

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a common cause of renal failure that is due to mutations in two genes, PKD1 and PKD2. Vascular complications, including aneurysms, are a well recognized feature of ADPKD, and a subgroup of families exhibits traits reminiscent of Marfan syndrome (MFS). MFS is caused by mutations in fibrillin-1 (FBN1), which encodes an extracellular matrix protein with homology to latent TGF-β binding proteins. It was recently demonstrated that fibrillin-1 deficiency is associated with upregulation of TGF-β signaling. We investigated the overlap between ADPKD and MFS by breeding mice with targeted mutations in Pkd1 and Fbn1. Double heterozygotes displayed an exacerbation of the typical Fbn1 heterozygous aortic phenotype. We show that the basis of this genetic interaction results from further upregulation of TGF-β signaling caused by Pkd1 haploinsufficiency. In addition, we demonstrate that loss of PKD1 alone is sufficient to induce a heightened responsiveness to TGF-β. Our data link the interaction of two important diseases to a fundamental signaling pathway.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic form of inherited renal failure worldwide.1 The disorder is caused by mutations in either PKD1 or PKD2, which encode two integral membrane proteins, polycystin-1 and polycystin-2, respectively.2,3 Polycystin-1 is a large nonkinase membrane receptor, whereas polycystin-2 is a calcium permeable, transient receptor potential (TRP)-like channel.1 These two proteins interact via their carboxy-termini and participate in a common signaling pathway that has yet to be clearly elucidated.4,5
Although most patients come to medical attention because of renal disease, ADPKD is a systemic disorder with an array of extrarenal manifestations.6 In particular, ADPKD is associated with an approximately 5-fold increase in the prevalence of intracranial aneurysms.7–10 There are also reports in the literature of patients with aneurysms and dissections of other large arteries, including the aorta.11–15
The connection between ADPKD and these vascular phenotypes has led to the hypothesis that polycystin signaling plays a role in maintaining vascular integrity.16,17 Several lines of experimental data are consistent with this concept. Genetic reporter studies in mice show that Pkd1 is highly expressed throughout the cardiovascular system and all major blood vessels including the aortic outflow tract.18 In addition, both polycystins are expressed in the two major cell types that comprise vessel walls, vascular smooth muscle cells (VSMCs) and endothelial cells.19–22 Finally, mice harboring a hypomorphic Pkd1 allele, which results in significantly reduced polycystin-1 expression, are able to survive into the postnatal period but develop dissecting aortic aneurysms.23,24 The aortic wall in these mice exhibits regional accumulation of matrix components between elastic lamellae. It was proposed that abnormal matrix coupled with VSMC proliferation weakens the aortic media, resulting in aneurysmal dilation and dissection.
We have been intrigued by multiple case reports in the literature describing individuals with ADPKD and features reminiscent of Marfan syndrome (MFS), an autosomal dominant connective tissue disorder characterized by aortic root aneurysm and rupture.25–27 MFS is caused by mutations in Fbn1, which encodes fibrillin-1, a large extracellular matrix protein with homology to latent TGF-β binding proteins.28 Until recently, the pathophysiology of MFS was thought to be due to a structural failure of the extracellular matrix. A number of studies, however, now demonstrate that increased signaling through the TGF-β pathway is of primary importance in the pathogenesis of the major manifestations of MFS, including aortic aneurysm as well as defects in pulmonary septation and myxomatous degeneration of cardiac atrioventricular valves.29–31 The current model is that fibrillin-1 binds and sequesters TGF-β, thereby limiting the availability of the active TGF-β molecule.32 Accordingly, fibrillin-1 deficiency results in increased levels of activated TGF-β and upregulation of canonical and noncanonical downstream signaling pathways.33,34 In the canonical pathway, the TGF-β receptor complex phosphorylates Smad2 and/or Smad3. Once activated, these Smads form a complex with Smad4, which results in translocation to the nucleus and transcription of specific target genes.35 The defects associated with fibrillin-1 deficiency in mice can be ameliorated by treatment with TGF-β antagonists, including TGF-β neutralizing antibodies and angiotensin II type 1-receptor blockers.30,31
The overlap between ADPKD and MFS in some families prompted us to investigate whether there might be a genetic interaction between Pkd1 and Fbn1. Here we show that mice that are double heterozygotes for mutations in Pkd1 and Fbn1 exhibit an exacerbation of the typical Fbn1 heterozygous aortic phenotype. In addition, our findings suggest that the likely basis of this genetic interaction is further upregulation of TGF-β signaling by Pkd1 deficiency.
Results
Genetic Interaction between Pkd1 and Fbn1
To investigate whether there might be a genetic interaction between Pkd1 and Fbn1, we bred mice heterozygous for a null mutation in Pkd1 (Pkd1KO/+) with a murine line heterozygous for a Fbn1 allele encoding a cysteine substitution in an epidermal growth factor (EGF)–like domain (Fbn1C1039G/+) that is representative of a common MFS mutation.36 Mice that are homozygous for mutations in Pkd1 die in utero but heterozygotes survive and are phenotypically unaffected. Pkd1 heterozygotes do not develop aortic wall abnormalities (Figure 1). In contrast, Fbn1C1039G/+ mice develop progressive aortic root dilation beginning at 2 weeks of age but aortic dissection is rare before 12 months of age.31,36 We found that double heterozygotes (Pkd1KO/+; Fbn1C1039G/+) displayed more severe aortic root abnormalities, including reduction in elastic fibers, elastic fiber fragmentation, and disorganization of the media, compared with single heterozygotes (Figure 1). This effect was detected at 2 months but was statistically significant by 6 months of age. Four independent observers who were blinded to the genotype of the animals performed the analyses (Supplemental Methods).
Figure 1.

Heterozygosity for Pkd1 exacerbates the aortic phenotype of Fbn1 mutant mice. (A–J) Representative ascending aortic sections are stained with Verhoeff–Van Gieson to highlight elastic fiber architecture. (A, B, F, and G) Wild-type (WT) and heterozygous Pkd1 (Pkd1KO/+) aortas have normal elastic fiber architecture at 2 months (A and B) and at 6 months of age (F and G). (C, H) Compared with WT, Fbn1C1039G/+ mice display increasing elastic fiber fragmentation between 2 months (C) and 6 months (H) of age. (D and I) These findings are aggravated in the Pkd1KO/+; Fbn1C1039G/+ double heterozygotes. (E and J) Average aortic wall architecture scores (±SEM) of the proximal ascending aorta at 2 months (E) and at 6 months (J). Four independent observers blinded to genotype graded the aortic wall architecture in four representative areas on a scale from 1 to 4. By 6 months, the aortic architecture score is significantly worse in the Pkd1KO/+; Fbn1C1039G/+ double heterozygotes compared with Fbn1C1039G/+ single heterozygotes (n=5). *P<0.001. Scale bars, 25 µm.
Fbn1 deficiency results in increased levels of activated TGF-β. We speculated that the genetic interaction between Fbn1 and Pkd1 might be based on a further up regulation of TGF-β activity in the double heterozygotes. To evaluate this possibility, we stained aortic sections from 6-month-old mice with antibodies to phosphorylated Smad2 (pSmad2), which reflects activation of the intracellular arm of the canonical TGF-β pathway. We observed increased levels of nuclear pSmad2 in the double heterozygotes (Pkd1KO/+; Fbn1C1039G/+) compared with littermates harboring either targeted mutation alone (Pkd1KO/+ or Fbn1C1039G/+) (Figure 2, A–E). The double heterozygotes also exhibited increased aortic staining for connective tissue growth factor (CTGF), a prototypical TGF-β responsive gene (Figure 2, F–J). Neither pSmad2 nor CTGF levels were elevated above wild type in the Pkd1KO/+ heterozygotes. Therefore, loss of one Pkd1 allele appears to increase the level of TGF-β activity only in the context of Fbn1 deficiency.
Figure 2.

Accentuation of canonical TGF-β signaling in Pkd1KO/+; Fbn1C1039G/+ double heterozygous aortas. (A–D) Aortic sections from 6-month-old mice stained with antisera to pSmad2, a marker of canonical TGF-β signaling. Increased nuclear pSmad2 is observed in the Fbn1C1039G/+mice (C) relative to wild-type mice (A) and Pkd1KO/+(B). (D) Staining for pSmad2 is further increased in Pkd1KO/+;Fbn1C1039G/+ aortas. (E) Blinded quantitative histologic evaluation of phosphorylated Smad2 expression (±SEM) (n=5). (F–I) Aortic sections from 6-month-old mice stained with antisera to CTGF, a prototypical TGF-β response gene. There is increased CTGF expression in Pkd1KO/+; Fbn1C1039G/+ mice (I) compared with all of the other genotypes (F–H). (J) Blinded quantitative histologic evaluation of CTGF expression (±SEM) (n=5). *P<0.05; **P<0.01. Scale bars, 25 µm.
One explanation for these observations is that haploinsufficient Pkd1KO/+ VSMCs in the aortic media have a heightened response to elevated levels of activated TGF-β that are produced in the Fbn1C1039G/+ background.29 To explore this possibility, we isolated aortic VSMCs from Pkd1KO/+ mice and littermate controls. At baseline, there was no TGF-β activation in either genotype but the response to exogenous TGF-β1 was increased in the Pkd1KO/+ VSMCs compared with the control (Figure 3). This was not due to differences in the levels of TGF-β receptor 1 or 2 (Supplemental Figure 1).
Figure 3.
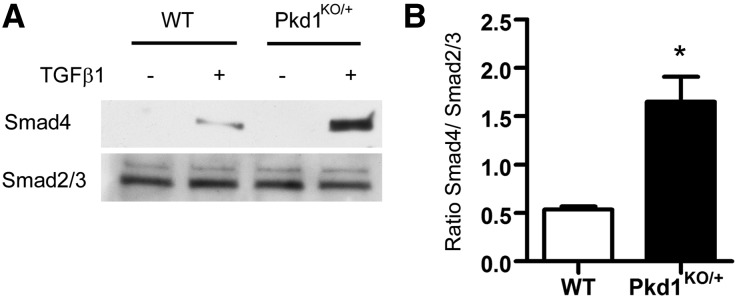
Pkd1KO/+ VSMCs show an increased response to TGF-β1. (A) VSMCs are isolated from Pkd1KO/+ and wild-type control (WT) aortas. The cells are serum starved overnight and treated with 4 µg/ml TGF-β1 for 1 hour. Cell lysates are immunoprecipitated with antibody to total Smad2/3, and then immunoblotted with anti-Smad4. Membranes are stripped and reprobed with anti-Smad2/3 as a loading control. (B) Quantification of Smad4 recruitment (±SEM). At baseline, there is no TGF-β activation in either sample but the response to TGF-β1 is increased in the Pkd1KO/+ heterozygotes compared with WT (n=3). *P<0.01.
Pkd1 Modulates TGF-β Activation in Multiple Cell Types
To determine whether the effect of Pkd1 on TGF-β activation could be extended to other cell types, we isolated murine embryonic fibroblasts (MEFs) from Pkd1KO/KO and Pkd1+/+ embryos. When MEFs were stimulated with TGF-β1, the degree of canonical pathway activation, reflected by recruitment of Smad4 to the Smad2/3 complex, was significantly greater in Pkd1KO/KO cells compared with wild type (Figure 4, D and E). Conversely, overexpression of Pkd1 in NIH3T3 cells resulted in decreased responsiveness to exogenous TGF-β1 compared with vector and Pkd1 mutant transfected cells (Figure 4, A–C). Similarly, MDCK cells stably expressing PKD1 showed decreased transcriptional activation of a TGF-β responsive luciferase reporter when the cells were treated with TGF-β1 (Supplemental Figure 2).
Figure 4.

Pkd1 modulates responsiveness to TGF-β. (A–C) NIH3T3 cells are transiently transfected with flag-tagged human PKD1, flag-tagged P89 (a truncating PKD1 mutation, R4227×), or vector control (V) and then stimulated with 4 µg/ml TGF-β1. (A and B) Cell lysates are immunoprecipitated with an antibody to total Smad2/3, and then immunoblotted with anti-Smad4 (top). Membranes are stripped and reprobed with anti-Smad2/3 as a loading control (bottom). A representative blot is shown in A. Quantification (±SEM) in B shows that PKD1 expressing cells exhibit decreased recruitment of Smad4 to the Smad2/3 complex compared with vector control and P89 (n=3 independent experiments). *P=0.01; **P=0.03. (C) A portion of the cell lysate is immunoprecipitated with M2 beads and Western blots are probed with an antibody to the C-terminus (top) or N-terminus (bottom) of PC1. As expected, P89 is slightly smaller than wild-type PC1 but transfection efficiencies are comparable. (D and E) MEFs derived from Pkd1KO/KO or wild-type littermate controls are serum starved overnight and then incubated with 4 µg/ml TGF-β1. At baseline, there is little activation of canonical TGF-β signaling. After stimulation with TGF-β1, however, Pkd1KO/KO MEFs exhibit increased recruitment of Smad4 to the Smad2/3 complex compared with controls. (E) Quantification (±SEM) of three experiments. *P<0.01. PC1, polycystin-1; FL, full-length PC1; CTF, C-terminal cleavage fragment; NTF, N-terminal cleavage fragment.
Deletion of Pkd1 in VSMCs Results in Aortic Wall Abnormalities
Because Pkd1 null mice die in utero, they cannot be used to study adult phenotypes. Therefore, to test whether complete deficiency of Pkd1 would recapitulate a Marfan-like phenotype, we used the Sm22α-Cre recombinase to direct deletion of Pkd1 in VSMCs of the aortic media.37 When we crossed Pkd1cond/cond and Pkd1KO/+; Sm22α-Cre+ mice, we found that Pkd1cond/KO; Sm22α-Cre+ animals (referred to as Pkd1VSMC−) were born at expected Mendelian ratios and survived to at least 1 year of age (Supplemental Table 1). On necropsy, however, they frequently had large pancreatic cysts and occasionally kidney cysts, which is likely due to leaky Cre recombinase expression. Nonetheless, this Sm22α Cre line displays strong activity in aortic VSMCs, with near complete deletion of Pkd1 (Supplemental Figure 3). This is similar to what has been reported by others using the same Cre recombinase to delete Pkd1.38
Three independent observers who were blinded to genotype graded aortic histopathology in cohorts of mice (n=8) at 2 and 6 months of age (Supplemental Methods). We focused our analysis on the proximal ascending aorta because this is the area that is most susceptible to dilation in Fbn1 mutant mice.31 At 2 months of age, there was no appreciable difference in aortic histology between Pkd1VSMC− mice and littermate controls. However, by 6 months of age, we found a reproducible increase in elastic fiber fragmentation and disorganization in the mice lacking VSMC Pkd1 (Figure 5). We found no significant difference in aortic histopathology in more distal areas of the aorta.
Figure 5.

Pkd1 deletion in VSMCs results in abnormal proximal ascending aortic architecture. (A–D) Ascending aortic sections from 6-month-old mice stained with Verhoeff–Van Gieson. (A) Elastic fiber architecture is intact in the Pkd1VSMC+ control aorta. (B–D) In contrast, aortic sections from Pkd1VSMC− mice demonstrate diffuse fragmentation of elastic fibers and increased extracellular matrix deposition. (E) Blinded quantitative histologic evaluation of aorta architecture (±SEM) scored by three observers (n=8). *P<0.001. Scale bars, 25 µm.
Another cohort of male mice (n=4) underwent an echocardiographic evaluation at 6 months of age. The Pkd1VSMC− animals were found to have enlargement of the aortic root and ascending aorta compared with littermate controls (Supplemental Table 2).
Pkd1 Deficiency Results in Increased TGF-β Signaling
We compared activation of the canonical TGF-β signaling in Pkd1VSMC− aortas versus littermate controls (Figures 6 and 7). At 6 months of age, Pkd1VSMC− aortas exhibited increased nuclear staining of pSmad2 and a downstream effector, CTGF (Figure 6). To quantify this, we prepared lysates from another set of Pkd1VSMC− aortas and controls and conducted immunoprecipitation with antisera to Smad2/3. Western blots were then probed with anti-Smad4 as an indicator of canonical TGF-β activation (Figure 7A). There was significantly more recruitment of Smad4 to the Smad2/3 complex in Pkd1VSMC− compared with controls. In addition, Pkd1VSMC− aortas had elevated levels of CTGF on Western blot analysis (Figure 7C).
Figure 6.

Pkd1VSMC− aortas have evidence of increased canonical TGF-β signaling. Representative proximal aortic sections from 6-month-old animals stained with antisera to pSmad2 (A and B) and CTGF (D and E). Pkd1VSMC− aortas (B and E) exhibit increased staining for each marker of TGF-β signaling compared with a control aorta (A and D). (C and F) Blinded quantitative histologic evaluation of pSmad2 and CTGF expression (±SEM) (n=8). *P<0.05. Scale bars, 25 µm.
Figure 7.

Pkd1 deletion in VSMCs results in activation of TGF-β signaling. (A and B) Western blot analysis of Smad4 recruitment in Pkd1VSMC− versus control (Pkd1VSMC+) aortas. Homogenized aortic tissue lysates are immunoprecipitated with an antibody to total Smad2/3, and then immunoblotted with anti-Smad4. Membranes are stripped and reprobed with anti-Smad2/3 as a loading control. (A) Representative Western blot. (B) Quantification of four littermate pairs (±SEM) demonstrates increased recruitment of Smad4 to the Smad2/3 complex in Pkd1VSMC− aortas versus Pkd1VSMC+ controls, indicating increased activity of canonical TGF-β signaling. (C and D) Western blot analysis of CTGF expression in Pkd1VSMC− and Pkd1VSMC+ control aortas. Quantification in three pairs (±SEM), shows increased expression in Pkd1VSMC− aortas versus controls. *P=0.02; **P=0.002.
Ascending Aortic Aneurysm from a Patient with ADPKD Demonstrates Evidence of Increased TGF-β Signaling
We identified an aortic sample harvested from a 19-year-old with a family history of ADPKD (Figure 8). The patient presented at age 18 years with hypertension and abdominal imaging revealed cystic kidneys. An echocardiogram at the time of diagnosis unexpectedly demonstrated an ascending aortic aneurysm measuring nearly 4.7 cm. The decision was made to repair the aneurysm preemptively and at surgery the individual had a bicuspid aortic valve with no evidence of aortic dissection. We prepared aortic tissue sections from this patient and from an archived sample harvested from a 19-year-old undergoing cardiac transplantation. Movat’s staining clearly demonstrated that the elastic fibers in the ADPKD aorta were smaller in diameter and more fragmented compared with the control aorta (Supplemental Figure 4). These findings are characteristic of the TGF-β–related aortopathies. In addition, the ADPKD aorta showed increased nuclear accumulation of pSmad2 as well as CTGF compared with the control (Figure 8, C and D, and Supplemental Figure 2). The increase in pSmad2 was not as dramatic, however, as that observed in the aorta from a patient with Loeys–Dietz syndrome (LDS). LDS is caused by heterozygous mutations in type 1 or type 2 TGF-β receptors and is associated with aortic aneurysms and upregulation of TGF-β signaling.32,39
Figure 8.

TGF-β activation in a human ADPKD aortic aneurysm specimen. (A) Three-dimensional magnetic resonance angiogram showing an ascending aortic aneurysm (red arrow) in a 19-year-old patient with ADPKD. (B) Magnetic resonance imaging confirms bilateral cystic kidneys from the same individual. (C) An aortic specimen after resection is stained for pSmad2. This ADPKD aorta shows increased nuclear staining for pSmad2 compared with a control aorta. As a positive control, an aortic sample from a patient with LDS shows intense staining for pSmad2. (D) Quantification of nuclear pSmad2 staining. We analyze 48 fields for each sample that includes 343, 550, and 401 nuclei for control, LDS, and ADPKD aortas, respectively. The total number of nuclei and brown-staining (pSMAD2+) nuclei are counted for each grid area and then summed. The percentage of pSMAD2+ nuclei is determined by dividing brown nuclei by total nuclei.
Discussion
There is relatively little understood about the pathogenesis of vascular complications in ADPKD. The description of cardiovascular phenotypes reminiscent of MFS in some ADPKD families suggested to us that there might be an overlap in the signaling pathways involved in the development of the vascular abnormalities that are common to these disorders. Over the past several years, a series of seminal studies has implicated dysregulated TGF-β signaling rather than structural wall weaknesses in the pathogenesis of inherited aortopathies.32 Although there has been controversy with respect to the inactivating nature of mutations in TGF-β pathway components, aortic tissues from individuals with syndromic aortic aneurysms including MFS consistently display an increase in canonical TGF-β signaling.39–43 To explore the relationship between ADPKD and MFS, we bred mice with targeted alleles of Pkd1 and Fbn1 and found that loss of one Pkd1 allele worsened the aortic phenotype of Fbn1 mutant mice. Moreover, this genetic interaction was associated with further upregulation of canonical TGF-β signaling.
These initial experiments prompted us to posit that heterozygosity for Pkd1 might result in further upregulation of TGF-β signaling, thereby exacerbating the MFS phenotype. In support of this idea we found that both VSMCs isolated from Pkd1KO/+ aortas and Pkd1 null MEFs exhibited increased responsiveness to exogenous TGF-β1. Conversely, overexpression of Pkd1 in fibroblasts diminished this response. Interestingly, we found that there was no evidence of TGF-β activation in Pkd1 deficient cells at baseline, suggesting that a source of TGF-β is required in order for the signal to be augmented. In the context of Pkd1/Fbn1 double heterozygous aortas, Fbn1 deficiency is predicted to result in higher levels of activated TGF-β.
The TGF-β signaling cascade has been previously evaluated in cystic kidney disease. Microarray studies of human ADPKD kidneys revealed that TGF-β and its pathway members are highly expressed in cystic epithelium.44 In mouse models in which Pkd1 can be conditionally inactivated, TGF-β signaling was not different from baseline in the early stages of cystogenesis; however, in later phases, cysts lining epithelial cells exhibited increased nuclear staining for pSmad2 along with increased expression of TGF-β target genes such as PAI-1.45 Our cell culture data suggest that increased TGF-β ligands in the setting of ADPKD might result in an amplified response that could contribute to fibrosis and disease progression. Interestingly, a retrospective study of patients with MFS demonstrated an increased incidence of renal and hepatic cysts compared with an age/sex-matched control population.46
In this work, we used the Cre lox system to efficiently delete Pkd1 in the aortic wall of mice harboring a Pkd1 conditional allele. Over a 6-month period, this resulted in mild but reproducible degeneration in elastic fiber morphology in the proximal ascending aorta, similar to what has been observed in Fbn1 mutant mice. In addition, we were able to detect upregulation of canonical TGF-β signaling in these mutant aortas. Taken together, our findings suggest that loss of polycystin-1 plays a role in augmenting TGF-β signaling and that this may be important in the pathogenesis of aortic aneurysms associated with ADPKD. Whether TGF-β also plays a role in intracranial aneurysms associated with ADPKD remains to be determined.
Our findings of structural aortic wall abnormalities in Pkd1VSMC− mice are different from those of another group that used similar genetic manipulations but did not detect any obvious histologic or functional aortic defects.38,47 Although these reports document abnormalities in the myogenic tone of mesenteric arteries in Pkd1VSMC− mice, these groups did not focus on the proximal ascending aorta, which is where we observed reproducible morphologic changes. The predominance of degenerative changes in this area can be explained by the embryonic origin of VSMCs in this part of the aorta.48 VSMCs in the proximal ascending aorta originate from specialized cardiac mesoderm progenitors (also called the second heart field) as well as the ectodermally derived cardiac neural crest.49,50 In contrast, VSMCs in the descending abdominal aorta arise from the paraxial mesoderm.48 Ectodermal VSMCs have been shown to exhibit increased responsiveness to exogenous TGF-β1 compared with mesodermal VSMCs that populate the abdominal aorta.51 These findings are consistent with the idea that there are regional differences in susceptibility to aortic aneurysm formation.32,52
The relatively mild phenotype of mice with isolated VSMC Pkd1 deletion is somewhat surprising. This is unlikely to be due to incomplete Sm22α Cre recombinase activity because we found close to 100% inactivation of Pkd1 in VSMCs isolated from the aorta. In contrast, mice that are homozygous for a hypomorphic Pkd1 allele exhibit a dramatic vascular phenotype by 1 month of age with widespread aortic wall abnormalities, including dissections of the thoracic and abdominal aorta.23,24 Because aortic dissection in Pkd1 hypomorphic mice occurs in the presence of significant renal cystic disease, it is possible that severe hypertension (as a “third” hit) is required for full expression of this phenotype.
Although there is consensus that ADPKD is linked with an increased risk of intracranial aneurysms, there has been debate as to whether there is a connection with aortic aneurysms. Abdominal aortic aneurysms in the general population are typically associated with increasing age and comorbidities such as smoking, hypertension, and dyslipidemia. Individuals with ADPKD may have many of these risk factors so the contribution of a Pkd mutation in this setting is difficult to discern. In comparison, thoracic aortic aneurysms can occur at any age, in the absence of cardiovascular risk factors, and are often associated with hereditary conditions such as MFS, LDS, and vascular Ehlers–Danlos syndrome. There are several case reports in the literature linking ADPKD with ascending aortic aneurysms.14,25 The patient described in this report was also found to have an ascending aortic aneurysm at a remarkably young age and aortic tissue from this individual showed evidence of increased canonical TGF-β signaling. We recognize that it is difficult to draw a definitive conclusion from one case and therefore it would be instructive to expand the analysis of TGF-β signaling to additional human samples. Nonetheless, taken together, the data suggest that certain ADPKD patients might be susceptible to ascending thoracic aortic aneurysms.53
It is unknown why only a subset of ADPKD patients develops aneurysms. Several studies have documented striking familial clustering of vascular complications, suggesting that features of the germ line mutation may be a predisposing factor. In support of this idea there are reports of multiple independent families with aneurysms and the same 2 bp deletion in Pkd1 exon 15.54,55 Not all members of these families, however, present with vascular phenotypes. One possibility is that genetic modifiers that upregulate the TGF-β signaling pathway might also increase the chance of aortic aneurysm in affected individuals.
Finally, our results have implications for the treatment of vascular complications associated with ADPKD. Renal cyst expansion in ADPKD is associated with activation of the renin-angiotensin-aldosterone axis.56 Angiotensin II in turn induces the TGF-β pathway in multiple tissues including VSMC.57,58 Therefore inhibition of this pathway in ADPKD patients with aortic aneurysms may be indicated. Although blockade of either the angiotensin AT1 receptors or of angiotensin-converting enzymes should inhibit the renin-angiotensin-aldosterone axis, a recent study in Fbn1 mutant mice suggested that the former is more effective in slowing aneurysm progression.33 In fact, both agents decrease the activity of the canonical TGF-β pathway, but only the angiotensin receptor blocker allowed continued signaling through the protective AT2 receptor, which reduces TGF-β–mediated activation of extracellular signal regulated kinases. This noncanonical signaling cascade has also been shown to be involved in aneurysm pathogenesis in MFS.33,34 We have yet to formally explore this mechanism in the setting of ADPKD but testing and comparing these agents in a suitable ADPKD mouse model may be worthy of further investigation and would provide ultimate confirmation that TGF-β signaling plays a central role in aortic pathology.
Concise Methods
Mice
Mouse work was performed at the Johns Hopkins School of Medicine and at the University of Maryland School of Medicine. The Institutional Animal Care and Use Committee at the Johns Hopkins University School of Medicine and at the University of Maryland School of Medicine approved the murine protocols. Pkd1KO, floxed alleles of Pkd1 and FbnC1039G mutant alleles, and genotyping strategies were previously described.36,59 We generated Pkd1KO/+; Fbn1C1039G/+ compound heterozygotes by crossing Pkd1KO and FbnC1039G heterozygotes and only the F1 littermates of such crosses were analyzed. The FbnC1039G allele was maintained in a C57BL/6 background while the Pkd1KO allele was kept in a mixed background (C57BL/6 and 129/Sv). We inactivated Pkd1 in VSMCs using a transgenic mouse line carrying Cre recombinase under the control of the Sm22α promoter (stock number: 004746; Jackson Labs).37 Pkd1KO/+ heterozygotes were bred with Sm22α-Cre transgenic mice to generate Pkd1KO/+; Sm22α-Cre+ male mice. These males were crossed with Pkd1cond/cond female mice, resulting in Pkd1KO/cond; Sm22α-Cre+ (Pkd1VSMC−) mice. Pkd1 cond/+; Sm22α-Cre− animals served as controls (Pkd1VSMC+).
Microscopic Examination and Immunohistochemistry
For morphologic analysis and immunohistochemistry of murine aortas, latex dye was injected into the left ventricle until it filled the aorta and its branches. Aortas were fixed in 10% buffered formalin. Proximal aortic sections were dissected and transverse sections were arranged in 1.5% agar before paraffin embedding.36 Slides were stained with hematoxylin and eosin or Verhoeff–Van Gieson. Immunohistochemistry using antibodies directed against phosphorylated Smad2 and CTGF (Cell Signaling Technology and Abcam, respectively) were performed as previously described.29,39 Slides were examined with a Nikon Eclipse E600 microscope.
Aortic Morphology
The aortic morphology of mice for each genotype was analyzed at 2 and 6 months of age. A minimum of three independent observers blinded to genotype graded aortic architecture in four areas of each proximal ascending aorta using a scale from 1 to 4 and previously described criteria (see the Supplemental Material).31 Aortic sections for all genotypes were digitally scrambled and scores were averaged (±SEM) after the data were unblinded. Comparisons across genotypes were made using the t test.
Cell Culture
MEFs were isolated from E12.5 embryos and plated in P-100 culture dishes. VSMCs were isolated from 6-month-old Pkd1KO heterozygotes and wild-type littermates using established techniques.60 VSMCs at passage 3–6 were plated in P-150 culture dishes. After reaching confluence the cells were serum starved for 24 hours and incubated with 4 µg/ml TGF-β1 (BD Bioscience) for 60 minutes before harvesting for Western blot analysis.
Plasmid Constructs and Transfections
Flag-tagged, human PKD1 cDNA constructs were previously described.61,62 NIH3T3 cells were transfected using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were harvested for immunoblotting.
Immunoprecipitation and Western Blot Analyses
Aortas were dissected from Pkd1VSMC- and littermate controls. The tissue from a single aorta was homogenized with the Tissuelyser (Qiagen), in radioimmunoprecipitation assay lysis buffer and protease inhibitor cocktail (Roche). The lysate was immunoprecipitated overnight at 4°C with antibody to total Smad2/3 (Santa Cruz). The immunoprecipitation products were run on 4%–15% SDS-PAGE gels and probed with anti-Smad4 (Santa Cruz) and then stripped and reprobed with antisera against total Smad2/3 (Santa Cruz) as a loading control. To detect CTGF, the gels were immunoblotted with anti-CTGF (Abcam), stripped, and reprobed with an anti-glyceraldehyde 3-phosphate dehydrogenase antibody (Abcam). Densitometric quantification was performed using ImageJ software (National Institutes of Health).
Clinical Specimens
The Institutional Review Board of Johns Hopkins Hospital approved this study. Aortic samples, collected at the time of surgery, were obtained from the pathology archive. The control sample was obtained from a 19-year-old individual undergoing cardiac transplant surgery. We stained aortic sections with Movat’s stain and antisera to phosphorylated Smad2 or CTGF as previously described.39,63
Statistical Analyses
The t test was used to compare aortic architecture scores and Smad4 protein expression among different experimental groups.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Jing Zhang and Mingyi Wang (Laboratory of Cardiovascular Science, National Institute on Aging, National Institutes of Health) for assistance with immunohistochemistry, Norman Barker (Department of Pathology, Johns Hopkins School of Medicine) for assistance with photography, and Loretha Myers (Dietz Lab) for her generous assistance with pSmad2 immunohistochemistry.
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01-DK076017 and R01-DK095036 to T.W., and R37-DK48006 and 1ZIA-DK075042 to G.G.G.), National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01-AR41135 and P01-AR49698 to H.C.D.), Maryland Kidney Foundation (minigrant to D.L.), Doris Duke Charitable Foundation (2009040 to M.H.), Howard Hughes Medical Institute (to H.C.D.), National Marfan Foundation (to J.H. and H.C.D.), and the Smilow Center for Marfan Syndrome Research (to H.C.D.). These studies utilized resources provided by the NIDDK-sponsored Baltimore Polycystic Kidney Disease Research and Clinical Core Center (P30-DK090868).
Part of this work was presented at the 2005 and 2009 Annual Meetings of the American Society of Nephrology, held November 8–13, 2005, in Philadelphia, Pennsylvania, and October 27–November 1, 2009, in San Diego, California, respectively.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Present Addresses: Dr. Connie Wang, Division of Nephrology, Hennepin County Medical Center, Minneapolis, Minnesota; Dr. Javid Moslehi, Division of Cardiovascular Medicine, Brigham and Women's Hospital, and the Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts; and Dr. Gregory G. Germino, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012050486/-/DCSupplemental.
References
- 1.Harris PC, Torres VE: Polycystic kidney disease. Annu Rev Med 60: 321–337, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, Gamble V, Harris PC: The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10: 151–160, 1995 [DOI] [PubMed] [Google Scholar]
- 3.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S: PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272: 1339–1342, 1996 [DOI] [PubMed] [Google Scholar]
- 4.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG: PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16: 179–183, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Gallagher AR, Germino GG, Somlo S: Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 17: 118–130, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pirson Y: Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 17: 173–180, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Chapman AB, Rubinstein D, Hughes R, Stears JC, Earnest MP, Johnson AM, Gabow PA, Kaehny WD: Intracranial aneurysms in autosomal dominant polycystic kidney disease. N Engl J Med 327: 916–920, 1992 [DOI] [PubMed] [Google Scholar]
- 8.Ruggieri PM, Poulos N, Masaryk TJ, Ross JS, Obuchowski NA, Awad IA, Braun WE, Nally J, Lewin JS, Modic MT: Occult intracranial aneurysms in polycystic kidney disease: Screening with MR angiography. Radiology 191: 33–39, 1994 [DOI] [PubMed] [Google Scholar]
- 9.Huston J, 3rd, Torres VE, Sulivan PP, Offord KP, Wiebers DO: Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 3: 1871–1877, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Irazabal MV, Huston J, 3rd, Kubly V, Rossetti S, Sundsbak JL, Hogan MC, Harris PC, Brown RD, Jr, Torres VE: Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 1274–1285, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukunaga N, Yuzaki M, Nasu M, Okada Y: Dissecting aneurysm in a patient with autosomal dominant polycystic kidney disease. Ann Thorac Cardiovasc Surg 18: 375–378, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Kanagasundaram NS, Perry EP, Turney JH: Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 14: 183–184, 1999 [DOI] [PubMed] [Google Scholar]
- 13.Al-Hakim W, Goldsmith DJ: Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 79: 474–475, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J, Kim SM, Lee SY, Lee HC, Bae JW, Hwang KK, Kim DW, Cho MC, Byeon SJ, Kim KB: A case of severe aortic valve regurgitation caused by an ascending aortic aneurysm in a young patient with autosomal dominant polycystic kidney disease and normal renal function. Korean Circ J 42: 136–139, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobrie G, Brunet-Bourgin F, Alamowitch S, Coville P, Kassiotis P, Kermarrec A, Chauveau D: Spontaneous artery dissection: Is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 13: 2138–2141, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Arnaout MA: The vasculopathy of autosomal dominant polycystic kidney disease: Insights from animal models. Kidney Int 58: 2599–2610, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout MA: Polycystin 1 is required for the structural integrity of blood vessels. Proc Natl Acad Sci U S A 97: 1731–1736, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R: Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A 98: 12174–12179, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffin MD, Torres VE, Grande JP, Kumar R: Vascular expression of polycystin. J Am Soc Nephrol 8: 616–626, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Torres VE, Cai Y, Chen X, Wu GQ, Geng L, Cleghorn KA, Johnson CM, Somlo S: Vascular expression of polycystin-2. J Am Soc Nephrol 12: 1–9, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Qian Q, Li M, Cai Y, Ward CJ, Somlo S, Harris PC, Torres VE: Analysis of the polycystins in aortic vascular smooth muscle cells. J Am Soc Nephrol 14: 2280–2287, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, Watnick TJ, Zhou F, Germino GG: Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci U S A 99: 16981–16986, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, Deruiter MC, Breuning MH, de Heer E, Peters DJ: Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 13: 3069–3077, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Hassane S, Claij N, Lantinga-van Leeuwen IS, Van Munsteren JC, Van Lent N, Hanemaaijer R, Breuning MH, Peters DJ, DeRuiter MC: Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol 27: 2177–2183, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Somlo S, Rutecki G, Giuffra LA, Reeders ST, Cugino A, Whittier FC: A kindred exhibiting cosegregation of an overlap connective tissue disorder and the chromosome 16 linked form of autosomal dominant polycystic kidney disease. J Am Soc Nephrol 4: 1371–1378, 1993 [DOI] [PubMed] [Google Scholar]
- 26.Biermann CW, Gasser TC, Breuer C, Rutishauser G: [Marfan syndrome and cystic kidneys of the adult type]. Helv Chir Acta 59: 513–515, 1992 [PubMed] [Google Scholar]
- 27.Selgas R, Temes JL, Sobrino JA, Viguer JM, Otero A, Sánchez Sicilia L: [Polycystic renal disease in the adult associated with an incomplete form of Marfan’s syndrome (author’s transl)]. Med Clin (Barc) 76: 311–313, 1981 [PubMed] [Google Scholar]
- 28.Ramirez F, Dietz HC: Marfan syndrome: From molecular pathogenesis to clinical treatment. Curr Opin Genet Dev 17: 252–258, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC: Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 33: 407–411, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC: TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 114: 1586–1592, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC: Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312: 117–121, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindsay ME, Dietz HC: Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 473: 308–316, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC: Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 332: 361–365, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC: Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332: 358–361, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derynck R, Zhang YE: Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425: 577–584, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC: Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest 114: 172–181, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M: Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A 99: 7142–7147, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassane S, Claij N, Jodar M, Dedman A, Lauritzen I, Duprat F, Koenderman JS, van der Wal A, Breuning MH, de Heer E, Honore E, DeRuiter MC, Peters DJ: Pkd1-inactivation in vascular smooth muscle cells and adaptation to hypertension. Lab Invest 91: 24–32, 2011 [DOI] [PubMed] [Google Scholar]
- 39.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC: A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37: 275–281, 2005 [DOI] [PubMed] [Google Scholar]
- 40.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collée M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM: Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 43: 121–126, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Cardoso S, Robertson SP, Daniel PB: TGFBR1 mutations associated with Loeys-Dietz syndrome are inactivating. J Recept Signal Transduct Res 32: 150–155, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL: Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 44: 922–927, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inamoto S, Kwartler CS, Lafont AL, Liang YY, Fadulu VT, Duraisamy S, Willing M, Estrera A, Safi H, Hannibal MC, Carey J, Wiktorowicz J, Tan FK, Feng XH, Pannu H, Milewicz DM: TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res 88: 520–529, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y: Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18: 2328–2343, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Hassane S, Leonhard WN, van der Wal A, Hawinkels LJ, Lantinga-van Leeuwen IS, ten Dijke P, Breuning MH, de Heer E, Peters DJ: Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol 222: 21–31, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Chow K, Pyeritz RE, Litt HI: Abdominal visceral findings in patients with Marfan syndrome. Genet Med 9: 208–212, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJ, Honoré E: Polycystin-1 and -2 dosage regulates pressure sensing. Cell 139: 587–596, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Majesky MW: Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol 27: 1248–1258, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML: Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol 281: 78–90, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML: Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol 281: 66–77, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Topouzis S, Majesky MW: Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev Biol 178: 430–445, 1996 [DOI] [PubMed] [Google Scholar]
- 52.Majesky MW, Dong XR, Hoglund VJ: Parsing aortic aneurysms: More surprises. Circ Res 108: 528–530, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leier CV, Baker PB, Kilman JW, Wooley CF: Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 100: 683–688, 1984 [DOI] [PubMed] [Google Scholar]
- 54.Watnick T, Phakdeekitcharoen B, Johnson A, Gandolph M, Wang M, Briefel G, Klinger KW, Kimberling W, Gabow P, Germino GG: Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very-early-onset disease. Am J Hum Genet 65: 1561–1571, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rossetti S, Chauveau D, Kubly V, Slezak JM, Saggar-Malik AK, Pei Y, Ong AC, Stewart F, Watson ML, Bergstralh EJ, Winearls CG, Torres VE, Harris PC: Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 361: 2196–2201, 2003 [DOI] [PubMed] [Google Scholar]
- 56.Chapman AB, Stepniakowski K, Rahbari-Oskoui F: Hypertension in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 17: 153–163, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rüster C, Wolf G: Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol 22: 1189–1199, 2011 [DOI] [PubMed] [Google Scholar]
- 58.Fukuda N, Hu WY, Kubo A, Kishioka H, Satoh C, Soma M, Izumi Y, Kanmatsuse K: Angiotensin II upregulates transforming growth factor-beta type I receptor on rat vascular smooth muscle cells. Am J Hypertens 13: 191–198, 2000 [DOI] [PubMed] [Google Scholar]
- 59.Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, Germino GG: A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol 15: 3035–3043, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Ray JL, Leach R, Herbert JM, Benson M: Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci 23: 185–188, 2001 [DOI] [PubMed] [Google Scholar]
- 61.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG: Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408: 990–994, 2000 [DOI] [PubMed] [Google Scholar]
- 62.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino GG: Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell 6: 1267–1273, 2000 [DOI] [PubMed] [Google Scholar]
- 63.Maleszewski JJ, Miller DV, Lu J, Dietz HC, Halushka MK: Histopathologic findings in ascending aortas in individuals with Loeys-Dietz Syndrome (LDS). Am J Surg Pathol 33: 194–201, 2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.