

Abstract
Doxorubicin (DXR) and daunorubicin (DNR) inhibit hypoxia-inducible factor-1 (HIF-1) transcriptional activity by blocking its binding to DNA. Intraocular injections of DXR or DNR suppressed choroidal and retinal neovascularization (NV), but also perturbed retinal function as demonstrated by electroretinograms (ERGs). DXR was conjugated to novel copolymers of branched polyethylene glycol and poly(sebacic acid) (DXR-PSA-PEG3) and formulated into nanoparticles that when placed in aqueous buffer, slowly released small DXR-conjugates. Intraocular injection of DXR-PSA-PEG3 nanoparticles (1 or 10 μg DXR content) reduced HIF-1-responsive gene products, strongly suppressed choroidal and retinal NV, and did not cause retinal toxicity. In transgenic mice that express VEGF in photoreceptors, intraocular injection of DXR-PSA-PEG3 nanoparticles (10 μg DXR content) suppressed NV for at least 35 days. Intraocular injection of DXR-PSA-PEG3 nanoparticles (2.7 mg DXR content) in rabbits resulted in sustained DXR-conjugate release with detectable levels in aqueous humor and vitreous for at least 105 days. This study demonstrates a novel HIF-1-inhibitor-polymer conjugate formulated into controlled-release particles that maximizes efficacy and duration of activity, minimizes toxicity, and provides a promising new chemical entity for treatment of ocular NV.
Keywords: age-related macular degeneration, angiogenesis, diabetic retinopathy, nanoparticles
1. Introduction
Angiogenesis plays an important role in several disease processes including tumor growth, rheumatoid arthritis, corneal transplant rejection, neovascular age-related macular degeneration (NVAMD), and proliferative diabetic retinopathy (PDR). Vascular endothelial growth factor (VEGF) is an important stimulator of new vessel growth in each of these disease processes, and the development of specific inhibitors of VEGF has provided substantial benefits in patients with tumors (1, 2) or NVAMD (3). One strategy to achieve additional benefits is to use combination therapy to block other proangiogenic factors in addition to VEGF. Other well-validated stimulators of tumor and ocular neovascularization (NV) include platelet-derived growth factor-BB (PDGF-BB) (4, 5), stromal-derived factor-1 (SDF-1) (6–8), and angiopoietin-2 (ANGPT2) (9–12). Compared to selective blockade of VEGF, combined blockade of VEGF and PDGF provides greater benefits in models of tumor or ocular NV (13–16). Thus, one can envision the development of combination therapies in which specific inhibitors for several proangiogenic factors are used together to achieve improved efficacy.
The promoters for the genes encoding these proangiogenic factors contain a hypoxia response element (HRE) and they are transcriptionally activated by hypoxia-inducible factor-1 (HIF-1) (17,18). Thus, an alternative strategy to achieve “combination therapy” for neovascular diseases is to develop inhibitors of HIF-1. To achieve this goal, a cell-based reporter assay was developed to screen for drugs that inhibit HIF-1 transcriptional activity. This screen identified digoxin and other cardiac glycosides and the anthracycline chemotherapeutic agents doxorubicin (DXR) and daunorubicin (DNR) as potent inhibitors of HIF-1-mediated gene transcription (19,20). Digoxin acts by reducing HIF-1 levels, while DXR and DNR have no effect on levels and exert their effect by blocking the binding of HIF-1 to DNA. In tumor xenograft models, DXR and DNR suppressed the expression of multiple angiogenic factors and reduced tumor angiogenesis and tumor growth. This provides an explanation for the previous clinical observation that low-dose anthracyclines inhibit tumor angiogenesis, the basis for metronomic therapy (21). We previously demonstrated that digoxin prevents upregulation of several proangiogenic factors in ischemic retina and suppresses retinal and choroidal NV (22). In this study, we investigated the effects of DXR and DNR in models of ocular NV, including a nanoparticle-based controlled release strategy for delivery of DXR-polymer conjugates.
2. Methods
2.1. Animals
Pathogen-free C57BL/6 mice (Charles River, Wilmington, MA) and Dutch belted rabbits (Robinson Services Inc, Mocksville, NC) were treated in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the guidelines of the Johns Hopkins University Animal Care and Use Committee.
2.2. Synthesis of PSA-PEG3 polymer
Poly[(sebacic acid)-co-(polyethylene glycol)3] (PSA-PEG3) was synthesized by melt polycondensation. Briefly, sebacic acid (Sigma-Aldrich, St. Louis, MO) was refluxed in acetic anhydride (Sigma-Aldrich, St. Louis, MO) to form sebacic acid prepolymer (Acyl-SA). Citric-polyethylene glycol (PEG3) was prepared as previously described (41) using methoxy-poly(ethylene glycol)-amine (CH3O-PEG-NH2,) Mn 5,000 (Rapp Polymere GmbH, Tubingen, Germany). CH3O-PEG-NH2, 2.0 g, citric acid (Sigma-Aldrich, St. Louis, MO), 26 mg, dicyclohexylcarbodiimide (DCC, Acros Organics, Geel, Belgium), 83 mg, and 4-(dimethylamino)pyridine (DMAP; Acros Organics, Geel, Belgium), 4.0 mg, were added to 10 mL dichloromethane (DCM) (Fisher, Pittsburgh, PA), stirred overnight at room temperature, then precipitated and washed with anhydrous ether (Fisher, Pittsburgh, PA) and dried under vacuum. Next, Acyl-SA (90% w/w) and PEG3 (10% w/w) were placed into a flask under a nitrogen gas blanket and melted (180°C) and high vacuum was applied. Nitrogen gas was swept into the flask after 15 minutes. The reaction was allowed to proceed for 30 minutes. Polymers were cooled to ambient temperature, dissolved in chloroform (Sigma-Aldrich, St. Louis, MO), and precipitated into excess petroleum ether (Fisher, Pittsburgh, PA). The precipitate was collected by filtration and dried under vacuum to constant weight. Polymer structure was verified by 1H nuclear magnetic resonance (NMR) spectroscopy in CDCl3 (Bruker Avance 400 MHz FT-NMR, Madison, WI). The weight percentage of PEG estimated by 1H NMR was 10.5%. The PSA-PEG3 polymer was characterized by gel permeation chromatography (GPC) (JASCO, Easton, MD). The weight-average molecular weight (Mw) of the polymer was 26.7 kDa with a polydispersity index of 2.10.
2.3. Preparation of DXR-polymer particles
DXR-PSA-PEG3 particles were prepared using an oil-in-water emulsion method. First PSA-PEG3 and DXR (NetQem, Durham, NC) were dissolved in DCM (Fisher, Pittsburgh, PA) at defined ratios and concentrations. For nanoparticles, 80 mg PSA-PEG3 and 20 mg DXR were dissolved in 6 mL DCM and 2 mL dimethyl sulfoxide (DMSO) (Fisher, Pittsburg, PA). For microparticles, 200 mg PSA-PEG3 and 40 mg DXR were dissolved in 3 mL DCM and 1.5 mL DMSO. The mixture was incubated at 50°C for 2 hours before homogenizing (L4RT, Silverson Machines, East Longmeadow, MA) in 100 mL of an aqueous solution of 1 % polyvinyl alcohol (25 kDa, Polysciences, Warrington, PA). The speed and duration of homogenization for the synthesis of nanoparticles and microparticles were 8,000 rpm for 3 minutes and 3,000 rpm for 1 minute, respectively. Particles were hardened by allowing DCM to evaporate at room temperature while stirring for 2 hours. Nanoparticles and microparticles were collected by centrifugation at 4°C for 20 minutes at 20,000 × g and 9,000 × g, respectively, followed by washing thrice with cell culture grade/endotoxin-free water (HyClone, Fisher, Pittsburgh, PA).
The size of nanoparticles (0.65 μm) was determined by dynamic light scattering using a ZetaSizer Nano ZS (Malvern Instruments, Southborough, MA) at 25°C at a scattering angle of 90°. A Coulter MultiSizer 4 (Beckman Coulter, Inc., Miami, FL) was used to determine the size of the microparticles (23.0 μm). Particles were lyophilized and stored at −20°C until use. Particle morphology was characterized by cold cathode field emission scanning electron microscopy (SEM) (JEOL JSM-6700F, Peabody, MA).
The conjugation of DXR to PSA-PEG3 polymer within the particles was verified by GPC with a UV-visible light detector to measure DXR absorbance at 490 nm. No free DXR was dectected in the particles. To determine the drug loading, particles were dissolved in DCM/DMSO (1:1) and the total drug content in particles was calculated by measuring the UV absorbance at 490 nm and comparing to a calibration curve. Drug loading of the nanoparticles and microparticles was 23.6% and 17.2%, respectively, presented as DXR content by weight/particle weight ×100%.
2.4. DXR release from particles
Particles were suspended in phosphate buffered saline (PBS, pH 7.4) at 2 mg/mL and incubated at 37°C on a rotating platform (140 RPM). At selected time points, supernatant was collected by centrifugation (13,500 × g for 5 minutes) and particles were resuspended in fresh PBS. DXR-conjugate content was measured by absorbance at 490 nm. HPLC analyses of released drug demonstrated that the DXR-PSA drug conjugate was released from the particles. No free DXR was detected, demonstrating both that free DXR was not present in the particles and that free DXR was not released from the particles.
2.5. Intraocular injections
Murine intraocular injections were done under a dissecting microscope with a Harvard Pump Microinjection System and pulled glass micropipettes as previously described (42). In various models described below, mice received an intraocular injection of 1 μl of PBS or PBS containing 10 μg, 1 μg or 0.1 μg of free DNR or DXR, or DXR-PSA-PEG3 nanoparticles (10 μg, 1 μg or 0.1 μg DXR content).
2.6. Mouse model of laser-induced choroidal NV
Choroidal NV was induced by laser photocoagulation-induced rupture of Bruch’s membrane as previously described (23). Immediately after laser-induced rupture of Bruch’s membrane, mice were randomized to various treatment groups for intraocular injections. At 1, 4, 7 and 14 days after injection, fundus photographs were taken with a Micron III camera (Phoenix Research Laboratories, Inc., Pleasanton, CA). After 14 days, the mice were perfused with 1 ml of PBS containing 25 mg/ml of fluorescein-labeled dextran (2,000 kDa average molecular weight) (Sigma-Aldrich, St. Louis, MO) and choroidal flat mounts were examined by fluorescence microscopy. Images were captured with a Nikon Digital Still Camera DXM1200 (Nikon Instruments Inc., New York, NY). Image analysis software (Image-Pro Plus; Media Cybernetics, Silver Spring, MD) was used to measure the total area of choroidal NV at each rupture site with the investigator masked with respect to treatment group.
2.7. Oxygen-induced ischemic retinopathy
C57BL/6 mice placed in 75% oxygen at postnatal day (P) 7 and at P12 were returned to room air and given an intraocular injection of 1 μl of PBS or PBS containing DNR, DXR, or DXR-PSA-PEG3 nanoparticles. At P17, the area of retinal NV on the surface of the retina was measured as previously described (22,26). Briefly, mice were given an intraocular injection of 1 μl of rat anti-mouse platelet endothelial cell adhesion molecule-1 (PECAM-1) antibody (Pharmingen, San Jose, CA) and after 12 hours they were euthanized and eyes were fixed in phosphate-buffered formalin for 5 hours at 26°C. Retinas were dissected, washed, and incubated with goat-anti rat polyclonal antibody conjugated with Alexa 488 (Invitrogen, Carlsbad, CA) at 1:500 dilution at 26°C for 45 minutes and flat mounted. An observer masked with respect to treatment group measured the area of NV per retina by image analysis.
2.8. Measurement of VEGF, PDGF-BB, and SDF-1
Mice with ischemic retinopathy were given an intraocular injection of DXR-PSA-PEG3 nanoparticles or PBS at P12 and at P17 mice were euthanized, retinas were homogenized in lysis buffer (10 mM Tris-HCl, pH 7.2, 50 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, with proteinase inhibitor cocktail (Roche, Indianapolis, IN), and centrifuged at 6000 g for 10 minutes, 4°C. Supernatants were assayed for VEGF, PDGF-BB, and SDF-1 using ELISA kits following the manufacturer’s instructions (R&D Systems, Inc., Minneapolis, MN). Total protein concentrations were measured with a protein assay kit (BioRad, Hercules, CA).
2.9. Assessment of effects of nanoparticles on VEGF-induced retinal NV
Hemizygous rhodopsin/VEGF transgenic mice (P14) that express VEGF in photoreceptors (27, 28) were given an intraocular injection of 1 μl of PBS or PBS containing DNR, DXR, or DXR-PSA-PEG3 nanoparticles. At several time points after injection, mice were anesthetized, perfused with fluorescein-labeled dextran and retinal flat mounts were examined by fluorescence microscopy at 400X magnification, which provides a narrow depth of field, so that when NV along the outer edge of the retina is brought into focus, the remainder of the retinal vessels are out of focus, allowing easy delineation and quantification of the NV. Images were digitized with a three-color charge-coupled device video camera (Cool SNAPTM-Pro; Media Cybernetics, Silver Spring, MD) and a frame grabber. Image analysis software (Image-Pro Plus 5.0) was set to recognize fluorescently stained NV and used to calculate the total area of NV per retina. The investigator performing image analysis was masked with respect to treatment group.
2.10. Recording of electroretinograms (ERGs)
Scotopic and photopic ERGs were recorded using an Espion ERG Diagnosys machine (Diagnosys LLC, Littleton, MA) as previously described (43, 44). For scotopic recordings, mice were dark adapted overnight, and for photopic recordings, mice were adapted for 10 minutes to background white light at an intensity of 30 cd/m2. Recordings for both eyes were made simultaneously with electrical impedance balanced. Scotopic ERGs were recorded at 11 intensity levels of white light ranging from −3.00 to 1.40 log cd-s/m2. Six measurements were averaged for each flash intensity. Photopic ERGs were recorded at three intensity levels of white light ranging from 0.60 to1.40 log(cd-s/m2) with a 30 cd/m2 background. Five measurements were averaged for each flash intensity.
2.11. Measurement of outer nuclear layer (ONL) thickness
ONL thickness was measured as previously described (45). Mice were euthanized, a mark was placed at 12:00 at the corneal limbus, and eyes were removed and embedded in optimal cutting temperature compound. Ten micrometer frozen sections were cut parallel to the 12:00 or 9:00 meridian through the optic nerve and fixed in 4% paraformaldehyde. The sections were stained with hematoxylin and eosin, examined with an Axioskop microscope (Zeiss, Thornwood, NY), and images were digitized using a three charge-coupled device (CCD) color video camera (IK-TU40A;Toshiba, Tokyo, Japan) and a frame grabber. Image-Pro Plus software was used to outline the ONL. With the observer masked with respect to treatment group, ONL thickness was measured at six locations, 25% (S1), 50% (S2), and 75% (S3) of the distance between the superior pole and the optic nerve and 25% (I1), 50% (I2), and 75%(I3) of the distance between the inferior pole the optic nerve.
2.12. Pharmacokinetic studies in rabbits
Five Dutch-belted rabbits were anesthetized with an intramuscular injection of 25 mg/kg ketamine and 2.5 mg/kg xylazine and pupils were dilated with 1% tropicamide. After instillation of 0.5% proparacaine hydrochloride, 100 μL of DXR-PSA-PEG3 particles (2.7 mg DXR content) was injected in one eye and 100 μL of saline was injected in the fellow eye. On 1, 8, 18, 33, 57, 97, and 105 days after injection, the rabbits were anesthetized and a 30 gauge needle was inserted into the anterior chamber and 100 μL of aqueous was removed. On day 105, rabbits were euthanized, eyes were removed and a vitreous sample was obtained. Samples were stored at −80°C until assayed for drug concentration.
2.13. Quantitation of released drug conjugate
A total of 100 μL of aqueous humor sample or vitreous sample was mixed with 200 μL of methanol (Fisher, Pittsburgh, PA) and incubated at 4°C for 3 hr. After centrifugation (15,000 ×g, 10 minutes) and filtration through a 0.2 μm PTFE filter, 150 μL of the filtrate was injected into a Waters HPLC system equipped with a c18 reverse phase column (5 μm, 4.6×250 mm; Grace, Deerfield IL). Released drug conjugate was eluted by an isocratic mobile phase containing water and acetonitrile (60%:40%, v/v; Fisher, Pittsburgh, PA) at 1 mL/minute and detected using a fluorescence detector (excitation wavelength: 500 nm, emission wavelength: 551 nm). The estimated limit of detection was 10 ng/mL (17 nM). A series of DXR aqueous solutions at different concentrations were used as calibration standards. The data were analyzed using Empower 3 chromatography data software (Waters Corporation, Milford MA).
3. Results
3.1. Anthracyclines suppress choroidal and retinal NV
In a mouse model of choroidal NV (23) that is predictive of drug effects in patients with neovascular AMD (24, 25), intraocular injection of 10 μg of DXR suppressed choroidal NV, while injection of 1 or 0.1 μg had no significant effect (Figure 1A–C). In neonatal mice with oxygen-induced ischemic retinopathy, a model predictive of effects in PDR, intraocular injection of 1 μg of DXR, but not 0.1 or 0.01 μg, significantly reduced the area of retinal NV (Figure 1D–F). The NV is visualized on retinal flat mounts after in vivo immunofluorescent staining with anti-PECAM1, a technique that selectively stains NV and hyaloid vessels (larger vessels are hyaloid vessels and everything else stained green is NV (26)). The mean area of choroidal or retinal NV in fellow eyes was not significantly different from that in eyes of mice in which both eyes were injected with vehicle indicating that there was no systemic effect from intraocular injections of DXR. Another HIF-1 inhibitor, DNR, had similar effects to those of DXR (Supplemental Figure 1).
Fig. 1.

Doxorubicin suppressed choroidal and retinal NV but also had toxicity.
Fourteen days after laser-induced rupture of Bruch’s membrane and intraocular injection of 10 μg of doxorubicin (DXR) or PBS, the area of choroidal NV was much smaller in DXR-injected eyes (A) compared to PBS-injected eyes (B). The mean area of choroidal NV was significantly less in eyes injected with 10 μg, but not those injected with 1.0 or 0.1 μg (C). In mice with oxygen-induced ischemic retinopathy, at P17, five days after intraocular injection of 1 μg of DXR, there was almost complete elimination of retinal NV with only hyaloid vessels and some precipitated DXR (arrowheads) seen on retinal flat mounts (D). Doses of 0.1 or 0.01 μg of DXR did not significantly reduce retinal NV (F). C57BL/6 mice (n = 15 for each dose level) were given an intraocular injection of 1 μg of DXR or PBS. Scotopic and photopic ERGs were done 14 days after injection and mean b-wave amplitudes were plotted against log flash intensity. There were significant reductions in scotopic and photopic b-wave amplitudes at all flash intensities in DXR-treated eyes (G, H, respectively). Statistical comparisons were made by unpaired t-test; *p<0.05.
Five days after injection of 1 μg of DXR (Figure 1D, arrowheads) or DNR (Supplemental Figure 1D, arrowheads) precipitated drug was visualized on the surface of the retina. Due to this observation and because DXR and DNR are antimetabolites as well as HIF-1 inhibitors, we performed electroretinograms (ERGs) to assess retinal function. Fourteen days after intraocular injections of 1 μg of DXR (Figure 1G, H) or DNR (Supplemental Figure 1G, H) there was a significant reduction in mean scotopic and photopic b-wave amplitudes, while injections of 0.1 μg had no deleterious effect on retinal function (data not shown). These data indicate that while DXR and DNR strongly suppress ocular NV, bolus injections of 1 μg or more of free drug can cause retinal toxicity.
3.2. Conjugation of DXR to polymer and preparation of nanoparticles
Sustained release formulations provide a potential strategy to minimize risk when using drugs with a narrow therapeutic window. In addition, prolonging the duration of effect after intraocular injections is a major goal in the development of new treatments for ocular NV. To ensure reliable, slow release from an inaccessible depot, DXR was conjugated to PSA-PEG3 (Supplemental Figure 2A) to generate 0.65 μm particles containing 23.6% DXR. Particle morphology, chemical composition of the DXR-PSA-PEG3 polymer-drug conjugate, and conjugation of DXR to the PSA-PEG3 polymer, were verified by SEM, NMR (Supplemental Figure 2, B–E), and GPC analyses (data not shown). DXR was released as a conjugate with sebacic acid in a steady fashion for over seven days under infinite sink conditions in PBS at 37°C in vitro (Supplemental Figure 2F) with no initial rapid drug release phase (i.e., no “burst effect”). Free DXR was not detected in vitro, indicating both that no free DXR was contained in the nanoparticles and that all released DXR was conjugated to sebacic acid (DXR-PSA). Larger (23.0 μm) particles containing 17.2% DXR displayed more sustained in vitro drug-conjugate release, for over 30 days under sink conditions (Supplemental Figure 2G), demonstrating that the duration of drug release can be extended simply by increasing particle size.
3.3. Effect of DXR-polymer nanoparticles on ocular NV
The effect of intraocular injection of DXR nanoparticles was first tested in mice with choroidal NV due to laser-induced rupture of Bruch’s membrane. After rupture of Bruch’s membrane, C57BL/6 mice had intraocular injection of DXR-PSA-PEG3 nanoparticles (1 μg DXR content). Fundus photos showed a large orange aggregate of nanoparticles overlying the posterior retina 1 day after injection (Figure 2A, arrows) that decreased slowly over time and was still visible on days 7 and 14 (Figure 2B,C, arrows). In mice perfused with fluorescein-labeled dextran to visualize choroidal NV by fluorescence microscopy at day 14, the area of choroidal NV appeared smaller in eyes given an intraocular injection of DXR-PSA-PEG3 nanoparticles (Figure 2D) compared to fellow eyes injected with PBS (Figure 2E). Image analysis confirmed that compared to eyes injected with PBS, the mean area of choroidal NV was significantly less in eyes injected with DXR-PSA-PEG3 nanoparticles (10, 1.0, or 0.1 μg DXR content; Figure 2F). In mice with oxygen-induced ischemic retinopathy, intraocular injections of DXR-PSA-PEG3 nanoparticles (1 μg DXR content) at postnatal day 12 (P12) significantly reduced the mean area of retinal NV compared to PBS-injected fellow eyes at P17 (Figure 2G–I).
Fig. 2.

DXR-PSA-PEG3 nanoparticles inhibit choroidal and retinal NV.
After laser-induced rupture of Bruch’s membrane in 3 locations, C57BL/6 mice received DXR-PSA-PEG3 0.65 μm particles (1 μg DXR content) via an intravitreal injection. Between 1 and 14 days after injection the nanoparticles were seen as orange aggregates (A, Day 1, B, Day 7, C, Day 14, arrows) in the posterior vitreous in front of the optic nerve (ON). Areas of laser photocoagulation are also visible (white arrowheads). The area of choroidal NV at Bruch’s membrane rupture sites appeared smaller in eyes injected with DXR-PSA- PEG3 nanoparticles (D) compared to those injected with PBS (E) and image analysis showed a significant reduction in mean area of choroidal NV in eyes injected with DXR-PSA- PEG3 nanoparticles (0.1, 1.0, or 10 μg DXR content) compared to fellow eyes injected with PBS (F). At P12, mice with oxygen-induced ischemic retinopathy received DXR-PSA-PEG3 nanoparticles (1 μg DXR content) in one eye and PBS in the fellow eye and at P17, there was a significant reduction in retinal NV in the former (G–I).
3.4. DXR-polymer nanoparticles inhibit key HIF-1-regulated angiogenic effectors and mediate regression of preexisting choroidal NV
The levels of three key HIF-1-regulated pro-angiogenic factors, VEGF, PDGF-BB, and SDF-1, were measured at P17 in the retinas of mice with ischemic retinopathy that had been given an intraocular injection of DXR-PSA-PEG3 nanoparticles (1 μg DXR content) or PBS at P12. DXR-PSA-PEG3 significantly reduced levels of each of the pro-angiogenic factors confirming its ability to suppress HIF-1 transcriptional activity in the retina (Figure 3, A–C). In contrast to treatment with a VEGF antagonist alone, combination treatment with a VEGF antagonist and a PDGF-BB antagonist (15) or treatment with a kinase inhibitor that blocks both VEGF and PDGF receptors (16) cause regression of established choroidal NV. Since DXR-PSA-PEG3 nanoparticles suppress both VEGF and PDGF-BB, we tested their effect on established choroidal NV. The area of choroidal NV was measured in a cohort of mice 7 days after rupture of Bruch’s membrane to provide the baseline area and DXR-PSA-PEG3 nanoparticles (1 μg DXR content) or PBS were injected in the eyes of the remainder of the mice. After an additional 7 days, eyes treated with DXR-PSA-PEG3 nanoparticles had a mean area of choroidal NV that was significantly less than the baseline area or that seen in control eyes injected with PBS (Figure 3D). These data demonstrate that DXR-PSA-PEG3 nanoparticles cause regression of established choroidal NV.
Fig. 3.
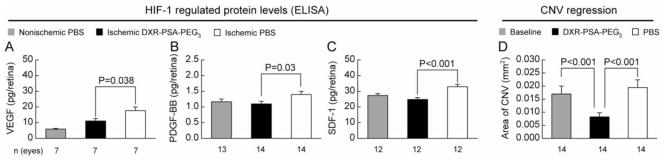
DXR-PSA-PEG3 nanoparticles inhibit HIF-1- regulated proangiogenic factors and mediate regression of preexisting ocular NV.
P12 mice with ischemic retinopathy received DXR-PSA-PEG3 nanoparticles (1 μg DXR content) in one eye and PBS in the fellow eye. Five days later (P17) protein was isolated from the retinas and analyzed by ELISA for the levels of HIF-1-regulated proteins, VEGF, PDGF-BB, and SDF-1, and compared to protein levels of normal, nonischemic retinas. A significant reduction in VEGF (A), PDGF-BB (B), and SDF-1 (C) was observed in particle-treated ischemic eyes compared to PBS-treated ischemic eyes. To evaluate regression of choroidal NV the mean area of choroidal NV was measured in a cohort of mice 7 days after rupture of Bruch’s membrane (baseline). The remaining mice were treated with DXR-PSA-PEG3 nanoparticles (10 μg DXR content) in one eye and PBS in the fellow eye. Seven days later, the mean area of choroidal NV was significantly less in nanoparticle-injected eyes than fellow eyes and also less than the baseline area of NV indicating regression of NV (D).
3.5. Intraocular injection of DXR nanoparticles did not cause toxicity as measured by ONL thickness or ERG amplitudes
The appearance of the retina was normal 14 days after intraocular injection of DXR-PSA-PEG3 (10 μg DXR content; Figure 4A) and was similar to that in eyes injected with PBS (Figure 4B). Compared to eyes injected with PBS, those injected with DXR-PSA-PEG3 showed no difference in mean outer nuclear layer thickness at several locations in the retina (Figure 4C) indicating that there was no reduction in photoreceptors in DXR-PSA-PEG3 – injected eyes. There was also no reduction in scotopic or photopic ERG b-wave amplitudes 14 days after intraocular injection of DXR-PSA-PEG3 nanoparticles (10 μg DXR content; Figure 4D, E) compared to eyes injected with PBS.
Fig. 4.
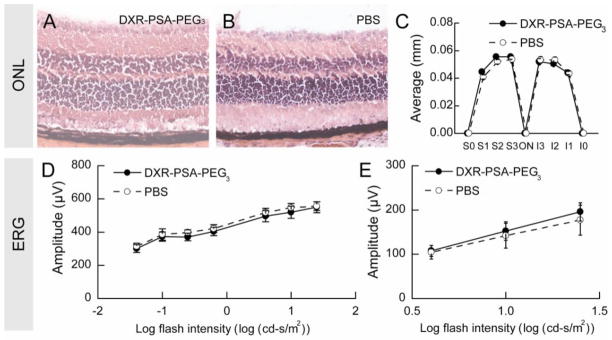
DXR-PSA-PEG3 nanoparticles do not cause retinal toxicity.
Two weeks following intraocular injection of DXR-PSA-PEG3 nanoparticles (10 μg DXR content) (A) or PBS (B) retinas appeared normal and there was no difference in outer nuclear layer (ONL) thickness at several locations in the retina (C). Electroretinograms (ERGs) 2 weeks after injection showed no difference in mean scotopic or photopic b-wave amplitudes over a range of stimulus intensities in eyes injected with DXR-PSA-PEG3 nanoparticles (10 μg DXR content) compared to those injected with PBS (D,E, respectively). Statistical comparisons were made by unpaired t-test.
3.6. Prolonged suppression of NV in rho/VEGF transgenic mice and sustained release in rabbits
Rho/VEGF transgenic mice, in which the rhodopsin promoter drives expression of VEGF in photoreceptors, have sustained expression of VEGF starting at P7 and provide an excellent model to test the duration of activity of a therapeutic agent (27,28). At P14, hemizygous rho/VEGF mice were given an intraocular injection of DXR-PSA-PEG3 nanoparticles (10 μg DXR content) in one eye and PBS in the fellow eye. At 4 (Figure 5A–C) or 5 weeks (Figure 5D–F), but not 6 weeks (Figure 5G–I) after injection, the mean area of subretinal NV was significantly less in DXR-PSA-PEG3 nanoparticle-injected eyes than vehicle-injected fellow eyes. This suggests that therapeutic levels were present for at least 5 weeks after injection.
Fig. 5.

Prolonged suppression of NV in rho/VEGF transgenic mice and sustained release in rabbits.
At P14 hemizygous rho/VEGF mice were given an intraocular injection of DXR-PSA-PEG3 nanoparticles (10 μg DXR content) in one eye and PBS in the fellow eye. At 4 (A–C) or 5 weeks (D–F), but not 6 weeks (G–I) after injection, nanoparticle-injected eyes had significantly less subretinal NV than fellow eyes injected with PBS. Aqueous samples obtained from 5 rabbits injected with DXR-PSA-PEG3 (2.7 mg DXR content) showed mean levels of DXR-conjugate that were well above the limit of detection (17 nM) for over 3 months (J). Rabbits were euthanized at day 105 and DXR-conjugate was still detectable in aqueous and much higher in vitreous (K). Scale bar = 100 μm.
To assess pharmacokinetics in an eye closer in size to a human eye than a mouse eye, DXR-PSA-PEG3 nanoparticles (2.7 mg DXR content) were injected into the vitreous cavity of rabbits and DXR-conjugate was assayed in aqueous humor samples at several time points (Figure 5J), and in the vitreous at study termination, Day 105 (Figure 5K). This dose was chosen, because considering the vitreous volumes of mice (0.0053 ml) versus rabbits (1.5 ml), it should provide a comparable post-injection drug-conjugate concentration of 1.8 mg/ml. One day after injection, mean aqueous level of the DXR-conjugate was 11.91 ± 4.13 μM. At Day 105 the aqueous level was 0.23 ± 0.31 μM, while the vitreous concentration was considerably higher at 11.91 ± 10.45 μM. Thus, following administration of DXR-PSA-PEG3 nanoparticles in the rabbit eye, DXR-conjugate release was sustained for well over three months.
4. Discussion
The two major types of ocular NV are choroidal NV and retinal NV. Choroidal NV occurs in diseases of the retinal pigmented epithelium/Bruch’s membrane complex, such as AMD, the most common cause of moderate and severe vision loss in individuals older than 60 (29). Retinal NV occurs in ischemic retinopathies, including diabetic retinopathy, retinopathy of prematurity, and retinal vein occlusions, which form a group of highly prevalent causes of severe vision loss. In fact, diabetic retinopathy is the most common cause of moderate and severe loss of vision in working aged Americans (30). Thus diseases complicated by retinal and choroidal NV are major public health problems.
VEGF is a key stimulator of choroidal (31) and retinal NV (32–34). Monthly intraocular injections of ranibizumab (Lucentis®, an antibody fragment that binds all isoforms of VEGF-A), causes improvement in visual acuity of 3 or more lines on a standardized eye chart (a doubling of the visual angle which is considered a substantial improvement) in 34–40% of patients with choroidal NV due to AMD (3,35). Injections of bevacizumab (Avastin®), a full length humanized monoclonal antibody that binds all isoforms of VEGF-A provides similar benefit (36). Injections of bevacizumab have also shown benefit in PDR and a severe complication of ischemic retinopathies, NV glaucoma (37,38). While VEGF antagonists have revolutionized the treatment of NV ocular diseases, there is still substantial room for improvement. In animal models, combined blockade of VEGF and PDGF-B may provide greater benefit than blockade of VEGF alone (15,16). Selective VEGF antagonists prevent growth of choroidal NV, but do not cause it to regress, whereas combined blockade of VEGF and PDGF causes regression (15,16). An early phase clinical trial confirmed that regression of choroidal NV commonly occurred with combined injections of VEGF-A and PDGF-BB antagonists, but not with the VEGF-A antagonist alone (39). It is likely that combination treatment with antagonists of many proangiogenic factors will be tested in the future.
Since HIF-1 upregulates many proangiogenic factors, including VEGF-A, PDGF-BB, and SDF-1 (18), suppression of HIF-1 is comparable to combination therapy with multiple specific inhibitors. In this study we found that DXR-PSA-PEG3 nanoparticles significantly reduce VEGF-A, PDGF-BB, and SDF-1 in ischemic retina. This confirms that the mechanism by which they work is by antagonism of HIF-1 and explains the strong efficacy and the ability to cause regression.
While the targeting of multiple gene products with HIF-1 antagonists has advantages with regard to efficacy for treatment of ocular NV, there may be potential hazards. Both DXR and DNR have poor aqueous solubility and form precipitates on the retinal surface after intraocular injection (Figure 1 and Supplemental Figure 1) which is likely to cause high local concentrations. This is associated with reduction in ERG amplitudes indicating reduced retinal function.
To stringently control DXR levels in the eye, DXR was covalently linked to PSA-PEG3 and the drug-polymer conjugate was formulated into nanoparticles. This novel approach allows for sustained delivery of low doses of released DXR-PSA conjugate to the retina with no substantial fluctuation in levels other than gradual reduction over the course of several months. There was no reduction in ERG amplitudes after intraocular injection of DXR-PSA-PEG3 (1 or 10 μg DXR content) and a single injection completely suppressed NV in transgenic mice with VEGF expression in photoreceptors for at least 35 days. To put this finding in perspective, in this same model, intraocular injection of 10 μg of ranibizumab or up to 25 μg of bevacizumab caused significant suppression of NV 14 days after injection, but the effect was completely lost by 21 days (40). Since strong efficacy was seen after intraocular injection of DXR nanoparticles (1 μg DXR content) and no toxicity was seen after injection of DXR nanoparticles (10 μg DXR content), there is a good therapeutic window. In rabbits, DXR-conjugate was present in aqueous humor and vitreous for at least 3 months after intraocular injection of nanoparticles (2.7 mg DXR content). Vitreous DXR-conjugate levels were 50-fold higher than those measured in the aqueous humor at Day 105 and well above the 200 nM DXR concentration shown to strongly inhibit HIF-1 activity in vitro (20).
Notably, the in vitro drug conjugate release rate greatly underestimated the duration of drug release in the eye. Under infinite sink conditions, in vitro drug release duration from the nanoparticles was seven days (Supplemental Figure 2F), while it was over 100 days in the rabbit eye (Figure 5J), and efficacy was sustained in mice for at least five weeks (Figure 5D,E). These data are consistent with other recent reports where it was observed that in vitro release into PBS was significantly faster than in vivo release in the vitreous of living animals (46–48). As the DXR-PSA released drug conjugate has a solubility approximately 300-fold lower than that of free DXR, the reduced solubility combined with the likelihood that infinite sink conditions do not exist in the eye, results in further hindrance of drug release in vivo. Therefore, drug-conjugate release in the eye displays a slower, more sustained release rate compared to in vitro release under infinite sink conditions.
Thus, this drug-conjugate nanoparticle approach provides a means to take advantage of the strong antiangiogenic activity of this small molecule HIF-1 inhibitor, prolong its duration of action, and prevent high intraocular levels which exceed the threshold for toxic effects. A sustained-release, small molecule-based HIF-1 inhibitory approach has considerable advantages compared to a biologic therapy, as proteins are ineffective against intracellular targets such as HIF-1. Furthermore, a single-agent that targets multiple pro-angiogenic effectors is expected to have substantial benefits from a clinical perspective and could provide a major step forward in the treatment of highly prevalent neovascular diseases of the eye.
5. Conclusions
Doxorubicin (DXR) is a HIF-1 inhibitor with potent antiangiogenic properties [20]. To improve its therapeutic window for the treatment of ocular neovascular diseases, a novel, sustained-released, fully biodegradable, nanoparticle formulation, DXR-PSA-PEG3, was developed. When delivered intraocularly to mice, DXR-PSA-PEG3 slowly released DXR- conjugates that significantly reduced HIF-1-responsive gene products and mediated both regression and long-term suppression of ocular NV without causing toxicity. Thus, DXR-PSA-PEG3 represents a promising small molecule-based HIF-1 inhibitory approach that simultaneously targets multiple pro-angiogenic effectors for the development of an improved therapy for neovascular eye diseases.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang JC, et al. A randomized trial of bevacizumab, an anti-vascular edntothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Eng J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld PJ, et al. Ranibizumab for neovascular age-related macular degeneration. N Eng J Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 4.Shih AH, et al. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Canc Res. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 5.Seo M-S, et al. Photoreceptor-specific expression of PDGF-B results in traction retinal detachment. Am J Pathol. 2000;157:995–1005. doi: 10.1016/S0002-9440(10)64612-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kryczek I, et al. CXCL12 and vascular endothelial growth factor synergtistically induce neoangiogenesis in human ovarian cancers. Canc Res. 2005;65:465–472. [PubMed] [Google Scholar]
- 7.Guleng B, et al. Blockade of the stomal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Canc Res. 2005;65:5864–5871. doi: 10.1158/0008-5472.CAN-04-3833. [DOI] [PubMed] [Google Scholar]
- 8.Lima e Silva R, et al. The SDF-1/CXCR4 ligand/receptor pair is an important contributor to several types of ocular neovascularization. FASEB J. 2007;21:3219–3230. doi: 10.1096/fj.06-7359com. [DOI] [PubMed] [Google Scholar]
- 9.Stratmann A, Risau W, Plate KH. Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol. 1998;153:1459–1466. doi: 10.1016/S0002-9440(10)65733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holash J, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 11.Hackett SF, Wiegand SJ, Yancopoulos G, Campochiaro P. Angiopoietin-2 plays an important role in retinal angiogenesis. J Cell Physiol. 2002;192:182–187. doi: 10.1002/jcp.10128. [DOI] [PubMed] [Google Scholar]
- 12.Oshima Y, et al. Angiopoietin-2 enhances retinal vessel sensitivity to vascular endothelial growth factor. J Cell Physiol. 2004;199:412–417. doi: 10.1002/jcp.10442. [DOI] [PubMed] [Google Scholar]
- 13.Bergers G, Song S, Meyer-Morse N, Bersland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erber R, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004;18:338–340. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 15.Jo N, et al. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol. 2006;168:2036–2053. doi: 10.2353/ajpath.2006.050588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi K, et al. The multi-targeted kinase inhibitor pazopanib causes suppression and regression of choroidal neovascularization. Arch Ophthalmol. 2009;127:494–499. doi: 10.1001/archophthalmol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly BD, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha sythesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K, et al. Antracycline chemotherpy inhibits HIF-1 transcriptional activity and tumor induced mobilizatio of circulating angiogenic cells. Proc Natl Acad Sci USA. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Hanahan D, Bergers G, Bergsland E. Less is more, regularly:metronomic dosing of cytoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105:1045–1047. doi: 10.1172/JCI9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida T, et al. Digoxin inhibits retinal ischemia-induced HIF-1alpha expression and ocular neovascularization. FASEB J. 2010;24:1759–1767. doi: 10.1096/fj.09-145664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tobe T, et al. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol. 1998;153:1641–1646. doi: 10.1016/S0002-9440(10)65753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saishin Y, et al. VEGF-TRAPR1R2 suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195:241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- 25.Heier JS, et al. The 1-year results of CLEAR-IT 2, a phase 2 study of vascular endothelial growth factor trap-eye dosed as needed after 12-week fixed dosing. Ophthalmology. 2011;118:1098–1106. doi: 10.1016/j.ophtha.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Shen J, et al. In vivo immunostaining demonstrates macrophages associate with growing and regressing vessels. Invest Ophthalmol Vis Sci. 2007;48:4335–4341. doi: 10.1167/iovs.07-0113. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto N, et al. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol. 1997;151(1):281–291. [PMC free article] [PubMed] [Google Scholar]
- 28.Tobe T, et al. Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci. 1998;39(1):180–188. [PubMed] [Google Scholar]
- 29.Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99:933–943. doi: 10.1016/s0161-6420(92)31871-8. [DOI] [PubMed] [Google Scholar]
- 30.Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. II Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch Ophthalmol. 1984;102:527–532. doi: 10.1001/archopht.1984.01040030405011. [DOI] [PubMed] [Google Scholar]
- 31.Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is an important stimulator in a model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41:3158–3164. [PubMed] [Google Scholar]
- 32.Aiello LP, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozaki H, et al. Intravitreal sustained release of VEGF causes retinal neovascularization in rabbits and breakdown of the blood-retinal barrier in rabbits and primates. Exp Eye Res. 1997;64(4):505–517. doi: 10.1006/exer.1996.0239. [DOI] [PubMed] [Google Scholar]
- 34.Ozaki H, et al. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol. 2000;156:679–707. doi: 10.1016/S0002-9440(10)64773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown DM, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Eng J Med. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- 36.Group CR, et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Eng J Med. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arevalo JF, et al. Intravitreal bevacizumab for proliferative diabetic retinopathy: 6 months follow-up. Eye. 2009;23:117–123. doi: 10.1038/sj.eye.6702980. [DOI] [PubMed] [Google Scholar]
- 38.Iliev ME, Doming D, Wolf-Schnurrbursch U, Wolf S, Sarra GM. Intravitreal bevacizumab in the treatment of neovascular glaucoma Am. J Ophthalmol. 2006;142:1054–1056. doi: 10.1016/j.ajo.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 39.Boyer DS Ophthotech Anti-PDGF in AMD Study Group. Combined inhibition of platelet-derived (PDGF) and vascular endothelial (VEGF) growth factors for the treatment of neovascular age-related macular degeneration (NV-AMD). Results of a phase 1 study. Invest Ophthalmol Vis Sci. 2009 Online ARVO abstract 1260. [Google Scholar]
- 40.Miki K, et al. Effects of intraocular ranibizumab and bevacizumab in transgenic mice expressing human vascular endothelial growth factor. Ophthalmology. 2009;116:1748–1754. doi: 10.1016/j.ophtha.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ben-Shabat S, Kumar N, Domb AJ. PEG-PLA block copolymer as potential drug carrier: preparation and characterization. Macromol Biosci. 2006;6:1019–1025. doi: 10.1002/mabi.200600165. [DOI] [PubMed] [Google Scholar]
- 42.Mori K, et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001;188:253–263. doi: 10.1002/jcp.1114. [DOI] [PubMed] [Google Scholar]
- 43.Okoye G, et al. Increased expression of BDNF preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J Neuosci. 2003;23:4164–4172. doi: 10.1523/JNEUROSCI.23-10-04164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komeima K, Rogers BS, Lu L, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2006;103:11300–11305. doi: 10.1073/pnas.0604056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213:809–815. doi: 10.1002/jcp.21152. [DOI] [PubMed] [Google Scholar]
- 46.Hartmann KI, Nieto A, Wu EC, Freeman WR, Kim JS, Chhablani J, Sailor MJ, Cheng L. Hydrosilylated porous silicon particles function as an intravitreal drug delivery system for danorubicin. J Ocular Pharm Ther. 2013;29:493–500. doi: 10.1089/jop.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu EC, Andrew JS, Cheng L, Freeman WR, Pearson L, Sailor MJ. Real time monitoring of sustained drug release using optical properties of porous silicon photonic crystal particles. Biomaterials. 2011;32:1957–1966. doi: 10.1016/j.biomaterials.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chhablain J, Nieto A, Hou H, Wu EC, Freeman WR, Sailor MJ, Cheng L. Oxidized porous silicon particles covalently grafted with daunorubicin as a sustained intraocular drug delivery system. IOVS. 2013;54:1268–1279. doi: 10.1167/iovs.12-11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.