L−1), and immediately used to inoculate the bioreactor. An additional sample of 10 mL was used for cell dry weight quantification, after filtration through a cellulose acetate filter (0.22 µm) and distilled water washing before drying at 95°C for 24 h.
Gas exchange
In bioreactors, liquid and gas phases were well mixed and gas diffusion was rapid such that both the CO2 Exhaust Rate (CER) and the O2 Uptake Rate (OUR) could be calculated at each time with online measurements:
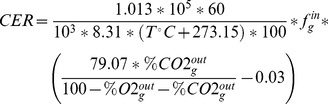 |
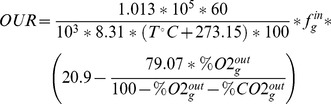 |
In these equations, it is assumed that the build-up of dissolved CO2 and N2 in the liquid phase was negligible. Flows (fgin) as well as mole fractions (% CO2, % O2) were measured with dehydrated air to avoid any mole fraction imbalance due to water vapour. CER and OUR were plotted versus time and summed in order to quantify total CO2 evolution and O2 consumption. The latter values are used in section Materials and methods 12.1. to determine YCO2/S and YO2/S respectively. All the abbreviations and symbols used in this study are listed in File S2.
Nucleic acids, proteins, antibiotics, triacylglycerol measurements and elemental composition
Nucleic acids were quantified after Norris and Ribbons [23] and proteins were quantified with the Bradford method [24]. Actinorhodin and undecylprodigiosin contents were determined spectrophotometrically according to Christiansen [25] and Tsao et al. [26] respectively. Production of CDA was detected using a bioassay adapted from Lautru et al. [27]. Triacylglycerol contents were determined by Fournier Transformed Infra Red Spectroscopy (FTIR) as described by Verleyen et al. [28] and Hong et al. [29]. FITR analyses were carried out at the Laboratoire de Chimie Chimie Physique, UMR 8000, CNRS/Université Paris Sud, Orsay, France (see File S1 for further details). The elemental composition (C, H, O, N) of S. coelicolor was determined with an elemental analyser (VarioCube, Elementar) on cells harvested during the exponential phase of growth. Elemental analyses were carried out at the Institut de Chimie des Substances Naturelles, UPR 2301, CNRS, Gif sur Yvette, France.
Proteins precipitation for metabolite analyses
Supernatants were de-proteinated by acid precipitation with HClO4 35% (0.1 mL (mL supernatant) −1) and then centrifuged 2 min at 6,000 g at room temperature. Supernatants were neutralized with cold KOH (7 mol L−1), centrifuged 2 min at 6,000 g at room temperature and filtered through a 0.22 µm polyether sulfone membrane.
Glucose and extracellular metabolites quantification
Glucose, α-ketoglutarate (2-oxoglutarate), pyruvate, L-lactate, acetate and ethanol were analysed from de-proteinated supernatants on an Agilent 1200 HPLC instrument with an Aminex HPX-87H column at 35°C using H2SO4 (5 mmol L−1) as the eluent (0.5 mL min−1). Metabolites were detected either with an Agilent SPD UV detector (210 nm) or an Agilent RID detector (40°C).
Ammonium quantification
The ammonium content was measured enzymatically in 20 µL of de-proteinated samples with glutamate dehydrogenase in 1 mL of an assay mixture made of Tris/HCl (0.1 mol L−1) pH 8.0, NADH (0.2 mmol L−1), ADP (0.6 mmol L−1), α-ketoglutarate (10 mmol L−1), and 5 units of glutamate dehydrogenase from bovine liver (Sigma #G2626). The A 340 was monitored until the end of the reaction (∼8 min).
Phosphate quantification
Phosphate was measured on 50 µL from de-proteinated samples using 100 µL of PiBlue (PiBlue Phosphate Assay Kit, Gentaur). The A 600 was monitored for 30 min at room temperature.
Enzymatic assays
40 mL of culture were harvested and bacterial cells were suspended in 10 mL Tris-Tricarballylic acid (15 mmol L−1) buffer pH 7.8, MgCl2 (10 mmol L−1), 10% glycerol. RNase A (1 mg mL−1) and DNase I (1 mg mL−1) were added when needed. The sample was sonicated 8 times with a Branson Sonifier 250 sonicator set at a power level = 2, during 20 seconds at 30 seconds intervals. Cell debris was removed by centrifuging at 10,000 g for 15 min at 4°C. The supernatant was used as the crude extract. The protein concentrations were measured by the Bradford method using BSA as a standard [24]. All enzyme activities were measured on a Beckmann DU7400 spectrophotometer at 28°C according to Sugimoto and Shiio [30], [31], Nachlas et al. [32] and Sauer et al. [33] with slight modifications (see File S1 for further details).
Sample preparation and GC-MS analyses
GC-MS analyses of amino acids to determine the 13C-labeling (see File S1 for further details) were carried out as described by Zamboni et al. [34]. Briefly, after 5 generations of exponential growth in the presence of 20% [1-13C] glucose, cells were collected, proteins were extracted and submitted to acidic hydrolysis, extracts were derivatized with methoxamine and N-methyl-N-(trimethylsilyl) trifluoroacetamide and injected on a GC-MS (Agilent 6890N gas chromatograph, time-of-flight mass spectrometer Pegasus III). Peak integration was performed using LECO Pegasus software. Because automated peak integrations have been occasionally unreliable, manual controls and/or corrections were systematically performed for each metabolite.
Five independent samples were collected along a 1.5-generation period and analysed in triplicate. During the sampling period, μ, qS, qO2, γCO2, YX/S, YACT/S, YCO2/S and YO2/S values were constant and at their maximal values. MID values for Glu, Thr, Asp, Ala and Val were identical, with less than 1% RDS. We therefore conclude that both metabolic and the isotopic steady-states were ensured in our experiment.
S. coelicolor central metabolism
Pathways considered
The central metabolic network of S. coelicolor was deduced primarily from genome annotation [3], transcriptome [7] and proteome analyses [8].
Glucose entry and phosphorylation through the Glk-GlcP permease complex in S. coelicolor were well described [35]–[37]. Consequently, we assumed that glucose phosphorylation was carried out by the ATP-dependent glucose kinase Glk.
All genes and enzymes involved in the Embden-Meyerhof-Parnas pathway (EMP), the tricarboxylic acid cycle (TCA), and the pentose phosphate pathway (PPP) were detected in S. coelicolor [8], [38]–[40].
The Entner-Doudoroff pathway (ED) has been shown to be non-functional in several Streptomyces species [39], [41]. Since (i) there was little growth with two carbon substrates and (ii) both isocitrate lyase and malate synthase were not detected in the S. coelicolor proteome [8], the glyoxylate shunt was considered to be negligible in our model.
In the present paper, comparison of the experimental and calculated γCO2 values, and analysis of 13C-labeling in valine, alanine, glutamate, glutamine and proline provided further evidence of these assumptions for S. coelicolor under our experimental conditions (see 3.4.).
PEP carboxylase was shown to be the sole anaplerotic enzyme in Streptomyces species [8], [38], [39], [42], [43] and was therefore considered in our model.
Few experimental data on the respiratory chain efficiency in Streptomyces species are available [44]. From consideration of genome mining, we assumed here that the respiratory chain of S. coelicolor could be branched, with a type I NADH dehydrogenase excreting 4 protons per NADH, an uncoupled type II NADH dehydrogenase and two terminal oxidases, the bd complex that excretes 2 protons per NADH and the bc1/aa3 supercomplex that excretes 6 protons per NADH [45]. The type I NADH dehydrogenase has been shown to account for about 70% of NADH dehydrogenase activity in aerobic conditions in Escherichia coli [46]. The supercomplex bc1/aa3 was shown to have a lower affinity for oxygen and to be the predominant complex in aerobic conditions [47]. Enzymes corresponding to both type I NADH dehydrogenase and to bc1/aa3 supercomplex were identified in S. coelicolor proteome [8]. Genome mining and proteome analyses [8] further revealed the presence of an F1F0-ATP synthase. Under the assumption that the synthesis of one ATP by F1F0-ATP synthase requires three to four protons, NADH re-oxidation via the bc1/aa3 branch yields 1.50–3.33 ATP. FADH re-oxidation was assumed to be due to succinate dehydrogenase [8], [44]. Depending on the pathway used by electrons, the transfer of two electrons from succinate to oxygen yields 0.50–2.00 ATP [48].
Considering all these data, a P/O value (moles of ATP produced per 0.5 mole of O2 consumed) of 2.0 could be reasonably considered for S. coelicolor. Nevertheless, our results on carbon flux distribution are not sensitive to the P/O value used.
Two genes of S. coelicolor (SCO7622 and SCO7623) putatively encode a PntAB-type nicotinamide nucleotide transhydrogenase that interconverts NADP + NADH to NADPH + NAD in E. coli [33], and thus the presence of a transhydrogenase was taken into account.
Amino acids, nucleic acids, peptidoglycan, lipids and small molecules biosyntheses
Amino acids, nucleic acids, small molecules, peptidoglycan, triacylglycerol and lipids biosynthetic pathways in S. coelicolor appeared to be identical to those present in other bacteria [42] and therefore their stoichiometry was assumed to be the same as that observed in other microorganisms [22], [49]–[61] (see File S3).
Antibiotic biosyntheses
The biosynthetic pathways for the 3 known antibiotics produced by S. coelicolor were reconstructed using S. coelicolor genome and published data [62]–[70]. For each antibiotic, all the steps of the biosynthesis were fused into one single reaction that includes precursors, reducing power and energy needs.
Biomass composition
To calculate carbon fluxes according to the stoichiometric approach of Holms [20], the biomass composition of S. coelicolor cells had to be determined. The respective amounts of the main polymers involved in the S. coelicolor biomass composition, namely proteins, DNA, RNAs, triacylglycerol, lipids and peptidoglycan were taken from Borodina et al. [71] with some modifications according to the experimental values we determined. These modifications are discussed in section Results 1.
Precursors, cofactors and energy requirements
Using the biomass composition, reactions associated with building-block biosynthesis (primary metabolism) and reactions for secondary metabolism, we calculated precursor, cofactor and energy requirements to synthesise 1 g of dry cells. On the basis of Ingraham's calculations for E. coli [22], the energy requirement for the polymerisation of macromolecules in S. coelicolor was calculated. We also calculated specific precursor, energy and reducing power requirements to synthesize 1 mole of antibiotics.
Flux calculations
Experimental kinetic parameter quantifications
For each experiment, we calculated the following graphs: ln(X(t)) versus t, X(t) versus S(t), ACT(t) versus S(t), RED(t) versus S(t), KET(t) versus S(t), PYR(t) versus S(t), CO2(t) versus S(t), O2(t) versus S(t), where X denotes biomass and S denotes source glucose. During exponential growth, each curve was linear, and the slope allowed us to determine growth rate constant μ (h−1) and the following yields: YX/S, YACT/S, Y RED/S, YCDA/S, YKET/S, YPYR/S, YCO2/S, YO2/S. For example, YX/S is the yield of biomass production with respect to glucose, etc. Note that yields are expressed in carbon equivalents, e.g. YX/S is in moles of biomass carbon per mole of glucose carbon. Using these values, the glucose consumption and productivity of metabolites (μmol (g cell dry weight)−1 h−1) were computed as (note that the following are algebraic and therefore can be negative):
Carbon balance (in %) was calculated as follows:
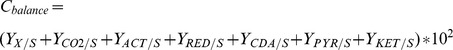 |
Anabolic fluxes calculation
Glucose-6-phosphate, fructose-6-phosphate, ribose-5-phosphate, erythrose-4-phosphate, glyceraldehyde-3-phosphate, 3-phosphoglycerate, phosphoenolpyruvate, pyruvate, acetyl-CoA, α-ketoglutarate and oxaloacetate are the 11 precursors from central metabolism. These precursors are sufficient to synthesize any macromolecule in the bacteria. That is, carbon flux to anabolic reactions can be represented by 11 individual fluxes from precursors to biomass and/or secreted metabolites (antibiotics, organic acids). Known molar requirements of biomass or antibiotic for precursors (for a given precursor i, they are denoted as pi for biomass, li for actinorhodin, mi for undecylprodigiosin, ni for the calcium dependent antibiotic expressed in μmol (g cell dry weight)−1 and μmol (μmol antibiotic)−1, respectively) and productivity (q and γ values, see Materials and Methods 12.1) allow one to calculate anabolic requirement fluxes Φi and the sum vi for any precursor i, as follows:
| (biomass) |
| (actinorhodin) |
| (undecylprodigiosin) |
| (calcium) |
| (organic) |
Metabolic Flux Analysis
Although anabolic fluxes Φi were calculated as described in Materials and Methods 12.2, catabolic fluxes in central metabolism calculations remained to be determined to obtain a full carbon-flux distribution. For this purpose, metabolic reactions were represented as a stoichiometric matrix (S) of size {m, n}. Each row of this matrix represented one unique compound amongst the m = 48 compounds and each column represented one reaction amongst the n = 46 lumped metabolic reactions. Note that the 46 lumped reactions correspond to 265 enzymatic reactions. S was a sparse matrix, with 0 as the most represented value. The flux through all reactions in the metabolic network was represented by the vector v (dimension n). The set of concentrations in metabolites was represented by the vector x (length m). The system dynamics is described by d x /dt = Sv. In the steady state, d x /dt = 0, that is, Sv = 0. In the present study, this equation is true as metabolism was studied at the steady state, that is, in the exponential growth phase in which turn-over fluxes are constant and therefore there is no build-up in intracellular metabolite concentration. Glucose specific consumption (qS) and the 11 anabolic fluxes (Φi) are used as external constraints. However, the system still has one degree of freedom. Data from labeling experiments were used to solve the system. O2 consumption, CO2 evolution and the respiratory quotient (QR) were not used as constraint here and were rather compared to the computed O2 and CO2 fluxes to check the reliability of our system, as discussed in section Results 2.
13C Metabolic Flux Analysis
Here, we took advantage of the software OpenFlux, which is a computational tool based on the Elementary Metabolite Units (EMU) framework [72], [73] to analyse 13C-labeling data. The EMU framework used here for the simulation is provided in Table S1. OpenFlux calculates fluxes from Mass Isotopic Distribution (MID, or isotopic pattern) of amino acids. [13C-1] glucose was used here as the labeling substrate and allowed us to determine the flux-ratio between the pentose phosphate pathway and glycolysis (PPP/Glyc ratio). In fact, when [13C-1] glucose was metabolized by the PPP, the 13C label was lost by decarboxylation, while the 13C label was incorporated into amino acids when it enters glycolysis. We recognize that reversible triose phosphate isomerase and aldolase activities could lead to a 13C-labeling in the C-6 atom position in fructose-1, 6-bisphosphate and then possibly in hexose phosphates [74]. If significant, this effect should be taken into account to calculate the relative commitment of PPP and glycolysis. The 13C-labeling in tryptophan and histidine should provide evidence of the labeling in C-6 because their -COOH group comes from this very atom position. While tryptophan was absent from the spectra since it is degraded to a considerable extent by acid hydrolysis [75], the 13C-labeling pattern in histidine (Table S2) showed that the C-6/C-1 isotopic exchange is negligible. Thus, the output of our calculations represent actual net fluxes.
MID data from mass-spectrometer were normalized to total mass-signals as follows, giving relative isotopic abundance:
Normalized MID were also corrected for natural isotope (C, H, N, O, Si) abundance during flux calculations. The glucose specific consumption qS, the 11 anabolic fluxes Φi and the normalized MID in glutamate (m 0/z = 348), threonine (m 0/z = 320), aspartic acid (m 0/z = 334), alanine (m 0/z = 218) and valine (m 0/z = 246) were combined to provide a unique solution for all carbon fluxes, thereby solving vector v.
For sensitivity analyses, carbon fluxes were estimated by minimizing the variance-weighted sum of squared residuals (SSR) between the simulated and model-predicted MIDs using non-linear least-squares régression [76]. In all cases, flux estimation was repeated at least 20 times starting with random initial values for all fluxes to find a global solution. At convergence, standard deviations and 95% confidence intervals for all fluxes were calculated using the parameter continuation technique described by Antoniewicz et al. [76]. This technique is based on evaluating the profile of SSR as a function of one flux, while the values for the remaining fluxes are optimized. The 95% confidence interval for an evaluated flux corresponds to flux values for which SSR increased by less than 3.84 [76]. A flux was considered non-observable if the 95% confidence interval was larger than the estimated flux value.
Metabolic fluxes were expressed as absolute (μmol (g cell dry weight)−1 h−1) or relative fluxes with respect to glucose uptake (μmol (μmol glucose) −1). Relative fluxes allowed the comparison between strains, as M145 and M1146 did have different glucose uptake rates.
Energy, reducing power and gas exchange fluxes calculations from carbon-flux distribution
With the formula used for precursors, the stoichiometry of reactions, including those for antibiotics syntheses (File S3), and the flux values (Materials and Methods 12.4), it was possible to calculate ‘intrinsic energy production’ (ATP), reducing power (NADH, NADPH) and gas exchange (O2, CO2). The total CO2 evolution rate was computed from catabolic CO2 production (glycolysis, TCA, PPP) and anabolic production as:
Then, this value was compared with our experimental CO2 production rate to verify the validity of the carbon-flux distribution (see section Results 4). In other words, experimental CO2 evolution was an independent data point not used as a constraint to calculate carbon-flux distribution. We assumed that NADPH requirement and production should have been equal since the metabolism was in steady state. If the production of NADPH was more important than the need, excess NADPH might have been converted to NADP+, thereby reducing NAD+ into NADH, by nicotinamide nucleotide transhydrogenase.
Similarly, the metabolic requirement for NADH should match NADH production; but here and quite generally, NADH production was larger than NADH oxidation. In order to satisfy our steady state condition, we assumed that excess NADH was re-oxidized by the respiratory chain. For O2 consumption, the anabolic demand was completed by the extra consumption required for excess NADH re-oxidation with the following stoichiometry: 1 mole of re-oxidized NADH consumed 0.5 mole of O2. For ATP as well, anabolic and catabolic fluxes were completed by the production of ATP associated with excess NADH re-oxidation. At this stage, knowledge of the actual P/O value (moles of ATP generated per mole of oxidized NADH) would be ideal. As stated in section Materials and Methods 11.1, we assumed that under 70% dissolved O2, the bc1/aa3 cytochrome pathway probably prevailed, and so a P/O value of 2 could be used to estimate ATP production from oxidative phosphorylation. Under this assumption, as for NADH, ATP production would be higher than ATP consumption. Excess ATP production would be likely compensated for by the consumption required for cell maintenance. Such a consumption is hereafter denoted as mATP, that is, energy requirements for homeostasis and turn-over of structural molecules as well as others processes that could not be quantified precisely (e.g. energy needed to excrete antibiotic).
Results
Composition of S. coelicolor biomass
As mentioned in section Materials and Methods 11.4, we mainly used here metabolite contents reported by Borodina et al. [71] with significant modifications for deoxynucleotides (for reasons that will become apparent below), the inclusion of bactoprenol (0.34 µmol (g cell dry weight)−1) in cell wall composition [50], and a modification of the triacylglycerol content in M1146. The biomass decomposition in building blocks is shown in Table 1. Using the elemental formula of individual metabolites and taking into account the loss of water during polymerization of macromolecules, the building block composition allowed us to recalculate the macromolecular composition of bacteria in proteins, DNA, RNAs and lipids (Table 2). Our estimates were in agreement with published values [77]–[80] except for phospholipids and DNA.
Table 1. Biomass composition of S. coelicolor M145 cells (μmol (g dry cells)−1).
| PROTEINS | PHOSPHOLIPIDS | SOLUBLE MOLECULES | |||
| Alanine | 556 | Phosphatidylethanolamine | 29 | NAD | 6 |
| Arginine | 145 | Phosphoglycerol | 6 | NADP | 5 |
| Asparagine | 162 | Cardiolipin | 1 | CoA | 5 |
| Aspartate | 160 | C14 | 10 | Menaquinone 8 | 4 |
| Cysteine | 68 | C15 | 31 | Tetrahydrofolate | 8 |
| Glutamate | 157 | C16 | 28 | FMN | 8 |
| Glutamine | 156 | C17 | 31 | FAD | 5 |
| Glycine | 830 | C18:1 | 6 | ATP | 1.5 |
| Histidine | 54 | TRIACYLGLYCEROLS | ADP | 0.5 | |
| Isoleucine | 196 | Glycerol-3-Phosphate | 22 | PEPTIDOGLYCAN | |
| Leucine | 305 | C14 | 1 | UDP-NAMb | 110 |
| Lysine | 205 | C15 | 30 | UDP-NAGa | 132 |
| Methionine | 91 | C16 | 7 | Alanine | 209 |
| Phenylalanine | 98 | C17 | 28 | Diaminopimelic acid | 125 |
| Proline | 173 | C18:1 | 0.2 | Glutamate | 11 |
| Serine | 189 | TEICHOIC ACIDS | Glycine | 107 | |
| Threonine | 191 | Teichoic acid | 16 | Bactoprenol Phosphate | 0.3 |
| Tryptophan | 24 | Lysine | 4 | DNA | |
| Tyrosine | 65 | RNA | dAMP | 27 | |
| Valine | 328 | AMP | 100 | dCMP | 70 |
| CELL WALL | CMP | 172 | dTMP | 27 | |
| UDP-NAGa | 83 | TMP | 110 | dGMP | 70 |
| Galactose | 167 | GMP | 138 | ||
UDP-N-Acetylglucosamine
UDP-N-Acetylmuramic acid
Table 2. Macromolecules content of S. coelicolor M145 cells.
| Macromolecules | Published dataa | Calculated datab | RSDc |
| Proteins | 41.2 | 41.1 | 0.17 |
| DNA | 3.6 | 6.0 | 35.36 |
| RNA | 16.7 | 16.7 | 0.00 |
| Phospholipids | 2.7 | 3.3 | 14.14 |
| Triacylglycerols | 1.8 | 2.0 | 7.44 |
| Soluble molecules | 3.0 | 2.6 | 10.10 |
| Peptidoglycan | 11.0 | 11.4 | 2.53 |
| Cell Wall | 4.4 | 4.4 | 0.00 |
| Teichoic acids | 6.6 | 6.8 | 2.11 |
| Ash | 9.0 | 5.7 | 31.75 |
| Total | 100.0 | 100.0 | 0.00 |
Biomass composition (% dry cell weight) from [71].
Calculated from our composition of building blocks (% dry cell weight)
Relative standard deviation (%) between published and calculated values.
The difference in phospholipid content was considered to be acceptable (0.42% of total biomass) since it was previously shown that such a low difference in biomass composition had a negligible impact on carbon flux determination [81].
The experimental DNA content obtained here was 6.0% (0.06 g (g cell dry weight)−1). As the mass of the S. coelicolor genome is 1.86-fold that of E. coli, which has a well-accepted DNA content of 3.1% [82], the value of 6% is much more accurate than that previously published for S. coelicolor [79].
The chemical elemental formula calculated from the data in Table 1 (with a correction for water content from 4 to 5% H2O excess [83]) was CH1.62 O0.45 N0.25 (normalized with respect to carbon), with 5.7% of ash. This is close to those found in other microorganisms [78], [84]–[87] and in good agreement with the elemental formula determined experimentally by combustion/reduction-based elemental analysis. We thus considered that the biomass composition described in Table 1 was accurate.
A difference in the triacylglycerol content was found in the biomass composition of M145 and M1146, with a triacylglycerol content of 0.02 g (g cell dry weight)−1 for M145 and 0.03 g (g cell dry weight)−1 for M1146 (M. Vergne, personal communication).
Using (i) the biomass composition (Table 1, Table 2), (ii) the stoichiometry of reactions describing the biosynthesis of metabolites (File S3) and (iii) the energy requirements re-calculated for S. coelicolor on the basis of published energy requirements for the polymerisation of macromolecules in E. coli [22], we calculated precursors, cofactors and energy requirements to synthesise 1 g of dry cell matter (Table 3).
Table 3. Precursors, energy and reducing power needs for 1 g dry biomass synthesis in S. coelicolor.
| Precursors, Energy, Reducing Power | μmol (g cell dry mass)−1 | |
| M145 | M1146 | |
| Glucose-6-phosphate | 327 | 327 |
| Fructose-6-phosphate | 357 | 357 |
| Ribose-5-phosphate | 1,344 | 1,344 |
| Erythrose-4-phosphate | 199 | 199 |
| Glyceraldehyde-3-phosphate | 281 | 292 |
| 3-Phosphoglycerate | 1,701 | 1,701 |
| Phosphenolpyruvate | 508 | 508 |
| Pyruvate | 2,600 | 2,629 |
| Acetyl Coenzyme A | 2,043 | 2,234 |
| α-Ketoglutarate | 754 | 754 |
| Oxaloacetate | 2,773 | 2,802 |
| CO2 | −1,433 | −1,491 |
| O2 | 17 | 17 |
| ATP | 34,606 | 34,849 |
| NADPH | 13,491 | 13,993 |
| NADH | −3,387 | −3,415 |
Positive values correspond to metabolite consumptions and negative values correspond to metabolite productions.
Enzymatic activities in S. coelicolor
In Streptomyces lividans and other Actinomycetes, glucose-6-phosphate dehydrogenase was shown to be NADP-dependent whereas 6-phosphogluconate dehydrogenase was shown to be NAD-dependent [39], [88]. Because of its importance for redox mass-balance, the nature of the cofactors was verified here. We found that glucose-6-phosphate dehydrogenase was strictly NADP-dependent, with a specific activity of 144.6±2.7 µmol min−1 (mg protein)−1 and no detectable activity with NAD. Furthermore, we found that 6-phosphogluconate dehydrogenase was strictly NADP-dependent, with a specific activity of 236.4±6.4 µmol min−1 (mg protein)−1 and no detectable activity with NAD. We also verified that isocitrate dehydrogenase was strictly NADP-dependent, with a specific activity of 132.7±4.3 µmol min−1 (mg protein)−1 and no activity at all with NAD.
The S. coelicolor genome contains two genes (SCO7622 and SCO7623) encoding a PntAB-like nicotinamide nucleotide transhydrogenase, but no UdhA-encoding gene. In E. coli, PntAB reduces NADH using NADP as a reductant in vivo while UdhA catalyses the reverse reaction [33]. By enzymatic measurement, we found a clear transhydrogenase activity, that catalysed the reduction of thio-NAD using NADPH with a specific activity of 90.4±3.7 µmol min−1 (mg protein)−1, and no activity at all with thio-NADP and NADH. Since no evident NADPH phosphatase-encoding gene was found in the S. coelicolor genome, transhydrogenase was then introduced in our model as the enzyme that balanced NADPH overproduction.
Growth and metabolite excretion of S. coelicolor strins
Batch-cultivation of the strains M145 and M1146 yielded a bacterial density of 8 g (cell dry weight) L−1 and 9 g (cell dry weight) L−1, respectively. The stationary growth phase began when the medium turned to be nitrogen-depleted, glucose and phosphate being limiting approximately 9 h after nitrogen depletion for both strains.
During the exponential phase in bioreactors, the growth rate constant (μ) of M1146 was 13±0.1% higher compared to M145 while the glucose specific uptake rate was nearly 13±0.5% lower (Table 4). The respiratory activity (O2 consumption and CO2 production) was 20±2% lower in M1146 compared to M145 and furthermore, strain M145 produced 2.03±0.08 µmol (g cell dry weight)−1 h−1 actinorhodin (up to 400 mg L−1 at the end of the culture) while no antibiotic production was detected at all in M1146.
Table 4. Kinetic parameters and GC-MS analyses.
| S. coelicolor M145 | S. coelicolor M1146 | |||
| Values | RSD (%) | Values | RSD (%) | |
| μ (h−1) | 0.0394 | 0.54 | 0.0446 | 0.95 |
| q S μmol (g cell dry mass)−1 h−1 | 664 | 3.24 | 575 | 1.40 |
| qO2 μmol (g cell dry mass)−1 h−1 | 2,235 | 2.97 | 1,528 | 6.67 |
| γCO2 μmol (g cell dry mass)−1 h−1 | −2,295 | 3.24 | −1,372 | 0.86 |
| γACT μmol (g cell dry mass)−1 h−1 | −2 | 5.87 | 0 | 0.00 |
| Glu-TMS (m/z = 348) m 0 | 0.54614 | 0.24 | 0.57610 | 0.30 |
| Glu-TMS (m/z = 349) m 1 | 0.30993 | 0.79 | 0.28750 | 0.34 |
| Glu-TMS (m/z = 350) m 2 | 0.14393 | 0.97 | 0.13640 | 0.83 |
| Thr-TMS (m/z = 320) m 0 | 0.57694 | 0.11 | 0.60627 | 0.14 |
| Thr-TMS (m/z = 321) m 1 | 0.29529 | 0.31 | 0.27454 | 0.45 |
| Thr-TMS (m/z = 322) m 2 | 0.12776 | 0.53 | 0.11919 | 0.98 |
| Asp-TMS (m/z = 334) m 0 | 0.57730 | 0.22 | 0.60477 | 0.08 |
| Asp-TMS (m/z = 335) m 1 | 0.29536 | 0.22 | 0.27410 | 0.37 |
| Asp-TMS (m/z = 336) m 2 | 0.12733 | 0.52 | 0.12113 | 0.96 |
| Ala-TMS (m/z = 218) m 0 | 0.71483 | 0.19 | 0.72507 | 0.15 |
| Ala-TMS (m/z = 219) m 1 | 0.20701 | 0.51 | 0.19744 | 0.52 |
| Ala-TMS (m/z = 220) m 2 | 0.07817 | 1.72 | 0.07749 | 0.85 |
| Val-TMS (m/z = 246) m 0 | 0.64474 | 0.36 | 0.66780 | 0.25 |
| Val-TMS (m/z = 247) m 1 | 0.26438 | 0.74 | 0.24531 | 0.70 |
| Val-TMS (m/z = 248) m 2 | 0.09088 | 0.45 | 0.08689 | 0.62 |
Kinetic parameters were experimental values obtained as described in 2.19.1. Mass Isotopic Distributions were normalized as described in section 2.19.4. Values were means of two independent experiments and RSD their relative standard deviations. Glu: Glutamic acid; Thr: Threonine; Asp: Aspartic acid; Ala: Alanine; Val: Valine.
For qS, γACT, qO2 and γCO2, positive values correspond to metabolite consumption and negative values correspond to metabolite production.
The carbon balance during the exponential phase was similar in both strains (92±2% in M145 and 87±1% in M1146) and slightly lower than 100%, suggesting that a certain carbon amount was not recovered. HPLC, LC-MS analyses and bioassays on culture supernatants did not allow the identification of extracellular metabolites that could account for this apparent loss of carbon. Nevertheless, these carbon balances are similar to the 94±9% value found by Borodina et al. [41] for S. coelicolor M145 and to the 90% value found by Bruheim et al. [89] for S. lividans during actinorhodin production in N-limited conditions. The carbon imbalance probably indicates the synthesis of one or several yet unidentified secondary metabolites in S. coelicolor under N-limitation.
During the stationary phase, both strains produced α-ketoglutarate (82 µmol±4 (g cell dry weight)−1 h−1 in M145 and 162 µmol±6 (g cell dry weight)−1 h−1 in M1146) and pyruvate (21 µmol±1 (g cell dry weight)−1 h−1 in M145 and 167 µmol±11 (g cell dry weight)−1 h−1 in M1146). There was, therefore, a clear difference in carbon allocation between the two strains, with M1146 liberating more pyruvate and α-ketoglutarate in the medium, likely as a result of carbon re-direction in the absence of growth and antibiotic production, as discussed in section 4.4.
Flux patterns of S. coelicolor strains
The use of stoichiometric coefficients and biomass composition to infer metabolic fluxes (Metabolic Flux Analysis) could not be carried out properly since an uncertain flux value remained at the PPP/glycolysis branching-point. We thus used 13C-labeling ([13C-1] glucose) and 13C Metabolic Flux Analysis in the isotopic steady-state to estimate the relative commitment to PPP as opposed to glycolysis. Isotopic patterns obtained by GC-MS in amino acids from protein hydrolysates are shown in Table 4. In the amino acids considered (Glu, Thr, Asp, Ala and Val), there was a lower 13C-labeling (mass m 0 proportionally more abundant) in the strain M1146, clearly showing a lower commitment of the 13C-label to glycolysis and the TCA (from which those amino acids are synthesized) and thus a larger commitment to PPP.
Analysis of 13C-label patterns in alanine and valine clearly showed the absence of 13C label in the C-1 atom position in both amino acids (Table S2). This demonstrated that the C-1 atom position in pyruvate was not labelled and thus the absence of an active Entner-Doudoroff pathway under our conditions. Moreover, a discrepancy between γCO2 and 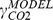 would have been detected if either the Entner-Doudoroff pathway or the glyoxylic shunt had been active in our conditions, but this is clearly not the case (see also below). Similarly, the 13C-labeling in C-1 in glutamate, glutamine and proline showed the activity of the anaplerotic phosphoenolpyruvate carboxylase and examination of the 13C-labeling at the C-5 position in glutamate, glutamine and proline indicated that the glyoxylic shunt was inactive (Table S2).
would have been detected if either the Entner-Doudoroff pathway or the glyoxylic shunt had been active in our conditions, but this is clearly not the case (see also below). Similarly, the 13C-labeling in C-1 in glutamate, glutamine and proline showed the activity of the anaplerotic phosphoenolpyruvate carboxylase and examination of the 13C-labeling at the C-5 position in glutamate, glutamine and proline indicated that the glyoxylic shunt was inactive (Table S2).
Using further the data of Table 4, fluxes were numerically estimated with OpenFlux, which is based on optimization of weighted least squares (see section Materials and Methods12.4).
We analyzed the statistical significance and confidence of estimated fluxes in S. coelicolor M145 and M1146, with [13C-1] glucose as a tracer and the sensitivity test described by Antoniewicz et al. [76]. Small sensitivities, i.e., large changes in the flux value resulting in small changes in the minimized sum of squared residuals, indicate that the flux cannot be estimated precisely. Large sensitivities, on the other hand, indicate that the flux is well determined. In the present study, sensivity tests fulfilled large sensitivity (96%) with a 95% confidence level in all cases. According to Wiechert et al. (1997), our flux maps are representative descriptions of the metabolic processes in S. coelicolor [90].
In M145 and M1146, the flux through the PPP was found to be 22±0.3% and 38±0.2% of available glucose-6-phosphate, respectively.
Our results from Metabolic Flux Analysis and isotopic labeling were combined to draw a carbon flux map in M145 and M1146 during the exponential phase of growth (Fig. 1, Fig. 2). Fluxes were expressed as μmol (g cell dry weight)−1 h−1 or normalized to glucose uptake (μmol (μmol glucose)−1). Consistently, the committed step of PPP (glucose-6-phosphate dehydrogenase) was found to be associated with a flux of 0.372±0.002 µmol (μmol glucose)−1 in M1146, which is 1.7-fold higher than in M145 (0.214±0.003 µmol (μmol glucose)−1). In contrast, the commitment to the TCA was 1.019±0.02 µmol (μmol glucose)−1 in M145, that is, 1.4-fold higher than in M1146 (0.730±0.018 µmol (μmol glucose)−1). Precursor abstraction from primary carbon metabolism to biomass synthesis was larger in M1146 (total 1.019±0.018 µmol (μmol glucose)−1, giving 60±1% (molC mol−1C) of carbon uptake allocated to biosyntheses) compared to M145 (0.813±0.02 µmol (μmol glucose)−1, 47±1%). There was a much larger commitment of phosphoenolpyruvate to carboxylation by the PEP carboxylase in M1146 and therefore, a larger anaplerotic oxaloacetate production to sustain the TCA.
Figure 1. Carbon flux distribution in S. coelicolor M145.

Grey arrows represent carbon fluxes within the central metabolism of S. coelicolor M145. Arrow widths are proportional to carbon fluxes. Flux values in the central metabolism are included in boxes with solid lines, flux values to biomass are included in boxes with dotted lines. The upper numbers represent average actual fluxes (μmol (g dry mass)−1 h−1), the lower number represent average normalized fluxes (μmol (μmol glucose)−1). Positive values correspond to consumption and negative values to production.
Figure 2. Carbon flux distribution in S. coelicolor M1146.

Grey arrows represent carbon fluxes within the central metabolism of S. coelicolor M1146. Arrow widths are proportional to carbon fluxes. Flux values in the central metabolism are included in boxes with solid lines, flux values to biomass are included in boxes with dotted lines. The upper numbers represent average actual fluxes (μmol (g dry mass)−1 h−1), the lower number represent average normalized fluxes (μmol (μmol glucose)−1). Positive values correspond to consumption and negative values to production. Boxes with a dark grey shading indicate values significantly higher in M1146 than in M145. Boxes with a light grey shading indicate values significantly lower in M1146 than in M145.
Summing anabolic and catabolic fluxes for CO2 gave the total calculated CO2 evolution rate (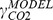 ) (Fig. 1, Fig. 2, right hand side). Our calculated fluxes may be compared to experimental values (Table 4, last line). There was good agreement between them, with a difference of 3.8±1% (M1146) and 2±1% (M145) only. Similarly, the total O2 consumption rate (
) (Fig. 1, Fig. 2, right hand side). Our calculated fluxes may be compared to experimental values (Table 4, last line). There was good agreement between them, with a difference of 3.8±1% (M1146) and 2±1% (M145) only. Similarly, the total O2 consumption rate (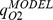 ) was computed from calculated fluxes and matched experimental qO2 values very well (difference of less than 2.7±0.5%). Experimental and computed CO2 and O2 exchange rates gave respiratory quotients (γCO2/qO2) of 0.982±0.002 for both strains, as would be expected during aerobic growth.
) was computed from calculated fluxes and matched experimental qO2 values very well (difference of less than 2.7±0.5%). Experimental and computed CO2 and O2 exchange rates gave respiratory quotients (γCO2/qO2) of 0.982±0.002 for both strains, as would be expected during aerobic growth.
Energy and redox balance
NADPH production was similar in both strains (Fig. 1, Fig. 2, right hand side). However, NADPH requirement for biomass biosynthesis was significantly higher for M1146 while in both, NADPH production exceeded needs. The production of NADH from catabolism and anabolism was lower (30±3%) in M1146 because the TCA was less active in that strain (see section Discussion 4). In M1146, the production of ATP from catabolism was lower (29±2%) than in M145 for the same reason. Contrary to M145, the production of ATP from catabolism was insufficient to cover ATP needs for biomass synthesis in M1146 (which are 13±0.1% higher than in M145, see section 4.3.). Nevertheless, a NADH-to-ATP conversion efficiency of 0.25 by the respiratory chain in M1146 would still cover ATP needs in that strain.
Discussion
Carbon fluxes in the central metabolism of S. coelicolor
The aim of this work was to reconstruct a comprehensive model of carbon flux distribution of primary carbon metabolism in S. coelicolor strains M145 and M1146, so as to better understand the consequences of the absence of antibiotic biosynthesis on central carbon metabolism. We used here both Metabolic Flux Analysis and 13C Metabolic Flux Analysis to solve fluxes across central carbon metabolism. Our analysis took advantage of published and experimental data on composition and reaction stoichiometries. As stated in section Results 4, this model seems valid since observed respiration rates (qO2 and γCO2 values, Table 4) matched computed CO2 release and O2 consumption, with a respiratory quotient of nearly 1.
Nevertheless, Metabolic Flux Analysis is usually viewed as unlikely to give a completely faithful representation of the actual flux distribution, and consideration of areas in which thecarbon flux-balance model diverges from known metabolic behaviour may be potentially more informative. For example, the oxidative reactions of the oxidative pentose phosphate pathway typically carry no flux in flux-balance solutions of heterotrophic metabolism in plants [91], [92]. This discrepancy arises because mass-balance on NADPH can model the provision of NADPH by other possible pathways such as, for exemple, isocitrate dehydrogenase. Here, isotopic labeling with [13C-1] glucose was carried out to infer the metabolic commitment to the PPP, which is a source of NADPH. There was a very clear 13C-enrichment in TCA-derived amino acids (m 1 and m 2 much larger that they should be at natural 13C-abundance), allowing to quantify reliably the 13C-commitment to glycolysis and the TCA versus the PPP.
Using this approach, we determined a contribution of PPP to glucose catabolism in S. coelicolor M145 (22±0.3%, Fig. 1) close to that reported by Naiempoor and Mavituna (25.1%) for S. coelicolor during actinohodin production when the growth rate (μ) was near 0.04 h−1 [18].
In M1146, our estimate of the PPP contribution is 38±0.2% (Fig. 2), similar to that found at similar growth rates (μ near 0.04 h−1) in S. lividans (35%) during actinorhodin production [93], Streptomyces noursei (33%) during nystatin production [94] as well as Streptomyces C5 (30%) during ε-rhodomycinone production [39].
We postulate that the higher flux through the PPP in M1146 is the consequence of a higher NADPH demand to sustain both a higher growth rate (as previously suggested for S. coelicolor [93], [94]) and an increased triacylglycerol biosynthesis. Parenthetically, the higher NADPH demand might also contribute to coping with oxidative stress (see Discussion 4).
Reductive power balance
It is believed that NADPH requirements and production should be balanced as cell metabolism reaches the steady state [95]. Here, in both strains, NADPH production exceeded NADPH needs (Fig. 1, Fig. 2, right hand side). If the NADPH content were steady, then the NADPH excess would have to be oxidized to NADP. In S. coelicolor, this might involve a nicotinamide nucleotide transhydrogenase, since our assays showed a thio-NAD reduction at the expense of NADPH. In their studies on actinorhodin production in an hyper-producing strain of S. lividans, Bruheim et al. [96] also suggested the involvement nicotinamide nucleotide transhydrogenase to balance excess NADPH production under conditions in which actinorhodin was synthesized.
Similarly, NADH production was larger than NADH needs and we assumed that this was compensated for by supplemental NADH oxidation through the respiratory chain. Other metabolic reactions that oxidise NADH might have also occurred, such as L-lactate dehydrogenase or alcohol dehydrogenase. However, such enzymatic activities are not likely since bacterial metabolism was aerobic under our conditions. In addition, we found no L-lactate and no ethanol at all in our experiments (data not shown).
ATP needs, mATP and P/O
In strain M145, ATP generated by catabolism (4.000±0.043 µmol ATP (μmol glucose)−1) would cover ATP needs for anabolism (3.927±0.052 µmol ATP (μmol glucose)−1). To the contrary, an apparent ATP deficit (1.118±0.013 µmol ATP (μmol glucose)−1) would exist in strain M1146.
For M145 and M1146, 5.643±0.113 µmol NADH (μmol glucose)−1 and 4.453±0.089 µmol NADH (μmol glucose)−1 were generated in excess by metabolism and from putative NADPH transhydrogenation, respectively.
As the carbon balance do not close, we are aware that the calculation of ATP needs for anabolism may be affected if ATP is required for biosynthesis of the unidentified metabolites. This should not be the case for NADH, as NADH needs for anabolism are negligible (Table 3).
The amount of ATP generated by NADH reoxidation by the respiratory chain obviously depends upon the P/O ratio. Here, even a P/O ratio as low as 0.25 would have been sufficient to cover ATP needs in M1146.
If P/O ratios larger than 0.25 were taken into account, both the oxidation of total NADH to NAD by the respiratory chain and intrinsic metabolic phosphorylation (pyruvate kinase, etc.) would have produced an apparent ATP excess in both strains.
This apparent ATP excess, which is certainly balanced by ATP consumption for cell maintenance, may be denoted as mATP. mATP represents the energy demand for essential, non-growth-related processes, including maintenance of ionic potential across the cytoplasmic membrane, metabolite transport, cells homeostasis, macromolecules turn-over and others processes [82]. Since 80% of the actinorhodin pool in M145 were found in the extracellular compartment, a substantial part of mATP was probably dedicated to its excretion.
Calculated mATP strongly depends upon the P/O value used and in practice, the P/O value may vary substantially depending on growth conditions [97]. However, the P/O ratio chosen here has no consequence on the carbon flux distribution found in the present study, that is, any value of the P/O ratio above 0.25 would yield the same carbon flux pattern.
Here, taking into account a P/O value of 2, mATP values (on a biomass basis) would be 7.545±0.3 and 4.482±0.2 mmol ATP (g cell dry weight)−1 h−1 in M145 and M1146, respectively, that is, fell well within the range (4.52–9.64 mmol ATP (g cell dry weight)−1 h−1) of those found in various bacteria [98]–[102] and S. coelicolor [71] grown on glucose as a carbon source.
A standard way to estimate maintenance energy is to plot specific glucose uptake rate (mmol glucose (g cell dry weight)−1 h−1) as a function of the dilution rate D (h−1) in a chemostat [103], the y intercept being the maintenance energy value [104]. From consideration of published dilution rates [10], [19], [71], [93], [105], one may calculate qS values in Streptomyces and a value of 0.425±0.05 mmol glucose (g cell dry weight)−1 h−1 is found. Using our own experimental data, maintenance energy should correspond to 0.498±0.019 mmol glucose (g cell dry weight)−1 h−1 in M145 and 0.341±0.015 mmol glucose (g cell dry weight)−1 h−1 in M1146. Even though such values agree reasonably well with the average value from literature, an experimental determination of energy maintenance under our conditions remains to be determined.
Alterations of metabolic fluxes in M1146 and their consequences
Since M145 produced solely actinorhodin from acetyl-CoA during its growth under our conditions, the genetic deletion of the cluster for actinorhodin biosynthesis in M1146 expectedly resulted in visible metabolic changes (Fig. 1, Fig. 2). Since acetyl-CoA is the main precursor of actinorhodin, acetyl-CoA accumulation could have been postulated in M1146. Nevertheless, acetyl-CoA accumulation is unlikely to occur for a number of biochemical [106], physiological [107], [108] and thermodynamical reasons.
Other evidences for the absence of acetyl-CoA accumulation came from the excretion of larger amounts of pyruvate in M1146 (167±11 µmol (g cell dry weight)−1 h−1) than in M145 (21±1 µmol (g cell dry weight)−1 h−1) during the stationary phase. In the absence of growth, acetyl-CoA could not be redirected to biomass in M1146 and accumulation was simply avoided by excreting its direct, freely diffusible precursor pyruvate.
During the exponential growth of M145, 80.4±1 µmol acetyl-CoA (g cell dry weight)−1 h−1 are dedicated to biomass production and 32.5±1 µmol acetyl-CoA (g cell dry weight)−1 h−1 to actinorhodin production. In M1146, with no other modification in glucose uptake and carbon flux distribution, a complete redirection of 32.5±1 µmol acetyl-CoA to biomass would result in a 40% increase of the specific growth rate (0.0552 h−1), a 40% increase of the anaplerotic flux and a decrease of qO2 (1,581 µmol (g cell dry weight)−1 h−1) and γCO2 (1,534 µmol (g cell dry weight)−1 h−1) values in M1146.
However, the specific growth rate of M1146 was only 13±0.1% higher than that of M145 and the rate of glucose consumption was 13±0.5% lower. In M1146, there was therefore a slightly lower flux to acetyl-CoA synthesis due to the lower glucose consumption rate and as a result, the flux through the TCA and ATP production were lower (Fig. 2). Accordingly, qO2 (1,475±0.05 µmol (g cell dry weight)−1 h−1) and γCO2 (1,448±0.05 µmol (g cell dry weight)−1 h−1) values were also lower.
The reasons of the limited increase in M1146 growth rate are not straightforward since S. coelicolor could grow with μ values up to 0.16 h−1, a high qS value (2 mmol (g cell dry weight)−1 h−1) and a low actinorhodin production rate (γACT = 6 nmol (g cell dry weight)−1 h−1) [19]. Three mechanisms at the origin of growth restriction are plausible:
First, in Streptomyces peuceticus var. caesius and S. coelicolor, a transcriptional regulation of glucose uptake rate has been demonstrated [36], [109], [110]. In M1146, a limitation of glucose entry mediated by an as yet unidentified regulatory protein (with acetyl-CoA or triacylglycerol as co-regulators) might have occurred.
Second, the absence of actinorhodin synthesis in M1146 might also cause growth limitation as a side effect. Indeed, quite recently, Shin et al. [111] demonstrated that the extracellular pool of actinorhodin was involved in the activation of the SoxR regulon in S. coelicolor. This regulon encodes proteins involved in the transport or the turn-over of small redox-active toxic compounds [112]. As described in E. coli by Fridovitch [113], such compounds might alter catalytic [4Fe-4S] centers of various dehydratases such as fumarase and aconitase in the TCA, or the dihydroxyacid dehydratase involved in the synthesis of abundant amino acids (Ile, Leu and Val), pantothenic acid and coenzyme A. Thus, under 70% of dissolved oxygen, the lack of actinorhodin production in M1146 might have lead to an increased intracellular oxidative stress, which in turn impeded the TCA, ATP synthesis, biomass synthesis and glucose consumption. The involvement of oxidative stress would be consistent with the increase in NADPH production by the PPP in M1146 (see Discussion 2).
Third, the carbon metabolism in M1146 partly compensated for the reduced growth rate (lower μ) and antibiotic synthesis by accumulating neutral lipidic compounds (triacylglycerol, see Results 1). In a similar molecular context, that is, in the bldA mutant of S. coelicolor unable to produce actinorhodin and undecylprodigiosin, Plaskitt and Chater [114] also demonstrated a higher triacylglycerol content. In fact, there seems to be a compromise between antibiotic and triacyglycerol production since (i) a higher actinorhodin production has been observed in the sco0958 mutant impaired in triacylglycerol synthesis [115] and (ii) the simultaneous deletion of the gene clusters encoding 5-polyketide synthase and addition of a plasmid comprising the gene cluster encoding actinorhodin biosynthesis, resulted in a dramatic increase of actinorhodin production in S. coelicolor [116].
M1146 as a super-host
M1146 was successfully used as a super-host for the production of actinorhodin, congocidin, chloramphenicol and caprazamycin after introduction of the corresponding gene clusters [10], [11]. However, the use of M1146 did not allow the efficient production of clorobiocin and coumermycin A1 [10].
The metabolic pathways associated with congocidin [117], chloramphenicol [118], caprazamycin [119], clorobiocin and coumermycin A1 production [120], [121] have all been elucidated. Precursor metabolites for the production of congocidin (3-phospho-glycerate, N-acetyl-glucosamine, sedoheptulose-7-phosphate and cytosine), chloramphenicol (shikimic acid, dichloro-acetyl-CoA), and caprazamycin (uridine, glycine, S-adenosyl-methionine, 3-methyl glutaryl-CoA, fatty acyl-CoA, per-methylated rhamnose) are all derived from glucose-1-phosphate, molecules from the pentose phosphate pathway, 3-phospho-glycerate and acetyl-CoA.
In the case of clorobiocin and coumermycin A1, precursors are, in that order of incorporation, prephenate (derived from both erythrose-4-phosphate and phosphoenolpyruvate), noviose (derived from glucose-1-phosphate) and 3- or 5-methyl-pyrrole-2-carboxylic acids. These pyrrole groups originate from threonine (precursor oxaloacetate) or proline (precursor α-ketoglutarate). Flinspach et al. [10] have clearly shown that clorobiocin and coumermycin A1 production was unsuccessfull in M1146, the main products beeing aminocoumarin derivatives without any pyrrole group or not the expected ones.
Although we are aware of possible pleiotropic effects due to the introduction of foreign genes in a given genetic background, these results seem consistent with our carbon flux description in M1146, i.e., an enhanced flux through PPP, a moderately decreased glycolytic flux to acetyl-CoA, an increased acetyl-CoA extraction for triacylglycerol synthesis and a lower flux in the TCA. In clorobiocin and coumermycin A1 biosyntheses, the consumption of erythrose-4-phosphate and phosphoenolpyruvate may exacerbate the flux impairement through the TCA, leading to the impossibility to produce a sufficient pyrrole content.
Conclusion and perspectives
In this paper, we demonstrated that combining Metabolic Flux Analysis and 13C Metabolic Flux Analysis yielded an accurate description of carbon fluxes in S. coelicolor under steady-state conditions. Our work further suggested the key role of nicotinamide nucleotide transhydrogenase for redox homeostasis in S. coelicolor.
The main metabolic difference between M145 and M1146 consisted in an enhanced flux through the PPP in M1146, likely to face higher needs for NADPH in this strain. The metabolic differences between M145 and M1146 also highlighted the energetic cost for actinorhodin production and excretion in M145.
In other words, there would be a metabolic trade-off between instantaneous growth and the production of antibacterial compounds, which are likely to provide a competitive advantage [122] or to be involved in quorum sensing [112], [123], [124]. In other words, antibiotics are believed to help to defend the food source and to face bacterial competition in natural environments.
Our results also suggest a plausible competition between polyketide biosynthetic pathways for their common precursor, acetyl-CoA. Therefore, the deletion of polyketide antibiotic pathways might be instrumental in increasing acetyl-CoA availability and thus triacylglycerol, which is an important source of renewable oleochemicals. Conversely, the impairement of triacylglycerol synthesis might be of interest to improve polyketide antibiotics production yield.
In Streptomyces, antibiotics are usually produced in small amounts at the transition phase when bacterial growth slows down as a result of nutrient exhaustion, that is, not in steady-state conditions. New rapid sampling techniques [125], new modelling approaches and highly sensitive mass spectrometers make possible to carry out non-steady 13C Metabolic Flux Analysis [126], [127]. Together with our knowledge of S. coelicolor metabolism under steady-state conditions, it is now possible to study metabolic carbon fluxes during non-exponential growth stages. This will offer a better insight into the understanding of secondary metabolite biosyntheses under such conditions.
Supporting Information
Atom carbon transitions.
(DOC)
Additional GC-MS analyses.
(DOC)
Supplementary Material and Methods.
(DOC)
Abbreviations and symbols.
(DOC)
The metabolic network of S. coelicolor.
(DOC)
Acknowledgments
The S. coelicolor strains came from the John Innes Centre (Norwich) and were a gift from MJ Bibb and JP Gomez-Escribano. The authors would also like to thank Monique Bolotin Fukuhara for her investment and continuous encouragement from the beginning of this work, MaximeVergne for FITRS analyses, Sylvie Lautru for additional HPLC analyses and Michaël Dubow for proofreading of the manuscript.
Funding Statement
Fabien Coze was supported by a grant (Grant ABG-25680/BDI SDV24) from the French National Center for Scientific Research (http://www.cnrs.fr/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hawkey PM, Jones AM (2009) The changing epidemiology of resistance. J Antimicrob Chemother 64 : suppl. 1, i3–i10. [DOI] [PubMed] [Google Scholar]
- 2. Mahajan GB, Balachandran L (2012) Antibacterial agents from actinomycetes-a review. Front Biosci 1: 240–253. [DOI] [PubMed] [Google Scholar]
- 3. Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, et al. (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2). Nature 417: 141–147. [DOI] [PubMed] [Google Scholar]
- 4. Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, et al. (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis . Nature Biotechnol 21: 526–531. [DOI] [PubMed] [Google Scholar]
- 5. Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, et al. (2008) Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol 190: 4050–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bibb M, Hesketh A (2009) Analyzing the regulation of antibiotic production in Streptomycetes. Methods Enzymol 458: 93–116. [DOI] [PubMed] [Google Scholar]
- 7. Nieselt K, Battke F, Herbig A, Bruheim P Wentzel A, et al. (2010) The dynamic architecture of the metabolic switch in Streptomyces coelicolor . BMC Genomics 11: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas L, Hogson DA, Wentzel A, Nieselt K, Ellingsen TE, et al. (2012) Metabolic switches and adaptations deduced from the proteomes of Streptomyces coelicolor wild type and phoP mutant grown in batch culture. Mol Cell Proteomics 11: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wright LF, Hopwood DA (1976) Actinorhodin is a chromosomally-determined antibiotic in Streptomyces coelicolor A3 (2). J Gen Microbiol 96: 289–297. [DOI] [PubMed] [Google Scholar]
- 10. Flinspach K, Westrich L, Kaysser S, Gomez-Escribano JP, Bibb M, et al. (2010) Heterologous expression of the biosynthetic gene clusters of coumermycin A(1), clorobiocin and caprazamycins in genetically modified Streptomyces coelicolor strains. Biopolymers 93: 823–832. [DOI] [PubMed] [Google Scholar]
- 11. Gomez-Escribano JP, Bibb MJ (2011) Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb Biotechnol 4: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jones AC, Ottilie S, Eustáquio AS, Edwards DJ, Gerwick L, et al. (2012) Evaluation of Streptomyces coelicolor A3 (2) as a heterologous expression host for the cyanobacterial protein kinase C activator lyngbyatoxin A. FEBS J 279: 1243–1251. [DOI] [PubMed] [Google Scholar]
- 13. Kim HB, Smith CP, Micklefield J, Mavituna F (2004) Metabolic flux analysis for calcium dependent antibiotic (CDA) production in Streptomyces coelicolor . Metab Eng 6: 313–325. [DOI] [PubMed] [Google Scholar]
- 14. Bibb M (1996) 1995 Colworth Prize Lecture. The regulation of antibiotic production in Streptomyces coelicolor A3 (2). Microbiology 142: 1335–1344. [DOI] [PubMed] [Google Scholar]
- 15. Hobbs G, Obanye AIC, Petiy J, Mason JC, Barratt E, et al. (1992) An integrated approach to studying regulation of production of the antibiotic methylenomycin by Streptomyces coelicolor A3 (2). J Bacteriol 174: 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alam MT, Merlo ME, The STREAM Consortium, Hodgson DA, Wellington EMH, et al. (2010) Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor . BMC Genomics 11: 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doull JL, Vining LC (1990) Nutritional control of actinorhodin production by Streptomyces coelicolor A3 (2): suppressive effects of nitrogen and phosphate. Appl Microbiol Biotechnol 32: 449–454. [DOI] [PubMed] [Google Scholar]
- 18. Naeimpoor F, Mavituna F (2000) Metabolic flux analysis in Streptomyces coelicolor under various nutrient limitations. Metab Eng 2: 140–148. [DOI] [PubMed] [Google Scholar]
- 19. Melzoch K, Teixeira de Mattos MJ, Neijssel OM (1997) Production of Actinorhodin by Streptomyces coelicolor A3 (2) grown in chemostat culture. Biotechnol Bioeng 54: 577–582. [DOI] [PubMed] [Google Scholar]
- 20. Holms WH (1986) The central metabolic pathways of Escherichia coli: relationship between flux and control at a branch point, efficiency of conversion to biomass, and excretion of acetate. Curr Top Cell Regul 28: 69–105. [DOI] [PubMed] [Google Scholar]
- 21. Egli T, Fiechter A (1981) Theoretical Analysis of Media Used in the Growth of Yeasts on Methanol. Microbiology 123: 365–369. [Google Scholar]
- 22.Ingraham JL, Maaløe O, Neidhardt FC (1983) Growth of the bacterial cell. Sinauer Associates, Inc. 435 p. [Google Scholar]
- 23.Norris JR, Ribbons DW (1970) Methods in Microbiology. Academic Press, Inc. 506 p. [Google Scholar]
- 24. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 25.Christiansen P (1970) Isolierung, Strukturmittlung und absolute konfiguration neuer actinorhodine [Doctoral dissertation]. Universität Göttingen, Germany.
- 26. Tsao SW, Rudd BAM, He XG, Chang CJ, Floss HG (1985) Identification of a red pigment from Streptomyces coelicolor A3 (2) as a mixture of prodigin derivatives. J Antibiot 38: 128–131. [DOI] [PubMed] [Google Scholar]
- 27. Lautru S, Oves-Costales D, Pernodet JL, Challis G (2007) MbtH-like protein-mediated cross-talk between non-ribosomal peptide antibiotic and siderophore biosynthetic pathways in Streptomyces coelicolor M145. Microbiology 153: 1405–1412. [DOI] [PubMed] [Google Scholar]
- 28. Verleyen T, Verhe R, Cano A, Huyghebaertet A, De Greyt W (2001) Influence of triacylglycerol characterics on the determination of free fatty acids in vegetable oils by Fourier transform infrared spectroscopy. J Amer Oil Chem Society 78: 981–984. [Google Scholar]
- 29. Hong K, Sun S, Tian W, Chenet GQ, Huang W (1999) A rapid method for detecting bacterial polyhydroxyalkanoates in intact cells by Fourier transform infrared spectroscopy. Appl Microbiol Biotechnol 51: 523–526. [Google Scholar]
- 30. Sugimoto S, Shiio I (1987a) Regulation of glucose-6-phosphate dehydrogenase in Brevibacterium flavum . Agric Biol Chem 51: 101–108. [Google Scholar]
- 31. Sugimoto S, Shiio I (1987b) Regulation of 6-phosphogluconate dehydrogenase in Brevibacterium flavum . Agric Biol Chem 51: 1257–1263. [Google Scholar]
- 32. Nachlas MM, Davidson MB, Goldberg JD, Seligman AM (1963) Colorimetric method for the measurement of isocitric dehydrogenase activity. J Lab Clin Med 62: 148–158. [PubMed] [Google Scholar]
- 33. Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E (2004) The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli . J Biol Chem 279: 6613–6619. [DOI] [PubMed] [Google Scholar]
- 34. Zamboni N, Fendt SM, Rühl M, Sauer U (2009) 13C-based metabolic flux analysis. Nature Protocols 4: 878–892. [DOI] [PubMed] [Google Scholar]
- 35. Ikeda H, Seno ET, Bruton CJ, Chater KF (1984) Genetic mapping, cloning and physiological aspects of the glucose kinase gene of Streptomyces coelicolor . Mol Gen Genet 196: 501–507. [DOI] [PubMed] [Google Scholar]
- 36. van Wezel GP, Mahr K, König M, Traag BA, Pimentel-Schmitt EF, et al. (2005) GlcP constitutes the major glucose uptake system of Streptomyces coelicolor A3 (2). Mol Microbiol 55: 624–636. [DOI] [PubMed] [Google Scholar]
- 37. van Wezel GP, König M, Mahr K, Nothaft H, Thomae AW, et al. (2007) A new piece of an old jigsaw: glucose kinase is activated posttranslationally in a glucose transport-dependent manner in Streptomyces coelicolor A3(2). J Mol Microbiol Biotechnol 12: 67–74. [DOI] [PubMed] [Google Scholar]
- 38. Dekleva ML, Strohl WR (1988a) Activity of phosphoenolpyruvate carboxylase of an anthracycline-producing Streptomycete. Can J Microbiol 34: 1241–1246. [DOI] [PubMed] [Google Scholar]
- 39. Dekleva ML, Strohl WR (1988b) Biosynthesis of epsilon-rhodomycinone from glucose by Streptomyces C5 and comparison with intermediary metabolism of other polyketide producing Streptomycetes. Can J Microbiol 34: 1235–1240. [DOI] [PubMed] [Google Scholar]
- 40. Salas JA, Quirós LM, Hardisson C (1984) Pathways of glucose catabolism during germination of Streptomyces spores. FEMS Microbiol Lett 22: 229–233. [Google Scholar]
- 41. Borodina I, Siebring J, Zhang J, Smith CP, van Keulen G, et al. (2008) Antibiotic overproduction in Streptomyces coelicolor A3 (2) mediated by phosphofructokinase deletion. J Biol Chem 283: 25186–25199. [DOI] [PubMed] [Google Scholar]
- 42. Hodgson DA (2000) Primary metabolism and its control in Streptomycetes: a most unusual group of bacteria. Adv Microb Physiol 42: 47–238. [DOI] [PubMed] [Google Scholar]
- 43. Bramwell H, Nimmo HG, Hunter IS, Coggins JR (1993) Phosphoenolpyruvate carboxylase from Streptomyces coelicolor A3 (2): purification of the enzyme, cloning of the ppc gene and over-expression of the protein in a Streptomycete. Biochem J 293: 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niederpruem DJ, Hackett DP (1961) Respiratory chain of Streptomyces . J Bacteriol 81: 557–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholls DG, Fergusson SJ (2002) Bioenergetics 3. Academic Press. 297 p. [Google Scholar]
- 46. Calhoun MW, Oden KL, Gennis RB, de Mattos MJ, Neijssel OM (1993) Energetic efficiency of Escherichia coli: effects of mutations in components of the aerobic respiratory chain. J Bacteriol 175: 3020–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kana BD, Weinstein EA, Avarbock D, Dawes SS, Rubin H, et al. (2001) Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis . J Bacteriol 183: 7076–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bott M, Niebisch A (2003) The respiratory chain of Corynebacterium glutamicum . J Biotechnol 104: 129–153. [DOI] [PubMed] [Google Scholar]
- 49.Bailey JE, Ollis DF (1986) Biochemical Engineering Fundamentals. McGraw Hill, Inc. 984 p. [Google Scholar]
- 50. Barreteau H, Magnet S, El Ghachi M, Touze T, Arthur M, et al. (2009) Quantitative high performance liquid chromatography analysis of the pool levels of undecaprenyl phosphate and its derivatives in bacterial membranes. J Chromatogr B Analyt Technol Biomed Life Sci 877: 213–220. [DOI] [PubMed] [Google Scholar]
- 51. Denoya CD, Fedechko RW, Hafner EW, McArthur HA, Morgenstern MR, et al. (1995) A second branched-chain alpha-keto acid dehydrogenase gene cluster (bkdFGH) from Streptomyces avermitilis: its relationship to avermectin biosynthesis and the construction of a bkdF mutant suitable for the production of novel antiparasitic avermectins. J Bacteriol 177: 3504–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fischer M, Bacher A (2008) Biosynthesis of vitamin B2: Structure and mechanism of riboflavin synthase. Arch Biochem Biophys 474: 252–265. [DOI] [PubMed] [Google Scholar]
- 53.Gottschalk G (1986) Bacterial Metabolism. Springer-Verlag. 359 p. [Google Scholar]
- 54. Kaneda T (1991) Iso- and anteiso-fatty acids in bacteria: biosynthesis, function, and taxonomic significance. Microbiol Rev 55: 288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuzuyama T (2002) Mevalonate and nonmevalonate pathways for the biosynthesis of isoprene units. Biosci Biotechnol Biochem 66: 1619–1627. [DOI] [PubMed] [Google Scholar]
- 56. Matsuhashi M, Dietrich CP, Strominger JL (1947) Biosynthesis of the peptidoglycan of bacterial cell walls. III. The role of soluble ribonucleic acid and of lipid intermediates in incorporation in Staphylococcus aureus . J Biol Chem 242: 3191–3206. [Google Scholar]
- 57.Neidhardt FC, Ingraham JL, Brooks Low K, Magasanik B, Schaechter M, et al. (1987) Escherichia coli and Salmonella: Cellular and molecular biology, vol 1, American Society for Microbiology. 797 p. [Google Scholar]
- 58.Nelson DL, Cox MM (2004) Lehninger Principles of Biochemistry. WH.Freeman and Company. 1119 p. [Google Scholar]
- 59.Stanier RY, Ingraham JL, Wheelis ML, Painter PR (1986) The Microbial World. Prentice-Hall. 689 p. [Google Scholar]
- 60.Stryer L (1988) Biochemistry. W H Freeman and Company. 1136 p. [Google Scholar]
- 61. Zhu K, Bayles DO, Xiong A, Jayaswal RK, Wilkinson BJ (2005) Precursor and temperature modulation of fatty acid composition and growth of Listeria monocytogenes cold-sensitive mutants with transposon-interrupted branched-chain alpha-keto acid dehydrogenase. Microbiology 151: 615–623. [DOI] [PubMed] [Google Scholar]
- 62. Das A, Khosla C (2009) Biosynthesis of aromatic polyketides in bacteria. Acc Chem Res 42: 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Finking R, Marahiel MA (2004) Biosynthesis of nonribosomal peptides 1. Annu Rev Microbiol 58: 453–488. [DOI] [PubMed] [Google Scholar]
- 64. Mo S, Sydor PK, Corre C, Alhamadsheh MM, Stanley AE, et al. (2008) Elucidation of the Streptomyces coelicolor pathway to 2-undecylpyrrole, a key intermediate in undecylprodiginine and streptorubin B biosynthesis. Chem Biol 15: 137–148. [DOI] [PubMed] [Google Scholar]
- 65. Moran GR (2005) 4-Hydroxyphenylpyruvate dioxygenase. Arch Biochem Biophys 433: 117–128. [DOI] [PubMed] [Google Scholar]
- 66. Valton J, Filisetti L, Fontecave M, Niviere V (2004) A two-component flavin-dependent monooxygenase involved in actinorhodin biosynthesis in Streptomyces coelicolor . J Biol Chem 279: 44362–44369. [DOI] [PubMed] [Google Scholar]
- 67. Valton J, Fontecave M, Douki T, Kendrew SG, Niviere V (2006) An aromatic hydroxylation reaction catalyzed by a two-component FMN-dependent monooxygenase. The ActVA-ActVB system from Streptomyces coelicolor . J Biol Chem 281: 27–35. [DOI] [PubMed] [Google Scholar]
- 68. Valton J, Mathevon C, Fontecave M, Niviere V, Ballou DP (2008) Mechanism and regulation of the Two-component FMN-dependent monooxygenase ActVA-ActVB from Streptomyces coelicolor . J Biol Chem 283: 10287–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Williamson NR, Fineran PC, Leeper FJ, Salmond GP (2006) The biosynthesis and regulation of bacterial prodiginines. Nat Rev Microbiol 4: 887–899. [DOI] [PubMed] [Google Scholar]
- 70. Worthington AS, Burkart MD (2007) The old is new again: asparagine oxidation in calcium-dependent antibiotic biosynthesis. ACS Chem Biol 2: 152–154. [DOI] [PubMed] [Google Scholar]
- 71. Borodina I, Krabben P, Nielsen J (2005) Genome-scale analysis of Streptomyces coelicolor A3 (2) metabolism. Genome Res 15: 820–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Antoniewicz MR, Kelleher JK, Stephanopoulos G (2007) Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab Eng 9: 68–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Antoniewicz MR, Stephanopoulos G, Kelleher JK (2006) Evaluation of regression models in metabolic physiology: predicting fluxes from isotopic data without knowledge of the pathway. Metabolomics 2: 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rollin C, Morgant V, Guyonvarch A, Guerquin-Kern JL (1995) 13C-NMR studies of Corynebacterium melassecola metabolic pathways. Eur J Biochem 227: 488–493. [DOI] [PubMed] [Google Scholar]
- 75. Matsubara H, Sasaki RM (1969) High recovery of tryptophan from acid hydrolysates of proteins. Bioch Biophys Res Comm 35: 175–181. [DOI] [PubMed] [Google Scholar]
- 76. Antoniewicz MR, Kelleher JK, Stephanopoulos G (2006) Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab Eng 8: 324–337. [DOI] [PubMed] [Google Scholar]
- 77. Olukoshi ER, Packter NM (1994) Importance of stored triacylglycerols in Streptomyces: possible carbon source for antibiotics. Microbiology. 140: 931–943. [DOI] [PubMed] [Google Scholar]
- 78. Roubos JA, Krabben P, de Laat WT, Babuska R, Heijnen JJ (2002) Clavulanic acid degradation in Streptomyces clavuligerus fed-batch cultivations. Biotechnol Prog 18: 451–457. [DOI] [PubMed] [Google Scholar]
- 79. Shahab N, Flett F, Oliver SG, Butler PR (1996) Growth rate control of protein and nucleic acid content in Streptomyces coelicolor A3 (2) and Escherichia coli B/r. Microbiology 142: 1927–1935. [DOI] [PubMed] [Google Scholar]
- 80. Zaretskaia M, Polin AN (1987) Polysaccharide composition of the cell wall of Streptomyces antibioticus RIA-594 (39), a producer of the antibiotic oleandomycin. Antibiot Med Biotekhnol 32: 529–533. [PubMed] [Google Scholar]
- 81. Daae EB, Ison AP (1999) Classification and sensitivity analysis of a proposed primary metabolic reaction network for Streptomyces lividans . Metab Eng 1: 153–165. [DOI] [PubMed] [Google Scholar]
- 82.Neidhardt FC, Ingraham JL, Schaechter M (1990) Physiology of the bacterial cell: a molecular approach. Sinauer Associates Inc. 506 p. [Google Scholar]
- 83. Lange HC, Heijnen JJ (2001) Statistical reconciliation of the elemental and molecular biomass composition of Saccharomyces cerevisiae . Biotechnol Bioeng 75: 334–344. [DOI] [PubMed] [Google Scholar]
- 84. Bushell ME, Fryday A (1983) The application of materials balancing to the characterization of sequential secondary metabolite formation in Streptomyces cattleya NRRL 8057. J Gen Microbiol 129: 1733–1741. [DOI] [PubMed] [Google Scholar]
- 85. Dominguez H, Rollin C, Guyonvarch A, Guerquin-Kern JL, Cocaign-Bousquet M, et al. (1998) Carbon-flux distribution in the central metabolic pathways of Corynebacterium glutamicum during growth on fructose. Eur J Biochem 254: 96–102. [DOI] [PubMed] [Google Scholar]
- 86. Erickson LE, Minkewich IG, Eroshin VK (1979) Utilization of mass-energy balance regularities in the analysis of continuous culture data. Biotechnol Bioeng 21: 575–591. [Google Scholar]
- 87. Roels JA (2009) Application of macroscopic principles to microbial metabolism. Biotechnol Bioeng 103: 2–59 discussion 1. [DOI] [PubMed] [Google Scholar]
- 88. Alves AMCR, Euverink GJW, Hektor HJ, Hessels GI, van der Vlag J, et al. (1994) Enzymes of glucose and methanol metabolism in the Actinomycete Amycolatopsis methanolica . J Bacteriol 176: 6827–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bruheim P, Sletta H, Bibb MJ, White J, Levine DW (2002) High-yield actinorhodin production in fed-batch culture by a Streptomyces lividans strain overexpressing the specific activator gene actII-ORF4. J Indus Microbiol Biotechnol 28: 103–111. [DOI] [PubMed] [Google Scholar]
- 90. Wiechert W, Siefke C, de Graaf AA, Marx A (1997) Bidirectional reaction steps in metabolic networks: II. Flux estimation and statistical analysis. Biotechnol Bioeng 55: 118–135. [DOI] [PubMed] [Google Scholar]
- 91. Hay J, Schwender J (2011) Computational analysis of storage synthesis in developing Brassica napus L. (oilseed rape) embryos: flux variability analysis in relation to 13C metabolic flux analysis. Plant J 67: 513–525. [DOI] [PubMed] [Google Scholar]
- 92. Williams TCR, Poolman MG, Howden AJ, Schwarzlander M, Fell DA, et al. (2010) A genome-scale metabolic model accurately predicts fluxes in central carbon metabolism under stress conditions. Plant Physiol 154: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Avignone Rossa C, White J, Kuiper A, Postma PW, Bibb M, et al. (2002) Carbon flux distribution in antibiotic-producing chemostat cultures of Streptomyces lividans . Metab Eng 4: 138–150. [DOI] [PubMed] [Google Scholar]
- 94. Jonsbu E, Christensen B, Nielsen J (2001) Changes of in vivo fluxes through central metabolic pathways during the production of nystatin by Streptomyces noursei in batch culture. Appl Microbiol Biotechnol 56: 93–100. [DOI] [PubMed] [Google Scholar]
- 95. Fuhrer T, Sauer U (2009) Different biochemical mechanisms ensure network-wide balancing of reducing equivalents in microbial metabolism. J Bacteriol 191: 2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bruheim P, Butler M, Ellingsen TE (2002) A theoretical analysis of the biosynthesis of actinorhodin in a hyper-producing Streptomyces lividans strain cultivated on various carbon sources. Applied Microbiol Biotechnol 58: 735–742. [DOI] [PubMed] [Google Scholar]
- 97. Andersen KB, von Meyenburg K (1980) Are growth rates of Escherichia coli in batch cultures limited by respiration? J Bacteriol 144: 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mulder MM, van der Gulden HM, Postma PW, van Dam K (1986) Energetic consequences of multiple K+ uptake systems in Escherichia coli . Biochim Biophys Acta 851: 223–228. [DOI] [PubMed] [Google Scholar]
- 99. Varma A, Palsson BØ (1994) Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol 60: 3724–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Stouthamer AH, Bettenhaussen CW (1975) Determination of the efficiency of oxidative phosphorylation in continuous cultures of Aerobacter aerogenes . Arch Microbiol 102: 187–192. [DOI] [PubMed] [Google Scholar]
- 101. Russel JB, Baldwin RL (1979) Comparison of maintenance energy expenditures and growth yields among several rumen bacteria grown on continuous culture. Appl Environ Microbiol 37: 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vallino JJ, Stephanopoulos G (1993) Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol Bioeng 41: 633–646. [DOI] [PubMed] [Google Scholar]
- 103. Schulze KL, Lipe RS (1964) Relationship between substrate concentration, growth rate, and respiration rate of Escherichia coli in continuous culture. Arch Mikrobiol 48: 1–20. [DOI] [PubMed] [Google Scholar]
- 104. Tempest DW, Neijssel OM (1984) The status of YATP and maintenance energy as biologically interpretable phenomena. Annu Rev Microbiol 38: 459–486. [DOI] [PubMed] [Google Scholar]
- 105. Kiviharju K, Moilanen U, Leisola M, Eerikäinen T (2007) A chemostat study of Streptomyces peuceticus var. caesius N47. Appl Microbiol Biotechnol 73: 1267–1274. [DOI] [PubMed] [Google Scholar]
- 106. Nemeria N, Volkov A, Brown A, Yi J, Zipper L, et al. (1998) Systemactic study of the six cysteines of the E1 subunit of the pyruvate dehydrogenase multienzyme complex from Escherichia coli: none is essential for activity. Biochemistry 37: 911–922. [DOI] [PubMed] [Google Scholar]
- 107. Chohnan S, Furukawa H, Fujio T, Nishihara H, Takamura Y (1997) Changes in the size and composition of intracellular pools of nonesterified coenzyme A and coenzyme A thioesters in aerobic and facultatively anaerobic bacteria. Appl Environ Microbiol 63: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lin H, Vadali RV, Bennett GN, San KY (2004) Increasing the acetyl-CoA pool in the presence of overexpressed phosphoenolpyruvate carboxylase or pyruvate carboxylase enhances succinate production in Escherichia coli . Biotechnol Prog 20: 1599–1604. [DOI] [PubMed] [Google Scholar]
- 109. Guzmán S, Carmona A, Escalante L, Imriskova I, López R, et al. (2005) Pleiotropic effect of the SCO2127 gene on the glucose uptake, glucose kinase activity and catabolite repression in Streptomyces peuceticus var. caesius . Microbiology 151: 1717–1723. [DOI] [PubMed] [Google Scholar]
- 110. Ramos I, Guzmán S, Escalante L, Imriskova I, Rodríguez-Sanoja R, et al. (2004) Glucose kinase alone cannot be responsible for carbon source regulation in Streptomyces peuceticus var. caesius . Res Microbiol 155: 267–274. [DOI] [PubMed] [Google Scholar]
- 111. Shin JH, Singh AK, Cheon DJ, Roe JH (2011) Activation of the SoxR regulon in Streptomyces coelicolor by the extracellular form of the pigmented antibiotic actinorhodin. J Bacteriol 193: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dietrich LEP, Teal TK, Price-Whelan A, Newman DK (2008) Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321: 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fridovitch I (1995) Superoxide radical and superoxide dismutases. Annu Rev Biochem 64: 97–112. [DOI] [PubMed] [Google Scholar]
- 114. Plaskitt KA, Chater KF (1995) Influences of developmental genes on localized glycogen deposition in colonies of a mycelial prokaryote, Streptomyces coelicolor A3 (2): A possible interface between metabolism and morphogenesis. Phil Trans R Soc Lond B 347: 105–121. [Google Scholar]
- 115. Arabolaza A, Rodriguez E, Altabe S, Alvarez H, Gramajo H (2008) Multiple pathways for triacylglycerol biosynthesis in Streptomyces coelicolor . Appl Environ Microbiol 74: 2573–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhou M, Jing X, Xie P, Chen W, Wang T, et al. (2012) Sequential deletion of all the polyketide synthase and nonribosomal peptide synthetase biosynthetic gene cluster and a 900-kb subtelomeric sequence of the linear hromosome of Streptomyces coelicolor . FEMS Microbiol Lett 333: 169–179. [DOI] [PubMed] [Google Scholar]
- 117. Lautru S, Song L, Demange L, Lombès T, Galons H, et al. (2012) A sweet origin for the key congocidine precursor 4-acetamidopyrrole-2-carboxylate. Angew Chem Int Ed 51: 1–6. [DOI] [PubMed] [Google Scholar]
- 118. He J, Margavey N, Piraae M, Vining LC (2001) The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non-ribosomal peptide synthetase gene. Microbiology 147: 2817–2829. [DOI] [PubMed] [Google Scholar]
- 119. Kaysser L, Siebenberg S, Kammerer B, Gust B (2010) Analysis of the liposidomycin gene cluster leads to the identification of new caprazamycin derivatives. Chembiochem 11: 191–196. [DOI] [PubMed] [Google Scholar]
- 120. Heide L (2009) The aminocoumarins: biosynthesis and biology. Nat Prod Rep 26: 1241–1250. [DOI] [PubMed] [Google Scholar]
- 121. Siebenberg S, Burkard N, Knuplesch A, Gust B, Grond S, et al. (2011) Two pathways for pyrrole formation in coumermycin A(1) biosynthesis: the central pyrrole moiety is formed from threonine. Chembiochem 12: 2677–2685. [DOI] [PubMed] [Google Scholar]
- 122. Challis GL, Hopwood DA (2003) Synergy and contigency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci USA 100: 14555–14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dietrich LEP, Price-Whelan A, Petersen A, Whiteley M, Newman DK (2006) The phenazine pyocianin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa . Mol Microbiol 61: 1308–1321. [DOI] [PubMed] [Google Scholar]
- 124. Pernodet JL, Alegre MT, Blondelet-Rouault MH, Guérineau M (1993) Resistance to spiramycin in Streptomyces ambofasciens, the producer organism, involves at least two different mechanisms. J Gen Microbiol 139: 1003–1011. [DOI] [PubMed] [Google Scholar]
- 125. Bolten CJ, Kiefer P, Letisse F, Portais JC, Wittmann C (2007) Sampling for metabolome analysis of microorganisms. Anal Chem 79: 3843–3849. [DOI] [PubMed] [Google Scholar]
- 126. Leighty RW, Antoniewicz MR (2011) Dynamic metabolic flux analysis (DMFA): a framework for determining fluxes at metabolic non-steady sate. Metab Eng 13: 745–755. [DOI] [PubMed] [Google Scholar]
- 127. Noh K, Wiechert W (2011) The benefits of being transient: isotope-based metabolic flux analysis at the short time scale. Appl Microbiol Biotechnol 91: 1247–1265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Atom carbon transitions.
(DOC)
Additional GC-MS analyses.
(DOC)
Supplementary Material and Methods.
(DOC)
Abbreviations and symbols.
(DOC)
The metabolic network of S. coelicolor.
(DOC)