

Abstract
Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy caused by uncontrolled activation of the alternative pathway of complement at the cell surface level that leads to microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney failure. In approximately one half of affected patients, pathogenic loss-of-function variants in regulators of complement or gain-of-function variants in effectors of complement are identified, clearly implicating complement in aHUS. However, there are strong lines of evidence supporting the presence of additional genetic contributions to this disease. To identify novel aHUS-associated genes, we completed a comprehensive screen of the complement and coagulation pathways in 36 patients with sporadic aHUS using targeted genomic enrichment and massively parallel sequencing. After variant calling, quality control, and hard filtering, we identified 84 reported or novel nonsynonymous variants, 22 of which have been previously associated with disease. Using computational prediction methods, 20 of the remaining 62 variants were predicted to be deleterious. Consistent with published data, nearly one half of these 42 variants (19; 45%) were found in genes implicated in the pathogenesis of aHUS. Several genes in the coagulation pathway were also identified as important in the pathogenesis of aHUS. PLG, in particular, carried more pathogenic variants than any other coagulation gene, including three known plasminogen deficiency mutations and a predicted pathogenic variant. These data suggest that mutation screening in patients with aHUS should be broadened to include genes in the coagulation pathway.
Hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney failure that is most frequently caused by Shiga-toxigenic Escherichia coli (STEC) serogroup O157. About 10% of HUS cases, however, are atypical and not caused by an antecedent STEC infection.1 The prevalence of atypical HUS (aHUS) is estimated at 1 per 2 and 1 per 3.3 million children in the United States and Europe, respectively.2,3 Until the recent availability of Eculizumab, aHUS carried significant morbidity and mortality, with 25% of patients dying of disease, 50% of patients progressing to end stage renal failure, and 40%–80% of patients experiencing disease recurrence after transplantation.1,4
Uncontrolled activation of the alternative pathway of complement at the level of the cell surface has been closely linked to disease.5,6 In about one half of aHUS patients, pathogenic mutations are identified in complement genes, including loss-of-function mutations in regulators of complement activity like complement factor H (CFH),7 complement factor I (CFI),8 and membrane cofactor protein (CD46)9,10 and gain-of-function mutations in effectors of complement like factor B (CFB)11 and complement component 3 (C3).12,13 In a recent large genetic screen of 794 aHUS patients, single mutations in CFH, C3, CFI, CFB, or CD46 were identified in 41% of patients, and combined mutations were found in 3% of patients.14 Additional genetic contributors include single nucleotide polymorphisms and haplotypes in CFH and CD46,14–17 copy number variation of the complement factor H-related three and one genetic intervals (homozygosity for deletion of CFHR3-CFHR1), and novel fusion genes of the factor H-related region caused by nonallelic homologous recombination.18–20
Although these studies clearly implicate mutations in complement genes in the pathogenesis of aHUS, additional genetic contributions to this disease are likely to exist. There are three lines of evidence that strongly support this hypothesis. First, in a large percentage of cases, no mutations are found in the commonly implicated complement genes. Second, there is an extremely high rate of incomplete penetrance in familial aHUS, which is consistent with the existence of additional modifying/contributory genetic factors. Third, current screens have narrowly focused on fewer than a dozen genes, although the complement system is substantially larger and integrated with numerous other signaling pathways.
In this study, we sought to address these limitations by using targeted genomic enrichment and massively parallel sequencing (TGE+MPS) to screen the coding sequence and splice sites of all genes in both the complement and coagulation pathways. We selected the coagulation pathway because of its role in thrombotic thrombocytopenic purpura (TTP), a thrombotic microangiopathy in which extensive microscopic thrombi form in the microvasculature. aHUS and TTP share the clinical features of microangiopathic hemolysis, thrombocytopenia, and renal involvement, but in TTP, patients also have fever and neurologic symptoms.21,22 The fact that the coagulation pathway warrants detailed scrutiny is also supported by the finding of pathogenic aHUS mutations in two anticoagulant-related genes, THBD23 and DGKE.24 THBD encodes thrombomodulin, which functions as a cofactor for thrombin to reduce blood coagulation and also regulates CFI-induced C3b inactivation.23,25 DGKE encodes diacylglycerol kinase-ε and is implicated in regulation of thrombus formation by modulating protein kinase C activity in endothelial cells and platelets.24
In total, we sequenced 85 genes in 36 European-American sporadic aHUS patients, and we filtered and prioritized variants based on frequency and functional effects. As expected, we found novel deleterious variants in multiple complement genes, but we also identified deleterious nonsynonymous rare variants in several genes that play important roles in coagulation, definitely implicating this pathway in aHUS. The most frequently mutated gene in the coagulation pathway was PLG, which encodes plasminogen, a zymogen that is secreted by the liver and converted into plasmin by a variety of enzymes on binding to clots.
Results
Patients
In total, 36 European-American patients with sporadic aHUS (24 men and 12 women) were recruited into this study from 2007 to 2011 (Figure 1). The diagnosis of aHUS was based on the presence of acute renal failure, thrombocytopenia, and microangiopathic hemolytic anemia after excluding antecedent STEC infection. All procedures were approved by the Institutional Review Board of the University of Iowa Carver College of Medicine.
Figure 1.

Diagnostic age distributes similarly in male and female aHUS patients. Although a trend toward higher diagnostic age was observed in women, this difference was not significant (Wilcoxon rank sum test P=0.067).
Sequencing Quality Metrics and Variant Calling
We performed TGE+MPS on 36 European-American aHUS patients using an RNA-bait panel that targets coding sequence of genes in the complement and coagulation pathways (hereafter referred to as capture and sequencing of complement-associated disease exons [CasCADE]). The mean total reads generated per sample approximated 9,000,000, with more than 97% of reads mapping to the reference genome and more than 50% of reads mapping to targeted regions. The average coverage per targeted base was 1670 reads; 99% of targeted regions were covered by more than 30 reads (Table 1).
Table 1.
Quality of sequencing results
| Sequencing Metrics | Mean ± SD | Median |
|---|---|---|
| Total reads | 9,195,907±1,294,156 | 9,176,081 |
| Mapped reads | 9,112,151±1,215,320 | 9,008,634 |
| Mapped reads, % | 97.84 | 98.39 |
| Reads overlapping targets | 5,025,594±875,441 | 5,053,139 |
| Reads overlapping targets, % | 54.84 | 55.92 |
| Average coverage | 1670.65±290.82 | 1677.48 |
| Bases covered 1×, % | 99.60 | 99.67 |
| Bases covered 10×, % | 99.17 | 99.35 |
| Bases covered 30×, % | 98.97 | 99.29 |
Of 7486 variants identified in 36 aHUS patients, 7071 (95.46%) variants passed quality control (Figure 2). Each subject carried a mean of 196 variants. After filtering by minor allele frequency and functional effect, 18 novel and 66 reported nonsynonymous rare variants (nsRVs) remained. All nsRVs were present in heterozygosity, with each patient carrying a mean of 2.33 nsRVs (range=0–5).
Figure 2.

Application of hard filtering uncovers pathogenic variants. Variants were filtered by focusing on nonsynonymous, frame shift, and indel changes. Rare variants are defined as variants with a minor allele frequency<1% in European-American and African-American populations based on data from the EVS database. Predicted deleterious mutations or rare variants are stop-gain mutations, indels, or missense variants with a high pathogenicity score (PS) (>4). PS for missense variants was calculated using PhyloP, SIFT, PolyPhen2, LRT, MutationTaster, and GERP++, and it ranged from zero to six. The mean PS of reported DRVs was 3.41; the mean PS of predicted deleterious missense variants was 4.57. *DRVs have been previously reported to be disease-causing or -associated. DRVs may or may not be recorded in public single nucleotide polymorphism databases. QC, quality control; UTR, untranslated region.
Reported Disease-Related Variants
Twenty-two of eighty-four nsRVs were reported disease-related variants (DRVs), most frequently being causally related to aHUS (15 DRVs in CD46, CFB, CFH, and CFI) (Table 2). Remarkably, however, there were three variants in PLG (c.112 A>G, p.Lys38Glu; c.2134 G>A, p.Gly712Arg; c.758 G>A, p.Arg253His) associated with plasminogen deficiency,26–28 which is a significant enrichment of DRVs in this gene compared with European-American data from the National Heart, Lung, and Blood Institute Exome Sequencing Project Exome Variant Server (EVS29; 3 DRV counts in 36 aHUS cases versus 76 DRV counts in 4300 EVS European-American subjects; Fisher exact test P=0.03). Other interesting ultrarare DRVs included a G-to-A transition that alters a canonical splice site in F12 (c.1681–1G>A) and leads to factor XII deficiency.30
Table 2.
Reported disease-related variants
| Gene | Nucleotide | AA Change | PSa | Related Disease | MAFb (EVS_EA) | dbSNP 137 |
|---|---|---|---|---|---|---|
| C9 | c.460 C>T | p.R154X | — | C9 deficiency50 | 0.0002 | rs144138616 |
| CD46 | c.692 C>G | p.P231R | 6 | aHUS51 | ND | ND |
| CD46 | c.648 G>C | p.W216C | 6 | aHUS51 | ND | ND |
| CD46 | c.565 T>G | p.Y189D | 5 | aHUS52 | ND | ND |
| CFB | c.967 A>C | p.K323Q | 2 | aHUS51 | ND | ND |
| CFH | c.1825 G>A | p.V609I | 1 | aHUS51 | 0.0006 | rs148165372 |
| CFH | c.1873 G>T | p.E625X | — | aHUS51 | 0.0002 | rs150694809 |
| CFH | c.2745 C>A | p.C915X | — | aHUS51 | ND | ND |
| CFH | c.3514 G>T | p.E1172X | — | aHUS53 | ND | rs121913060 |
| CFH | c.3583 G>T | p.E1195X | — | aHUS51 | ND | ND |
| CFH | c.3592 G>A | p.E1198K | 4 | aHUS54 | ND | ND |
| CFH | c.3643 C>G | p.R1215G | 4 | aHUS7 | ND | rs121913051 |
| CFH | c.3644 G>Ac | p.R1215Q | 2 | aHUS55 | ND | ND |
| CFI | c.355 G>A | p.G119R | 3 | aHUS51 | 0.0013 | rs141853578 |
| CFI | c.859 G>A | p.G287R | 5 | aHUS51 | 0.0001 | rs182078921 |
| F12 | c.1681–1 G>A | NA | — | Factor XII deficiency30 | 0.0006 | ND |
| PLG | c.112 A>G | p.K38E | 5 | Plasminogen deficiency26 | 0.0062 | rs73015965 |
| PLG | c.2134 G>A | p.G712R | 3 | Plasminogen deficiency27 | 0.0007 | ND |
| PLG | c.758 G>A | p.R253H | 5 | Plasminogen deficiency28 | 0.0006 | rs143034754 |
| THBD | c.1502 C>T | p.P501L | 3 | aHUS23 | 0.0029 | rs1800579 |
| VWF | c.2561 G>A | p.R854Q | 4 | Von Willebrand disease56 | 0.0056 | rs41276738 |
All variants are heterozygous. AA, African-American; EA, European-American; C9, complement component 9; F12, coagulation factor XII; VWF, Von Willebrand factor; NA, not available; ND, no data.
Pathogenicity score (PS) was calculated using PhyloP, SIFT, PolyPhen2, LRT, MutationTaster, and GERP++. The average PS of reported disease-related variants was 3.81.
Minor allele frequency (MAF) values in the European-American population are from the Exome Variant Server (EVS). Novel variants are those variants not reported in the EVS, 1000 Genomes, or dbSNP databases.
Mutation is carried by two patients.
Novel nsRV Identification
Among 18 novel variants that we identified, there were 15 missense, 2 frame-shift, and 1 splice site change variants (Supplemental Table 1). These variants included a G-to-C transversion in VWF, c.4165G>C (p.Glu1389Gln), that alters the same nucleotide as a DRV (c.4165G>A, p.Glu1389Lys), which is reported to cause type 2 von Willebrand disease by reducing amounts of high-molecular weight VWF and VWF-platelet binding.31 One novel variant, c.3221A>G, p.Asn1074Ser in F5, was shared by two aHUS patients and alters a potential N-link glycosylation site.
Prediction of Deleterious Variants
To assess possible pathogenicity of 62 variants not previously implicated in disease (58 missense, 1 splice site, and 3 frame-shift indels variants), we first used combined computational prediction methods (PhyloP, SIFT, PolyPhen2, LRT, MutationTaster, and GERP++) to calculate the mean pathogenicity score (PS) of the reported missense DRVs. Based on this PS, which was 3.81, we accepted as likely deleterious all novel and nsRVs with a PS of at least 4 (Table 2). The average PS of 16 missense novel and nsRVs meeting this minimal threshold was 4.57 (Table 3). Three frame-shift indels (c.356_357insG in C3AR1, c.443_453del in complement component 2, and c.1770delG in mannan-binding lectin serine protease 1 [MASP1]) and a splice site change (c.1160–2A>G in CFH) were also considered possibly damaging.
Table 3.
Predicted deleterious nsRVs
| Gene | Nucleotide | AA Change | PSa | MAFb (EVS_EA) | dbSNP 137 |
|---|---|---|---|---|---|
| Reported | |||||
| C1Sc | c.943 G>A | p.D315N | 6 | 0.0053 | rs117907409 |
| C3 | c.4855 A>C | p.S1619R | 4 | 0.0028 | rs2230210 |
| C3AR1 | c.356_357insG | p.D119fs | — | 0.0004 | ND |
| C8A | c.1331 G>A | p.R444H | 6 | 0.0050 | rs143908758 |
| CFHR5c | c.832 G>A | p.G278S | 4 | 0.0088 | rs139017763 |
| CR2 | c.524 C>T | p.P175L | 4 | 0.0074 | rs75282758 |
| FCN1 | c.866 A>G | p.N289S | 4 | 0.0016 | rs138055828 |
| MASP2 | c.467 G>A | p.C156Y | 6 | 0.0097 | rs41307788 |
| PHB | c.128 G>T | p.R43L | 4 | 0.0088 | rs2233665 |
| PLG | c.505 C>T | p.P169S | 4 | 0.0004 | rs143256245 |
| Novel | |||||
| C2 | c.443_453del | p.148_151del | — | ||
| C3 | c.3085 G>A | p.D1029N | 6 | ||
| CFH | c.595 A>G | p.S199G | 4 | ||
| CFH | c.1160–2 A>G | — | — | ||
| CR1 | c.4750 C>T | p.R1584W | 4 | ||
| MASP1 | c.1770delG | p.G590fs | — | ||
| MASP2 | c.1633 A>G | p.N545D | 4 | ||
| VWF | c.4165 G>Cd | p.E1389Q | 4 |
All variants are heterozygous. AA, African-American; EA, European-American; C1S, complement component 1 s subcomponent; C3AR1, complement component 3a receptor 1; C8A, complement component 8 alpha polypeptide; CR2, complement component receptor 2; FCN1, ficolin 1; PHB, prohibitin; C2, complement component 2; CR1, complement receptor 1; ND, no data.
Pathogenicity score (PS) was calculated using PhyloP, SIFT, PolyPhen2, LRT, MutationTaster, and GERP++. The mean PS of predicted deleterious missense nsSNVs was 4.57.
Minor allele frequency (MAF) values in the European-American population are from the Exome Variant Server (EVS). Novel variants are those variants not reported in the EVS, 1000 Genomes, and dbSNP databases.
Mutation is carried by two patients.
Mutation (c.4165 G>A) in VWF causing Von Willebrand disease was found at the same position (21094983) but leads to a different alteration in the amino acid sequence (p.E1389K versus p.E1389Q).
In aggregate, 42 deleterious variants (22 DRVs and 20 predicted deleterious variants) were identified in 25 patients (69.4%) (Figure 3). Not surprisingly, of these variants, 20 variants were found in genes commonly implicated in aHUS (CFH, CD46, C3, CFI, CFB, and THBD). However, 15 variants were found in other complement-related genes, and 7 variants were found in coagulation pathway genes. The greatest number of deleterious variants was found in CFH (11 variants in 10 patients), but the second greatest number was in PLG (4 variants in 4 patients), suggesting that this gene makes an important contribution to the aHUS phenotype.
Figure 3.

Distribution of reported and predicted deleterious variants per gene implicates PLG in aHUS pathogenesis. The second greatest number of deleterious variants was identified in PLG. This enrichment of deleterious variants is statistically significant (P<0.05). A detailed description of the variants is given in Supplemental Table 3.
Copy Number Variants in CFHRs
We identified copy number variants (CNVs) in the CFHR genomic region in 15 patients (41.7%). Homozygosity for delCFHR3-CFHR1 was found in three patients who carried no deleterious nsRVs. Ten patients were heterozygous for this deletion, and in two other patients, we identified heterozygous duplications in the CFHR1 and CFHR4 genes. The frequency of these variations was not statistically different than the frequency seen in 314 healthy controls (unpublished data; Fisher exact test P=0.16; validated Hardy–Weinberg Equilibrium in patients and controls).
Multiple Variants
Twelve patients (33.3%) carried two or more deleterious nsRVs (Supplemental Table 2). In six of these patients, the deleterious nsRVs were present only in complement genes, but in the remaining six patients, deleterious nsRVs were found in both complement and coagulation genes. Although not statistically significant, there was a trend to negative correlation between CNVs of the CFHR region and deleterious nsRVs (Figure 4). In five patients (13.9%), we were unable to identify genetic alterations in any complement or coagulation gene.
Figure 4.

Copy number of complement factor H related (CFHR) genes negatively correlates with number of deleterious variants in aHUS patients. Less frequent copy number variation of CFHR genes was observed in patients with more deleterious variants. nsRV, deleterious nonsynonymous rare variant; hom, homozygous; het, heterozygous; del, deletion; dup, duplication.
Discussion
In this study, we used TGE+MPS to screen 85 genes in the complement and coagulation pathways in 36 European-American patients with sporadic aHUS. After variant calling, quality control, and hard filtering, we identified 84 reported or novel nsRVs, 22 of which have been previously associated with disease. Using computational prediction methods (PhyloP, SIFT, PolyPhen2, LRT, MutationTaster, and GERP++), 20 additional variants were predicted to be deleterious. As expected and consistent with published data, nearly one half of these 42 variants (19; 45%) were found in genes implicated in the pathogenesis of aHUS (CFH, CD46, CFB, CFI, and C3). However, PLG, a gene in the coagulation pathway, also emerged as an important gene in the pathogenesis of aHUS.
The four variants that we identified in PLG included three reported plasminogen deficiency-related variants: c.112 A>G (p.Lys38Glu), c.758 G>A (p.Arg253His), and c.2134 G>A (p.Gly712Arg). Of these known variants, c.112 A>G, p.Lys38Glu (rs73015965; also referred to as Lys19Glu27) is noteworthy for being the first variant identified in sporadic plasminogen-deficient patients (two men and one woman) and being associated with low plasminogen levels in the unaffected parental carrier.26 Subsequent studies have shown that this missense variant is found in one third of patients with plasminogen deficiency type I and associated with less severe reduction in plasma plasminogen levels and milder disease.27,28 The fourth variant that we identified, c.505 C>T, p.Pro169Ser, is novel and predicted to be disease-causing based on genetic conservation, bioinformatics (PS=4), and frequency data (Figure 5 and Table 2).
Figure 5.
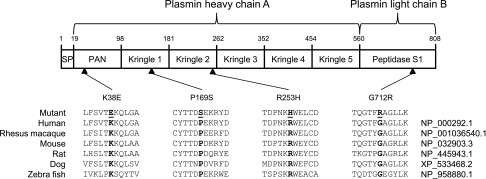
Deleterious variants of PLG are conserved. The four identified deleterious variants in PLG localize to different domains of plasminogen. The amino acids are highly conserved across multiple species, suggesting that these variants may impair normal function of the protein.
The enrichment of PLG for plasminogen deficiency DRVs is statistically significant (P<0.05) and suggests that subnormal activity of plasminogen or plasmin may compromise the degradation of thrombi in aHUS. We also identified other DVRs linked to thrombosis and fibrinolysis, including a variant in THBD (c.1502 C>T [p.Pro501Leu]) that has been associated with aHUS23 and thromboembolic disease.32 In vitro, this missense change reduces cell surface expression of thrombomodulin, which may contribute in vivo to suppression of the alternative pathway through CFI-mediated C3b inactivation23 and venous thrombosis.33 Another variant, c.1681–1 G>A in F12, alters a canonical splice acceptor, leading to a truncated transcript, reduced protein expression, increased partial thromboplastin time, and recurrent superficial venous thrombosis.30
The role of coagulation activity in aHUS patients has been evaluated at the mRNA level in one study, in which gene expression was quantitated in glomeruli from archival paraffin-embedded renal biopsies.34 Expression of antifibrinolytic, prothrombotic plasminogen activator inhibitor-1 and antithrombotic thrombomodulin were increased, and expression of profibrinolytic, antithrombotic tissue plasminogen activator was decreased compared with controls, suggesting that reduction of fibrinolysis is important in aHUS.34 Plasminogen-related studies on typical HUS, however, have yielded equivocal results.35–39 Some studies have reported that inhibition of fibrinolysis precedes and may be the cause of renal injury,35 with plasma levels of plasminogen activator inhibitor-1 inversely correlating with improvement in renal function.39 Other studies, however, report that evidence of impaired fibrinolysis in children with typical HUS is lacking.37
Novel deleterious variants were found in known complement genes as well as complement genes not previously associated with aHUS, like complement component 2, complement receptor 1, ficolin 1, and MASP1 and -2 (Supplemental Table 1). Although these findings need to be replicated, they do suggest that aHUS can be caused by a broad range of complement abnormalities. A single novel variant was also identified in ADAMTS13 (c.3056 T>C or p.M1019T), a finding that is noteworthy for the aforementioned clinical similarity between aHUS and TTP. This variant is predicted to have limited impact on protein structure and function (PS=0), which we expected given the aHUS phenotype of the patient. Functional studies would be required to determine whether the variant has a minor effect on ADAMTS13 activity. Interestingly, this patient also carries a deleterious variant in MASP2 (c.1633 A>G or p.N545D, PS=4).
Multiple variants in common aHUS genes were identified in two patients, which is in agreement with expected frequencies (3%–12%) (Supplemental Table 2).14 However, these numbers are almost certainly an underestimate, because one third of patients carried multiple deleterious nsRVs. We also found that most identified coagulation variants (seven of eight) were accompanied by one or two complement mutations (Supplemental Table 2). In all cases, these variants were found in heterozygosity, suggesting a role as disease modifiers. Our data also suggest that CNVs of the CFHR genetic region may contribute to the aHUS phenotype. Although it is well known that homozygosity for deletion of CFHR3-CFHR1 is more common in the aHUS patient population and correlates with the presence of CFH autoantibodies,40 our results show that del(CFHR3-CFHR1) is disease-contributory, whereas patients with aHUS who carry more mutations are less likely to have CFHRs deletions and vice versa.
Figure 6 shows a modified model of aHUS pathogenesis that integrates our results and recent findings in heme as a secondary hit.41 The key concept in this model is the dysregulation loop, which can be activated spontaneously or by trigger events at either point: overactivation of complement or formation of thrombi in microvessels. Overactivation of complement causes endothelial cells lysis, which leads to thrombi formation. Thrombi in microvessels cause mechanical damage to erythrocytes with release of heme. Heme directly activates the alternative pathway and represses membrane cofactor protein and decay-accelerating factor expression in endothelial cells,41 thus closing the loop. Dysregulation of complement and/or coagulation typically caused by genetic abnormalities maintains the cycle.
Figure 6.

Modified model implicates dysregulation of both the complement and coagulation pathways in the pathogenesis of aHUS. This model suggests three stages in the development of aHUS. (1) The disease is initiated by trigger events, such as nonenteric bacterial infections, viruses, drugs, malignancies, transplantation, pregnancy, and medical conditions like scleroderma, antiphospholipid syndrome, and systemic lupus erythematosus. The complement cascade is activated, and/or the coagulation pathway leads to thrombus formation. (2) A dysregulation loop develops, which is driven by overactivation of complement that causes endothelial cells stimulation and lysis. Lysis leads to thrombi formation, with altered function of the coagulation pathway caused by genetic variants in coagulation genes, such as PLG, PLAT (encoding tissue plasminogen activator, the main plasminogen activator), PLAU (encoding urokinase plasminogen activator, primarily responsible for pericellular plasmin activation), F12, ADAMTS13, and VWF. The consequence is enhanced formation or decreased degradation of thrombi, promoting thrombosis. Thrombi in small vessels cause mechanical damage to erythrocytes releasing peptides (such as heme41) or overexpression of other unidentified factors that activate the complement system. Genetic variants in complement genes and/or acquired factors like CFH autoantibodies lead to inadequate/ineffective complement regulation. (3) This dysregulation loop induces the clinical onset of aHUS.
We noted two important limitations in this study. First, the relatively small sample size reduces statistical power to uncover minor genetic contributions. Second, we focused on exonic variants and indels at the exclusion of intronic and untranslated region variants, which could be used to identify haplotypes that may contribute to aHUS. Although a more comprehensive genetic screen in large cohort of aHUS patients will remediate these shortcomings, this study is noteworthy for implicating the coagulation pathway and PLG in particular as important contributors to aHUS.
Concise Methods
DNA Extraction, Targeted Genomic Enrichment, and Massively Parallel Sequencing
Genomic DNA was extracted from peripheral blood using the Gentra Puregene Kit (Qiagen Inc., Valencia, CA), and quality was verified by 1% agarose gel electrophoresis and NanoDrop 1000 spectrophotometry (Thermo Scientific Inc., Wilmington, DE). Before sequence capture, DNA concentration was determined using the Qubit dsDNA BR Assay Kit (Invitrogen Inc., Carlsbad, CA).
Targeted genomic enrichment was performed using CasCADE, an RNA-bait panel based on the Agilent SureSelect Target Enrichment System (Agilent Technologies Inc., Santa Clara, CA) that targets coding sequence of genes in the complement and coagulation pathways. CasCADE was developed and optimized to capture 1071 exons of 85 complement and coagulation genes representing 250 kB genomic DNA (Supplemental Figure 1). In CasCADE versions 1 and 2, baits were balanced based on sequencing results of test samples to optimize efficiency and accuracy of targeted enrichment, and in this study, we used CasCADE version 3 to perform capture and enrichment of 2 µg genomic DNA using a solution-based targeted genomic enrichment protocol as previously described.42 Multiplexing indexes were used to barcode prepared libraries. Quality and concentration were evaluated using a Bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA). After pooling, libraries were sequenced with a 2× 100-bp paired-end module in one lane on a HiSeq 2000 (Illumina Inc., San Diego, CA) at the DNA Core Facility of the University of Iowa’s Institute of Human Genetics.
Variant Calling and Filtering
Sequence data were analyzed using a local installation of the open-source Galaxy software running on a high-performance computing cluster at the University of Iowa. A dedicated compute queue for Galaxy as well as shared compute resources were leveraged for minimized job wait time and continuous availability. We use a combination of custom and publicly available tools on the pipeline running in Galaxy: read mapping was performed with Burrows–Wheeler Alignment, duplicate removal was performed with Picard, local realignment and variant calling were performed with GATK, enrichment statistics were performed with NGSRich, and variant annotation was performed with ANNOVAR.43,44
During hard filtering, we used quality control metrics that included a phred-scaled quality score≥30, depth≥10, and Q/D≥5, and then focused on the following variants: nonsynonymous, frame shift, splicing site changes, and indels. Common variants from either the EVS29 or the 1000 Genomes Project (April of 2012)45 with a minor allele frequency value>1% in any population were excluded. We defined as novel those variants not reported in EVS, 1000 Genomes, National Center for Biotechnology Information dbSNP (build 137), aHUS mutation database (www.fh-hus.org), or our in-house database. To assign pathogenicity, we combined multiple functional prediction methods, including PhyloP, SIFT, PolyPhen2, LRT, MutationTaster, and GERP++. Prediction results for each variant were integrated to generate a pathogenicity score, which was the sum of tools predicting the variant to be deleterious. Missense nonsynonymous single nucleotide variants were considered likely deleterious when the PS≥4. Splicing site variants, nonsense mutations, and indels were not assigned a PS.
Multiplex Ligation-Dependent Probe Amplification
CNVs were evaluated over the contiguous CFH-CFHR genomic region using multiplex ligation-dependent probe amplification. Probes were designed according to MRC-Holland’s guidelines (MRC-Holland, Amsterdam, The Netherlands) or obtained from the literature.18,19,46,47 CNVs were assigned by data comparison of normalized peak areas generated in five control samples.48,49
Statistical Analyses
Data were analyzed using R (version 2.15.1). The Fisher exact test was used to compare allelic frequencies in cases and controls. All tests were two-tailed, and P values<0.05 were considered significant.
Disclosures
None.
Supplementary Material
Acknowledgments
This study was supported in part by the Foundation for Children with Atypical HUS.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013050453/-/DCSupplemental.
References
- 1.Noris M, Remuzzi G: Atypical hemolytic-uremic syndrome. N Engl J Med 361: 1676–1687, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Loirat C, Frémeaux-Bacchi V: Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 6: 60, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zimmerhackl LB, Besbas N, Jungraithmayr T, van de Kar N, Karch H, Karpman D, Landau D, Loirat C, Proesmans W, Prüfer F, Rizzoni G, Taylor MC, European Study Group for Haemolytic Uraemic Syndromes and Related Disorders : Epidemiology, clinical presentation, and pathophysiology of atypical and recurrent hemolytic uremic syndrome. Semin Thromb Hemost 32: 113–120, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Constantinescu AR, Bitzan M, Weiss LS, Christen E, Kaplan BS, Cnaan A, Trachtman H: Non-enteropathic hemolytic uremic syndrome: Causes and short-term course. Am J Kidney Dis 43: 976–982, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Le Quintrec M, Roumenina L, Noris M, Frémeaux-Bacchi V: Atypical hemolytic uremic syndrome associated with mutations in complement regulator genes. Semin Thromb Hemost 36: 641–652, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Kavanagh D, Goodship T: Genetics and complement in atypical HUS. Pediatr Nephrol 25: 2431–2442, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM, Turnpenny P, Goodship JA: Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 53: 836–844, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH: Complement factor I: A susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet 41: e84, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, Müslümanoğlu MH, Kavukcu S, Filler G, Pirson Y, Wen LS, Atkinson JP, Goodship TH: Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A 100: 12966–12971, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noris M, Brioschi S, Caprioli J, Todeschini M, Bresin E, Porrati F, Gamba S, Remuzzi G, International Registry of Recurrent and Familial HUS/TTP : Familial haemolytic uraemic syndrome and an MCP mutation. Lancet 362: 1542–1547, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, López-Trascasa M, Sánchez-Corral P, Morgan BP, Rodríguez de Córdoba S: Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A 104: 240–245, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frémeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, Moghal N, Kaplan BS, Weiss RA, Lhotta K, Kapur G, Mattoo T, Nivet H, Wong W, Gie S, Hurault de Ligny B, Fischbach M, Gupta R, Hauhart R, Meunier V, Loirat C, Dragon-Durey MA, Fridman WH, Janssen BJ, Goodship TH, Atkinson JP: Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112: 4948–4952, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sartz L, Olin AI, Kristoffersson AC, Ståhl AL, Johansson ME, Westman K, Fremeaux-Bacchi V, Nilsson-Ekdahl K, Karpman D: A novel C3 mutation causing increased formation of the C3 convertase in familial atypical hemolytic uremic syndrome. J Immunol 188: 2030–2037, 2012 [DOI] [PubMed] [Google Scholar]
- 14.Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, Pinto S, Goodship TH, Alberti M, Ribes D, Valoti E, Remuzzi G, Noris M, European Working Party on Complement Genetics in Renal Diseases : Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24: 475–486, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, López-Trascasa M, Sánchez-Corral P, Rodríguez de Córdoba S: Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet 14: 703–712, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Ermini L, Goodship TH, Strain L, Weale ME, Sacks SH, Cordell HJ, Fremeaux-Bacchi V, Sheerin NS: Common genetic variants in complement genes other than CFH, CD46 and the CFHRs are not associated with aHUS. Mol Immunol 49: 640–648, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, Moss J, Walport MJ, Cook HT, de Córdoba SR, Botto M: Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med 204: 1249–1256, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venables JP, Strain L, Routledge D, Bourn D, Powell HM, Warwicker P, Diaz-Torres ML, Sampson A, Mead P, Webb M, Pirson Y, Jackson MS, Hughes A, Wood KM, Goodship JA, Goodship TH: Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med 3: e431, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, Goodship JA, Licht C, Goodship TH, Skerka C: Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet 3: e41, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, Schmidt CQ, Staniforth SJ, Holmes LV, Ward R, Morgan L, Goodship TH, Marchbank KJ: Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 115: 379–387, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desch K, Motto D: Is there a shared pathophysiology for thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome? J Am Soc Nephrol 18: 2457–2460, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Motto D: Endothelial cells and thrombotic microangiopathy. Semin Nephrol 32: 208–214, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, Zoja C, Remuzzi G, Conway EM: Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 361: 345–357, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, Ji W, Overton JD, Mane SM, Nürnberg G, Altmüller J, Thiele H, Morin D, Deschenes G, Baudouin V, Llanas B, Collard L, Majid MA, Simkova E, Nürnberg P, Rioux-Leclerc N, Moeckel GW, Gubler MC, Hwa J, Loirat C, Lifton RP: Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45: 531–536, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadler JE: Thrombomodulin structure and function. Thromb Haemost 78: 392–395, 1997 [PubMed] [Google Scholar]
- 26.Schuster V, Seidenspinner S, Zeitler P, Escher C, Pleyer U, Bernauer W, Stiehm ER, Isenberg S, Seregard S, Olsson T, Mingers AM, Schambeck C, Kreth HW: Compound-heterozygous mutations in the plasminogen gene predispose to the development of ligneous conjunctivitis. Blood 93: 3457–3466, 1999 [PubMed] [Google Scholar]
- 27.Tefs K, Tait CR, Walker ID, Pietzsch N, Ziegler M, Schuster V: A K19E missense mutation in the plasminogen gene is a common cause of familial hypoplasminogenaemia. Blood Coagul Fibrinolysis 14: 411–416, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Tefs K, Gueorguieva M, Klammt J, Allen CM, Aktas D, Anlar FY, Aydogdu SD, Brown D, Ciftci E, Contarini P, Dempfle CE, Dostalek M, Eisert S, Gökbuget A, Günhan O, Hidayat AA, Hügle B, Isikoglu M, Irkec M, Joss SK, Klebe S, Kneppo C, Kurtulus I, Mehta RP, Ornek K, Schneppenheim R, Seregard S, Sweeney E, Turtschi S, Veres G, Zeitler P, Ziegler M, Schuster V: Molecular and clinical spectrum of type I plasminogen deficiency: A series of 50 patients. Blood 108: 3021–3026, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available at: http://evs.gs.washington.edu/EVS/ Accessed December 1, 2012
- 30.Schloesser M, Hofferbert S, Bartz U, Lutze G, Lämmle B, Engel W: The novel acceptor splice site mutation 11396(G—>A) in the factor XII gene causes a truncated transcript in cross-reacting material negative patients. Hum Mol Genet 4: 1235–1237, 1995 [DOI] [PubMed] [Google Scholar]
- 31.Chegeni R, Vickars L, Favaloro EJ, Lillicrap D, Othman M: Functional analysis of three recombinant A1-VWF domain mutants in comparison to wild type and plasma-derived VWF facilitates subtyping in type 2 von Willebrand disease. Thromb Res 127: 161–166, 2011 [DOI] [PubMed] [Google Scholar]
- 32.Ohlin AK, Norlund L, Marlar RA: Thrombomodulin gene variations and thromboembolic disease. Thromb Haemost 78: 396–400, 1997 [PubMed] [Google Scholar]
- 33.Ohlin AK, Marlar RA: Thrombomodulin gene defects in families with thromboembolic disease—a report on four families. Thromb Haemost 81: 338–344, 1999 [PubMed] [Google Scholar]
- 34.Modde F, Agustian PA, Wittig J, Dammrich ME, Forstmeier V, Vester U, Ahlenstiel T, Froede K, Budde U, Wingen AM, Schwarz A, Lovric S, Kielstein JT, Bergmann C, Bachmann N, Nagel M, Kreipe HH, Brocker V, Bockmeyer CL, Becker JU: Comprehensive analysis of glomerular mRNA expression of pro- and antithrombotic genes in atypical haemolytic-uremic syndrome (aHUS). Virchows Arch 462: 455–464, 2013 [DOI] [PubMed] [Google Scholar]
- 35.Chandler WL, Jelacic S, Boster DR, Ciol MA, Williams GD, Watkins SL, Igarashi T, Tarr PI: Prothrombotic coagulation abnormalities preceding the hemolytic-uremic syndrome. N Engl J Med 346: 23–32, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Proesmans W: The role of coagulation and fibrinolysis in the pathogenesis of diarrhea-associated hemolytic uremic syndrome. Semin Thromb Hemost 27: 201–205, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Van Geet C, Proesmans W, Arnout J, Vermylen J, Declerck PJ: Activation of both coagulation and fibrinolysis in childhood hemolytic uremic syndrome. Kidney Int 54: 1324–1330, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Kruez W, Linde R, Becker S, Seifried E: Successful treatment of haemolytic uraemic syndrome with recombinant tissue-type plasminogen activator. Lancet 341: 1665–1666, 1993 [DOI] [PubMed] [Google Scholar]
- 39.Bergstein JM, Riley M, Bang NU: Role of plasminogen-activator inhibitor type 1 in the pathogenesis and outcome of the hemolytic uremic syndrome. N Engl J Med 327: 755–759, 1992 [DOI] [PubMed] [Google Scholar]
- 40.Józsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, Zipfel PF, Skerka C: Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 111: 1512–1514, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT: Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 122: 282–292, 2013 [DOI] [PubMed] [Google Scholar]
- 42.Shearer AE, Hildebrand MS, Smith RJ: Solution-based targeted genomic enrichment for precious DNA samples. BMC Biotechnol 12: 20, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen R, Paul JS, Albrechtsen A, Song YS: Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet 12: 443–451, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goecks J, Nekrutenko A, Taylor J, Galaxy Team : Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA, 1000 Genomes Project Consortium : A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U: A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet 38: 1173–1177, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Schmid-Kubista KE, Tosakulwong N, Wu Y, Ryu E, Hecker LA, Baratz KH, Brown WL, Edwards AO: Contribution of copy number variation in the regulation of complement activation locus to development of age-related macular degeneration. Invest Ophthalmol Vis Sci 50: 5070–5079, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Ahn JW, Ogilvie CM, Welch A, Thomas H, Madula R, Hills A, Donaghue C, Mann K: Detection of subtelomere imbalance using MLPA: Validation, development of an analysis protocol, and application in a diagnostic centre. BMC Med Genet 8: 9, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.González JR, Carrasco JL, Armengol L, Villatoro S, Jover L, Yasui Y, Estivill X: Probe-specific mixed-model approach to detect copy number differences using multiplex ligation-dependent probe amplification (MLPA). BMC Bioinformatics 9: 261, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witzel-Schlömp K, Späth PJ, Hobart MJ, Fernie BA, Rittner C, Kaufmann T, Schneider PM: The human complement C9 gene: Identification of two mutations causing deficiency and revision of the gene structure. J Immunol 158: 5043–5049, 1997 [PubMed] [Google Scholar]
- 51.Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ: Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat 31: E1445–E1460, 2010 [DOI] [PubMed] [Google Scholar]
- 52.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey MA, Blouin J, Caudy A, Arzouk N, Cleper R, Francois M, Guest G, Pourrat J, Seligman R, Fridman WH, Loirat C, Atkinson JP: Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol 17: 2017–2025, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, Jozsi M, Neumann HP, Remuzzi G, Zipfel PF: Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest 111: 1181–1190, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ståhl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D: Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 111: 5307–5315, 2008 [DOI] [PubMed] [Google Scholar]
- 55.Jalanko H, Peltonen S, Koskinen A, Puntila J, Isoniemi H, Holmberg C, Pinomaki A, Armstrong E, Koivusalo A, Tukiainen E, Makisalo H, Saland J, Remuzzi G, de Cordoba S, Lassila R, Meri S, Jokiranta TS: Successful liver-kidney transplantation in two children with aHUS caused by a mutation in complement factor H. Am J Transplant 8: 216–221, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Gaucher C, Mercier B, Jorieux S, Oufkir D, Mazurier C: Identification of two point mutations in the von Willebrand factor gene of three families with the ‘Normandy’ variant of von Willebrand disease. Br J Haematol 78: 506–514, 1991 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.