

Abstract
Hypoxia is a significant feature of solid tumor cancers. Hypoxia leads to a more malignant phenotype that is resistant to chemotherapy and radiation, is more invasive and has greater metastatic potential. Hypoxia activates the hypoxia inducible factor (HIF) pathway, which mediates the biological effects of hypoxia in tissues. The HIF complex acts as a transcription factor for many genes that increase tumor survival and proliferation. To date, many HIF pathway inhibitors indirectly affect HIF but there have been no clinically approved direct HIF inhibitors. This can be attributed to the complexity of the HIF pathway, as well as to the challenges of inhibiting protein–protein interactions.
Hypoxia in cancer
Cancer is the second most common cause of death in the USA, with practically one in every four deaths due to cancer [301]. Moreover, the most recent data report that in 2008 there were over 12.5 million new cases of cancer diagnosed worldwide and that by 2030 this number is anticipated to swell to over 20 million [302]. Many cancers involve solid tumor formation, and during periods of rapid growth, tumors out-grow existing blood supply, leading to the development of hypoxic (partial oxygen pressure of less than 5 Torr) and anoxic regions [1]. Tumors remedy this by producing angiogenic factors that lead to the formation of tumor vasculature, although, with structural and functional anomalies. These include arteriovenous shunts, blind ends, occlusions, high angle branching patterns, and broken, leaky vessels [2]. Abnormalities in the tumor vasculature limit oxygen delivery, leading to acute hypoxia [3].
Hypoxic tumors are more resistant to radiation and chemotherapy, are more invasive, are genetically unstable, resist apoptosis and have greater metastatic potential; all of which leads to poorer prognosis overall for patients [4,5]. It has been demonstrated that tumor irradiation is three-times more effective when carried out under oxygen-rich versus anoxic conditions. Moreover, the effectiveness of anticancer therapeutics that target rapidly dividing cells is reduced against hypoxic cells due to the decreased rate of cell proliferation that increases with distance from vasculature [6]. Another factor in treatment resistance is due to the cancer stem-like cells (CSCs), which are a relatively rare subpopulation of tumor cells with self-renewal capacity. CSCs reside in hypoxic niches of tumors and are more resistant to radio- and chemo-therapy-induced DNA damage, allowing them to survive the treatment and repopulate the tumor with their progeny. Typically, prolonged hypoxia triggers cell apoptosis, but in tumors, it can lead to the selection of tumor cells with mutant p53, which are resistant to apoptosis and confer a more malignant phenotype [7]. Clinical studies have shown that many cancers with hypoxic tumors are more likely to be metastatic, including soft tissue sarcomas, squamous cell head and neck carcinomas, cervical carcinomas and malignant melanomas [8]. Therefore, hypoxia can either lead to cell death through apoptotic or necrotic pathways, or to cell proliferation through activation of various other pathways [3].
The hypoxia inducible factor pathway
The hypoxia inducible factor (HIF) pathway is a major mediator of the biological effects of hypoxia in tissues (Figure 1) [9]. HIFs are basic-loop-helix-loop motif heterodimeric transcription factors composed of two subunits: an oxygen-regulated α subunit (HIF-1α, -2α, or -3α) and the constitutively expressed HIF-1β (also called aryl hydrocarbon receptor nuclear translocator) [10]. Under normoxic conditions, HIF-α subunits are hydroxylated by Fe2+-dependent HIF prolyl hydroxylases (PHD), mostly PHD2, at two proline residues (402 and 564 in human HIF-1α) located in the oxygen-dependent degradation domain [11,12]. This dihydroxylated form of HIF-α is bound by VHL, which is an E3 ubiquitin ligase that leads to the ubiquitination of HIF-α and subsequent proteasomal degradation [13,14]. This interaction between VHL and HIF-1α has been shown in some instances to be promoted by acetylation of HIF by the ARD1 acetyltransferase at lysine 532, increasing ubiquitination and degradation [15]. However, the role of acetylation of HIF is still somewhat controversial and needs further exploration.
Figure 1. The hypoxia inducible factor-1.

Under normoxic conditions, HIF-1α is rapidly hydroxylated by prolyl hydroxylases, which mediates binding by the VHL ubiquitin ligase complex, addition of poly-Ub which tags it for proteosomal degradation. Under hypoxic conditions, prolyl hydroxylases cannot hydroxylate HIF-1α, preventing VHL binding which leads to HIF-1α stabilization. HIF-1α then heterodimerizes with HIF-1β, recruits the p3oo/CBP co-factors and forms an active HIF transcription complex in the nucleus that binds to HREs on target genes and activates their transcription.
HIF: Hypoxia inducible factor; HRE: Hypoxia response element; Ub: Ubiquitin chains; VHL: von Hippel-Lindau tumor suppressor protein.
Conversely, under hypoxic conditions, PHDs lose activity and HIF-α units are no longer degraded. As HIF-1α or HIF-2α accumulate, they bind to HIF1-β and form the HIF heterodimers, which translocate to the nucleus and, along with co-activators (p300 and CBP), form the transcriptional complexes that bind to hypoxic response elements (HREs) in the regulatory regions of many genes [16]. HREs are composite cis-acting elements, comprising the necessary but not sufficient HIF-binding site with the consensus sequence RCGTG (where R = purine A or G) and a HIF-ancillary sequence [17]. The trans activation potential of the HIF-α/p300 complexes is regulated via deacetylation of p300 by histone deacetylase [18].
HIF activity is also regulated in an oxygen-dependent manner by an asparaginyl hydroxylase, Factor Inhibiting HIF-1 (FIH1), which hydroxylates an asparagine in the C-terminal activation domain (CAD) of the α subunit (N-803 in human HIF-1α) under normoxia and mild hypoxia. Hydroxylation of N-803 blocks interactions between HIF-1α and p300/CBP coactivators preventing HIF activation [19]. Because FIH1 is less sensitive to oxygen drop than PHDs, it remains active under moderate hypoxia and keeps inactive the HIF-α molecules that avoid the PHD-mediated degradation that occurs in mild hypoxia [20].
PHDs and FIH1 are dioxygenases that, in addition to molecular oxygen, require Fe2+ and 2-oxoglutarate (2-OG) co-factors for activity [20]. This provides an opportunity for regulation of their activity and in turn HIF-1α levels/HIF activity. Iron chelation is frequently used for activation of HIF-1 in cell culture [20]. There has also been some evidence of mitochondrial involvement in HIF regulation in that, under moderately hypoxic conditions, mitochondria will produce reactive oxygen species (ROS) from complex III of the electron-transport chain, which inhibit the activity of PHDs by oxidizing Fe2+ to Fe3+, thus accelerating the stabilization of HIF [21,22]. 2-OG is an inter mediate in the tricarboxylic acid (TCA) cycle and structurally related compounds (e.g., dimethyloxalylglycine or other TCA cycle intermediates, fumarate and succinate) can inhibit activity of PHDs directly by reversibly competing for the active site with 2-OG or indirectly via increasing cellular levels of ROS [23]. Recently, heterozygous mutations in IDH1 or IDH2 genes were described in glioma and acute myeloid leukemia. Mutant proteins, acting in a dominant-negative fashion, are defective in their ability to oxidize isocitrate to 2-OG. An initial report suggested that in cells with mutant IDH HIF-1α is indirectly stabilized because reduced synthesis of 2-OG lessens PHD function [24]. However, further studies found comparable levels of 2-OG in both wild-type and mutant cells; instead, the mutant IDHs acquire a neomorphic activity, which reduces 2-OG to 2-hydroxyglutarate (2-HG) [25]. Moreover, it was reported that (R)-2-HG, the product of mutant IDH, can be utilized in place of 2-OG in the active center of PHDs. Here, (R)-2-HG (but not (S)-2-HG which acts as an inhibitor of PHDs) is oxidized into 2-OG, resulting in increased PHD activity and correspondingly decreased HIF-α levels and HIF activity [26]. More work will be required to finalize the functional consequences of IDH mutations on activity of the HIF pathway and tumor growth.
Finally, HIF-α is situated at the convergence of multiple oncogenic and tumor suppressor pathways, including the PI3K/AKT and MAPK/ERK pathways, which regulate HIF nonspecifically in an oxygen-independent manner. Activation of the PI3K/AKT pathway has been demonstrated to increase translation of HIF-1α mRNA and HIF-1α production [27,28]. Tumor suppressors (p53, GSK3β, and so forth) interfere with HIF function by decreasing HIF-1α stability or transcriptional activity [23]. One important type of regulation is that of HIF-1α phosphorylation, which can affect both HIF-1α stability and its transactivation potential. HIF-1α can be phosphorylated by GSK3 at three serine residues (551, 555 and 589) within the human HIF-1α N-terminal transactivation domain [29,30]. This recruits Fbw7 and USP28, which then mediates HIF-1α ubiquitination and subsequent VHL-independent proteasomal degradation [31]. PLK3 also destabilizes HIF-1α by phosphorylation at two serine resides (576 and 657) [32]. Conversely, phosphorylation of HIF-1α can have a stabilization effect. It has been demonstrated that ATM can phosphorylate HIF-1α at Ser-696, which increases its stability [33]. HIF-1α is also phosphorylated in its CAD by ERK1, which increases its transcriptional activity, but not its stability [34]. Thr-796 in HIF-1α is phosphorylated by CK2 and this phosphorylation is also important for its transactivation potential and not stability [35,36]. The p42 and p44 MAPK pathways regulate HIF-1α post-translationally by phosphorylating two serine residues (641 and 643). This phosphorylation promotes nuclear accumulation of HIF-1α, which leads to an increase of HIF-1-activated transcription [37,38]. In addition, HIF-1α Ser 247 phosphorylation coregulates the dimerization of HIF-1α/HIF-1β complex. HIF-1α can be phosphorylated on Ser-247 in the PAS-B domain by CK1, which destabilizes the HIF-1α/-1β complex, and thus diminishes its transcriptional activity [39]. HIF-1α/-1β dimerization is also regulated by means other than HIF-1α phosphorylation. For example, COMMD1 binds to the N-terminal domain of HIF-1α, competing with HIF-1β binding, and subsequently decreases the DNA binding and transcriptional activation of the complex [40]. Finally, the HIF-1α/p300 interaction is also post-translationally regulated. Both hydroxylation of asparagine 803 and S-nitrosylation of cysteine 800 in the C-TAD of HIF-1α decrease p300 binding, while phosphorylation of Thr-796 in the C-TAD does not affect p300 binding [41].
Distinct roles of HIF-α subunits
Among the three members of the HIF-α family, HIF-1α and HIF-2α (also called EPAS1, MOP2 or HLF) have been extensively studied, whereas significantly less is known about the third member, HIF-3α. HIF-α proteins exhibit high conservation in overall amino acid sequence, as well as similar domain structure, mechanisms of activation, heterodimerization with HIF-1β, and binding to the same HIF-binding site [20]. HIF-1α and HIF-2α each contain an N-terminal activation domain and CAD, and both act as positive transcriptional regulators. In contrast, the truncated HIF-3α lacks a CAD and acts as a dominant negative regulator of transcription and its expression is suppressed in renal cell carcinoma [23].
Despite the above-mentioned similarities, there is mounting evidence that HIF-1α and HIF-2α subunits are functionally distinct. The fact that neither HIF-1α−/− nor HIF-2α−/− embryos can survive suggests a lack of functional complementation between the two isoforms [42]. The ubiquitous expression of HIF-1α, as opposed to the more cell type-specific expression of HIF-2α, also suggests different physiological roles. Knockdown of HIF-1α or HIF-2α by siRNA elicits remarkably different cell-specific effects: in endothelial and breast cancer cells hypoxia-inducible gene expression has been demonstrated to be critically dependent on HIF-1α but not HIF-2α [43]; whereas in renal carcinoma cells, expression was critically dependent on HIF-2α [44]. HIF-α isoforms display unexpected reciprocal suppressive interactions in renal cell carcinoma: enhanced expression of HIF-2α suppresses HIF-1α and vice versa [44].
In certain cancers, HIF-1α and HIF-2α have been shown to play contrasting roles in tumorigenesis. Contrary to earlier conclusions, HIF-1α expression was found to correlate with lower disease stages in clear cell renal cell carcinoma (ccRCC), non-small-cell lung cancer, head and neck squamous cell carcinoma and neuroblastoma; whereas HIF-2α expression was found to correlate with more advanced stages and was consistently scored as a negative prognostic factor [45]. The most compelling evidence about the distinct roles of HIF-1α and HIF-2α has been accumulated in the VHL-deficient ccRCC: while ccRCC always overexpress HIF-2α and sometimes overexpress HIF-1α, only HIF-2α is required for their growth [46]. In addition, both HIF-1α and HIF-2α, stabilized upon VHL inactivation, can transcriptionally down-regulate transcription of HIF-1α in ccRCC through a mechanism that involves binding of either subunit to a reverse HRE in the HIF-1α proximal promoter followed by a series of repressive histone modifications [47]. This observation is in agreement with previous reports suggesting that in the early stages of ccRCC HIF-1α provides a metabolic advantage due to activating glycolytic enzymes, but hinders progression by inducing cell-cycle arrest or apoptosis. Inactivation of HIF-1α and its replacement with HIF-2α discontinues this inhibitory effect and ccRCC rapidly progresses [45,48,49]. Together, these data support the notion that HIF-2α has a greater oncogenic potential than HIF-1α in ccRCC [50].
Distinct functions of HIF-1α & -2α in certain types of cancer can be explained by differential direct & indirect transcriptional effects
HIF-1α and HIF-2α activate different sets of transcriptional targets (direct transcriptional effect). Several studies have shown that HIF-1α and HIF-2α differ in their capability to transactivate hypoxia-inducible genes. Some genes were transactivated exclusively by HIF-1α (notably glycolytic and proapoptotic genes) whereas others were transactivated by both isoforms [43,51]. HIF-2α, on the other hand, is a more efficient activator of genes encoding stem cell markers (see below). In a report that used siRNA methodology, HIF-2α preferentially regulated a small group of genes that had in common binding sites for the ETS family of transcription factors in their promoter regions. Knockdown of ELK-1, the most prominent member of the ETS family, significantly reduced hypoxic induction of the HIF-2α-dependent genes [52]. A number of regulatory elements (e.g., ATF-1, CREB-1, AP-1 and Sp1)have been described to cooperate with HREs, [16]; however data on whether their cognate factors preferentially engage in cooperation with HIF-1α or HIF-2α are scarce. Together, these studies suggest that, of the two isoforms, HIF-1α is a more universal activator [51], whereas HIF-2α is the more selective activator of transcription and the observed significant quantitative differences/specificity of activation could be accounted for by preferential cooperation of one isoform with transcription factors binding to the regulatory elements juxtaposed to HREs [23]. Sirtuins, a family of stress-responsive nicotin-amide adenine dinucleotide-dependent histone deacetylases that serve as sensors of the cellular redox state, also control transcriptional activity of HIF-1α and HIF-2α [23]. Sirt1 was reported to deacetylate both HIF-1α and HIF-2α with different functional outcomes: deacetylation of HIF-1α decreases its transcriptional activity [53], whereas deacetylation of HIF-2α resulted in enhanced transcriptional activity [54]. Thus, conditions of cellular redox/metabolic stress fine-tune activity of HIF-1α and HIF-2α.
HIF-1α and HIF-2α differentially modulate the activity of other transcription factors (indirect transcriptional effects). HIF-1α and HIF-2α have been reported to modulate (through direct or indirect interactions) the transcriptional activity of certain critical oncogenes/tumor suppressors with opposite outcomes. Notable in this respect is MYC, the function of which is antagonized by HIF-1α but enhanced by HIF-2α [46,55]. Differential regulation of the tumor suppressor protein p53 by HIF-1α and HIF-2α was also reported. On the one side, HIF-1α directly binds p53, inducing its stabilization, and eventually causing cell death [56,57]. Conversely, inhibition of HIF-2α has been shown to promote p53 activity and higher levels of HIF-2α contribute to increased radio- and chemoresistance [58]. In this way, HIF-1α and HIF-2α can achieve opposing effects on tumor behavior through indirect transcriptional effects.
HIF-1α and HIF-2α also have different effects on CSCs, also known as tumor-initiating or tumor-propagating cells, which are undifferentiated cells with the capacity to self-renew and reconstitute tumors in vivo that are phenotypically similar to the parental tumor [59]. An important feature of CSCs is their enhanced resistance to radio- and chemotherapy that limits DNA damage, allowing them to survive the treatment and repopulate the tumor with their progeny. CSCs thus represent an important therapeutic target and their complete elimination is expected to greatly enhance the treatment efficacy [59,60]. There are numerous reports implicating hypoxia and HIFs in CSC biology. For example, populations of cells enriched for CSCs can be isolated from glioblastoma tumors using the cell surface marker CD133 and hypoxia may promote the expansion of the CD133-positive cells through activation of HIF-1 [61]. Other reports highlight the prominent role of HIF-2α in CSCs: this isoform is selectively expressed in CSCs, whereas HIF-1α was expressed at comparable levels in both CSC and non-CSC (CD133-negative) populations [62]. In the same study, only targeting HIF-2α by shRNA decreased the growth of CSC populations in vitro, inhibited CSC-mediated angiogenesis and significantly prolonged the survival of mice intracranially implanted with CSCs. In a separate study, HIF-2α colocalized with neural crest markers in a subset of immature cells in neuroblastoma tumors and HIF-2α knockdown induced differentiation of these cells [63]. HIFs thus help maintain an undifferentiated state in some populations of CSCs as well as other stem and progenitor cells [60]. HIFs apparently control self-renewal and differentiation processes in these cells by means of trans-activating critical genes and the key transcription factors involved in these processes. In this category, interaction of HIF-1α with Notch1 helps maintain the undifferentiated cell state [64], whereas HIF-2α induces expression and transcriptional activity of the marker of the undifferentiated state – Oct4 [65]. HIF-2α also regulates the transcription factor Sox2 [66], which, in turn, controls pluripotency by direct regulation of Oct4 levels [67]. Recently, reprogramming of somatic cells into induced pluripotent cells was achieved through transduction of four defined transcription factors (c-MYC, KLF4, SOX2 and Oct4) all of which are transactivated by HIF [55,65,66]. Together, these reports provide evidence that HIFs directly control markers of the undifferentiated state (stemness) and are necessary for maintaining CSCs. Due to its critical role and selective activation, HIF-2α could be an important therapeutic target in CSCs.
Genes activated by HIF
Along with its co-activators, the HIF complex acts as a transcription factor for hundreds of genes, including VEGF, NOS, GLUT1, LDH, CA9 and MDR1 (Table 1) [69-107]. Many of these genes affect cancer progression through angiogenesis, erythropoiesis, increased glucose metabolism, immune evasion, immortalization, genetic instability, increased invasion and metastasis, pH regulation, drug resistance, CSC maintenance and proliferation.
Table 1. Selected hypoxia inducible factor target genes and their effects on cancer progression.
| Effect on cancer progression | Genes activated by hypoxia inducible factor-1 | Ref. |
|---|---|---|
| Angiogenesis | ANGPT2, C-MET, ID2, LEP, NOS, PDGF, SCF, SDF-1, VEGF | [74-82] |
| Drug resistance | MDR1 | [83] |
| Erythropoiesis | EPO | [84] |
| Genetic instability | DEC1, DEC2, MSH2, MSH6 | [85-86] |
| Glucose metabolism | GLUT1, GPI, HK1, HK2, LDH, MCT1, PGK1, PKM2 | [87-92] |
| Immortalization | TERT | [93] |
| Immune evasion | NT5E | [94] |
| Invasion | C-MET, EDN-1, FN-1, MMP-2, MMP-14, SDF-1 | [75,81,95-98] |
| Metastasis | C-MET, CXCR4, LOX, TWIST1, ZEB1 | [75,85,99-101] |
| pH regulation | CA-9, CA-12 | [102] |
| Proliferation | C-MYC, ID2, IGF-2, NOS, | [76,78,103,104] |
| Stem cell maintenance | ABCG2, JARID1B, OCT4, NANOG, Sox2, KLF4, c-myc, miR302 | [55,65-67,105-107] |
Two of the most essential functions of cancer biology that HIF-1 activates are angiogenesis and glucose metabolism [108];the former controls oxygen and nutrient delivery and the latter generates energy and synthetic intermediates for growth and survival. VEGF is an important growth factor in angiogenesis and vascularization and plays a significant role in hypoxic conditions and has been demonstrated to increase in vivo tumor size and vascularization [109]. VEGF also plays a critical role in embryonic development – deletion of a single allele of the gene in VEGF-knockout mice is lethal within two weeks of development. HIF-1 activates VEGF transcription by binding to its HRE and VEGF synthesis increases angiogenesis [110]. NOS produces nitric oxide, which promotes angiogenesis and cell proliferation, and increases cell survival by inhibiting apoptosis. HIF-1 binds to the NOS HRE and thus upregulates NOS expression and increases cancer progression [111]. Histological studies have associated increased malignancy and aggressiveness in cancers with increased HIF-1 and VEGF expression, and NOS expression has been confirmed in many cancers, including breast, head and neck, prostate and colorectal [111,112].
HIF-1 is a transcription factor for many genes involved in the glycolytic cascade, including GLUT1 and LDH, which allow tumors to grow under hypoxic conditions by metabolizing glucose to lactate through anaerobic glycolysis [92,113]. One result of anaerobic glycolysis is decreased production of ROS generated by oxidative phosphorylation, which can prevent cellular senescence, and thus remove a constraint on tumor growth [114]. Another consequence of glycolysis and lactate buildup is hypoxic acidosis, which, if not prevented, would lead to a decrease in intracellular pH, cell damage and death. However, HIF-1 activates the synthesis of monocarboxylate transporters (e.g., MCT) that extrude lactate into the extracellular space [115] and CA-9 and CA-12, which use CO2 to generate HCO3− to dampen the effects of acidosis and increase cell survival. Not surprisingly, higher GLUT1 levels are correlated with poor survival in many cancers – including breast, head and neck, esophageal, stomach, bladder, ovarian, colorectal, and non-small-cell lung carcinomas – and CA-9 is used as a marker for cancer progression [1,116].
In addition to activating gene expression for cancer progression, HIF-1 decreases the effectiveness of anticancer therapies such as radiation and chemotherapy. HIF-1 itself is stabilized by radiation, even doubling its activity in the two days following therapy, due to the re-oxygenation and release of ROS that follows radiation treatment [117,118]. The radiation-induced production of ROS increases HIF-1α accumulation by reducing its degradation through an AMP-activated protein kinase (AMPK)-dependent pathway, where ROS-activated AMPK inhibits the interaction between HIF-1α and VHL, thus preventing ubiquitination and subsequent degradation [119,120]. Increased HIF-1α levels have also been shown to decrease the effectiveness of chemotherapy in various types of cancer [121-124], due to factors such as HIF-mediated regulation of drug efflux, cell proliferation and survival, metabolic reprogramming and inhibition of DNA damage [125]. One specific mechanism by which HIF-1 can decrease effectiveness of therapy is by activation of MDR1 and the expression of the multiple drug resistance phenotype, which is present in the most aggressive cancers and correlates with the metastatic ability of multiple solid tumor cancers [126]. In addition, many anticancer therapeutics, such as doxorubicin, rely on the generation of ROS for cytotoxicity, but ROS stabilize HIF-1α [127]. Moreover, many therapeutics contain weakly basic moieties that can react with the acidic tumor microenvironment and lose efficacy [128].
Molecular targets of HIF-1 inhibitors
There are many different strategies for inhibiting the HIF-1 pathway. Small molecules have been shown to inhibit HIF activity through a variety of mechanisms including HIF-1α protein synthesis, HIF-1α protein stabilization, HIF-1α–HIF-1β dimerization, HIF-1 dimer DNA binding, and interactions with other proteins. There were two comprehensive reviews in 2012 by Semenza [129] and Xia et al. [130] that cover these mechanisms in detail and give fairly exhaustive lists of the small- molecules that inhibit at each level.
Many HIF-1 inhibitors affect the HIF-1 pathway upstream of HIF-1α synthesis (Figure 2). Cancer metabolism and regulation is a very complex system and it can be difficult to separate the desired effects on one pathway from another. This complexity can work for medicinal chemists in a synergistic manner by up- or down-regulating multiple pathways/targets by producing similar net results in the design and implementation of therapeutics. Conversely, the inhibitors may have unexpected counter-productive effects which may contribute to the high late-stage clinical failures for anticancer drugs, which is around 70% for Phase II trials and 60% for Phase III trials [131]. These high attrition rates can, moreover, be attributed to several factors, including the hypoxic and acidic tumor microenvironment; veracity and fidelity of in vitro preclinical models; drug absorption, distribution, metabolism, excretion, and toxicity; drug delivery in vivo; and translation to the clinic [132].
Figure 2. Mechanisms of hypoxia inducible factor inhibition.

HIF: Hypoxia inducible factor; HRE: Hypoxia response element; PHD: Prolyl hydroxylases; Ub: Ubiquitin chains; VHL: von Hippel-Lindau tumor suppressor protein.
In the field of HIF-1 inhibitors, the role of hypoxia is key. Tumor hypoxia has been touted as “the best validated target that has yet to be exploited in oncology” [5]. The definition of hypoxia has been well established at 0–5% oxygen, but there is a variance in the levels of oxygen defining normoxia. Atmospheric oxygen levels are approximately 20%, whereas tissues in the body have normal oxygen levels that range from 0 (bone marrow) to 14% (lung, liver, kidney and heart), with levels of approximately 10–12% in circulation [133]. Thus, in vitro assays that are carried out under ‘normoxic’ conditions, but are simply left open to the atmosphere ought to be considered ‘hyperoxic’ when compared to in vivo oxygen levels [134]. This difference in oxygen levels can contribute negatively to many preclinical studies and should be considered as a contributing factor to drug attrition.
Another factor in tumor hypoxia is the acidic extracellular environment due to the increased expression of the glycolytic phenotype. This creates a unique pH gradient for tumors that is the opposite of that of normal tissue [135]. It has been demonstrated that weakly basic anticancer therapeutics, such as doxorubicin, are excluded from acidic tumors [136]. This acidic micro environment can also contribute negatively to in vivo studies and should be considered as another factor contributing to drug failure.
HIF-1 inhibitors in the clinic: hits & misses
Transcription factors have, for a long time, been considered undruggable targets, and to date, no specific inhibitor of HIF has been brought to market. Multiple levels of regulation and the fact that multiple signaling pathways converge on HIF-α explain the diversity of compounds that, among other targets, inhibit HIF. Although there are many challenges to targeting HIF, there have been a number of compounds that have made it to clinical trials as anticancer therapeutics, which have also been shown to inhibit HIF activity. Some of these have failed their trials, while others have been US FDA approved for patient treatment. The following illustrates a few of these examples (Table 2) and what their outcomes say for the field as a whole.
Table 2. Small anticancer molecules with hypoxia inducible factor-1 inhibitory activity.
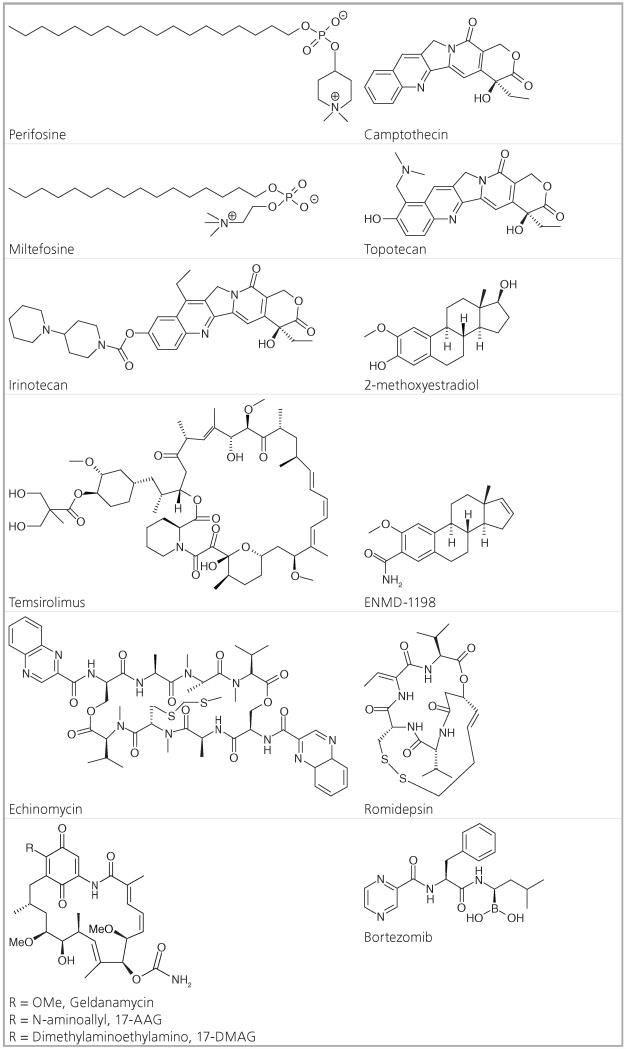
|
Camptothecins: camptothecin, topotecan, irinotecan
In the 1960s, camptothecin was first isolated from the bark of a native Chinese tree, Camptotheca acuminata, by Wall and Wani. By the mid 1970s, camptothecin was in clinical trials due to its anticancer activity, but it was terminated because of its serious side effects [137]. Research on camptothecin was not resumed until the discovery of DNA topo isomerase 1 (TOP1) as its primary target and the successful development of water-soluble derivatives of camptothecin: topotecan (Hycamtin®) and irinotecan (Camptosar, CPT-11) [138]. The camptothecins have a unique mechanism of action that targets TOP1, an essential human enzyme that relaxes DNA supercoiling by forming single strand breaks, unwinding, then religating the strands back together. The camptothecins act as irreversible inhibitors to the TOP1–DNA complex by intercalating into DNA at the protein–DNA interface, which then prevents the religation of the DNA strands [139]. Both irinotecan and topotecan have similar mechanisms of action as camptothecin. TOP1 is an upstream regulator of the HIF-1 pathway. TOP1 inhibition leads to HIF-1α down-regulation due to decreased protein accumulation and translation that is independent of oxygen or proteasomal degradation [140]. It has been suggested that this mechanism could be due to the activation of a novel antisense transcript from the 5′ end of HIF-1α and the removal of Pol II from the promoter-proximal pause site of the HIF-1α gene [141]. The mechanism of inhibition of HIF-1α translation by camptothecin and its derivatives is TOP1-dependent but DNA-damage independent. Irinotecan was approved for treatment of colorectal cancer in 1996 [142] and topotecan was approved for the treatment of Stage IVB recurrent carcinoma of the cervix in 2006 [219]. In 2007, the FDA approved Hycamtin for treatment of relapsed small cell lung cancer [304]. The successful development of these camptothecin derivatives would not have been possible without further elucidation of their mechanism of action. This case illustrates how drugs (and classes of drugs) that have unknown mechanisms of action and have previously failed clinical trials can be repurposed and brought to market once the mode of action is elucidated and the pharmacokinetics optimized.
Bortezomib
Bortezomib (Velcade®, PS-341) is a dipeptide boronic acid-containing compound that reversibly inhibits the chymotryptic activity of the 20S subunit of the 26S proteasome due to the high binding affinity and specificity between the boron atom and the 20S subunit [143]. The proteasome regulates protein expression and function by degradation of ubiquitinated proteins, and cleanses the cell of unfolded or mis-folded proteins. Bortezomib has been shown to directly inhibit proliferation and induce apoptosis in multiple myeloma cell lines and patient tumor cells resistant to conventional therapies [144]. Proteasome inhibition by bortezomib leads to disruption of intracellular protein metabolism. The downstream biological effects of proteasome inhibition are numerous, with direct effects on both multiple myeloma cells and their micro environment, including inhibition of cytokine secretion, suppression of adhesion molecule expression and inhibition of angiogenesis [145]. Because bortezomib’s primary target is the proteasome, it prevents the degradation of ubiquitinated HIFα and leads to the accumulation of HIF-1α under normoxia. However, the stabilized HIF-1(−2) is inactive due to upregulation of FIH activity, which inhibits recruitment of p300 [146]. There has been other evidence that bortezomib does not directly affect the formation of the HIF-1-p300 complex, but interferes with the C-terminal domain of HIF-1α [147]. More recently, bortezomib was shown to inhibit the PI3K/AKT/mTOR pathway upstream of the HIF pathway in prostate cancer cells [148]. Due to bortezomib’s remarkable clinical activity against multiple myeloma, it was rapidly approved by the FDA in 2003 to treat relapsed and refractory multiple myeloma [149]. It is unknown whether HIF inhibition is critical to the clinical activity of bortezomib. Bortezomib is an excellent example of an FDA-approved anticancer therapeutic that indirectly inhibits HIF expression and function and that has differing modes of action in different cancer models, which illustrates the complexity of the field.
Romidepsin
Romidepsin (Istodax®, FK228, FR901228) is an anticancer agent isolated from the bacterium Chromobacterium violaceum that was first reported by Fujisawa Pharmaceutical Company (now Astellas Pharma) in 1994 [65]. Its mechanism of action was demonstrated in 1998 [150,151]. As a prodrug with a disulfide bond, romidepsin undergoes reduction to release a zinc-binding thiol [152]. This thiol reversibly interacts with a zinc atom in the binding pocket of Zn-dependent histone deacetylases (HDACs) to block their activity, classifying romidepsin as a HDAC inhibitor (HDACI). HDACs affect gene expression by removal of acetyl groups from acetylated lysine residues in histones. HDACs also deacetylate nonhistone proteins, such as transcription factors. In vitro, romidepsin causes the accumulation of acetylated histones, thus inducing cell cycle arrest and apoptosis of some cancer cell lines with IC50 values in the nano molar range [153]. HDAC activity is crucial for the transactivation potential of HIF-1α and most HDACIs regulate acetylation of the HIF-1α/p300 complex [154]. It has also been suggested that the class II isozyme HDACs are involved in direct acetylation and ubiquitination of HIF-1α [155]. Romidepsin was approved as a treatment for cutaneous T-cell lymphoma in 2009 [305]. Other HDACIs are also in preclinical and clinical development, but many in the field have moved to target other mechanisms of action due to high adverse effects associated with the HDACIs.
Temsirolimus
Temsirolimus (Torisel®, CCI-779), the ester version of rapamycin, is the first FDA approved inhibitor of mammalian target of rapamycin (mTOR/TORC1), which is a serine/threonine-specific kinase in the phosphatidylinositol 3–kinase (PI3K) related protein family [156]. In RCC, temsirolimus was shown to inhibit tumor growth and HIF expression by inhibition of the mTOR-dependent kinase cascade needed for HIF mRNA translation [157]. Inhibition of PI3K and its downstream target mTOR was demonstrated in prostate cancer (PC-3) cells to decrease HIF-1-dependent gene expression and temsirolimus was shown to suppresses HIF-1 activation by increasing HIF-1α degradation rate in hypoxic PC-3 cells [158]. The FDA approved Torisel (temsirolimus) for the treatment of RCC in 2007 [306]. However, not all inhibitors of the P13K/AKT/mTOR pathway have had such success.
Perifosine
Perifosine is an alkylphospholipid analogue, which has demonstrated significant antiproliferative activity in several human tumor model systems both in vitro and in vivo. It has a similar structure to miltefosine, a drug that has been approved in Europe for the treatment of cutaneous lymphomas and metastasis from breast cancer. Perifosine’s activity is also due to its effect on the PI3K/AKT/mTOR pathway, an upstream regulator of the HIF pathway. Many growth factors that upregulate HIF activate the PI3K–AKT–mTOR pathway, which leads to increased HIF-1α protein translation and stability [159]. Perifosine, which inhibits AKT in a dose-dependent manner, entered Phase I clinical trials in 2003 [160] and, in 2010, reached Phase II trial due to significant growth inhibition in vitro and in vivo in the Waldenstrom macro globulinemia model [161]. In 2012, however, perifosine failed a late-stage Phase III study on colorectal cancer due to lack of efficacy but will continue in another Phase III study as part of combination therapy with bortezomib against multiple myeloma [307]. Perifosine is one of many compounds indirectly affecting HIF expression that showed great promise in preclinical models, but has failed to exhibit efficacy in human trials.
2-methoxyestradiol
2-methoxyestradiol (2ME2) is a natural metabolite of estradiol and is a potent antitumor agent, due to its antiproliferative, antimetastatic and antiangiogenic activity. These antitumor activities result from its pro-apoptotic activity, microtubule activity and production of superoxides [162]. Its main potency appears to be derived from disruption of cellular microtubules that are necessary for HIF-1α translocation to the nucleus. 2ME2 thus retards HIF-1 nuclear accumulation and activity in a manner that is both oxygen- and proteasome-independent [163]. It was subsequently found that microtubule inhibitors such as 2ME2 and taxol also inhibit HIF-1α mRNA translation [164]. A Phase I clinical trial of 2ME2 was successfully concluded in 2006 [165]. In 2011, 2ME2 nanocrystal dispersion failed to show a significant effect in a Phase II study against castration-resistant prostate cancer [166] and in 2012, in a second Phase II study against metastatic renal cell carcinoma due to both lack of objective effects and high toxicity. The clinicians recommended halting trials of 2ME2 in favor of a new 2ME2 analog in development [167]. Hopefully, this new analog, ENMD-1198, will have more favorable results.
Echinomycin
Echinomycin (quinomycin A, NSC 526417) is a cyclic peptide antibiotic agent of quinoxaline that was originally isolated from Streptomyces echinatus [168]. Echinomycin is known to bind DNA in a sequence-specific manner. Binding sites for echinomycin contain the central sequence 5′-CG-3′ and the key recognition elements are contained in the sequences 5′-ACGT-3′ and 5′-TCGT-3′, which are part of the DNA recognition motifs for several transcription factors [169]. Inhibition of HIF-1 binding to the HRE (RCGTG; where R = purine A or G), a step required for induction of transcription, is a potential mechanism by which echinomycin may inhibit HIF-1 activity [170]. Echinomycin was shown in chromatin immunoprecipitation experiments to selectively inhibit binding of HIF-1 to DNA [171]. Despite echinomycin’s in vitro apoptosis-inducing activity and the initial report of its in vivo antitumor effect in mice, echinomycin’s clinical development was halted in the late 1980s following extensive testing as a cytotoxic agent in Phase I-II trials, which failed to show significant activity [172].
Ansamycins: geldanamycin, 17-AAG, 17-DMAG
Geldanamycin (GA) is a macrocyclicpolyketide antibiotic containing a benzoquinone moiety and was originally isolated from Streptomyces hygroscopicus [173]. GA is a Hsp90 inhibitor, that acts by binding the N-terminal ATP-binding domain of Hsp90, leading to the destabilization and eventual degradation of Hsp90 client proteins [174-176]. Hsp90, one of the most abundant cellular proteins, assists in protein folding and degradation and is upregulated during cellular stress [177,178]. HIF is a client protein of Hsp90 and the inhibition of Hsp90 has been shown to destabilize HIF, leading to HIF degradation and decrease in transcriptional activity [179,180]. GA and its derivatives 17-AAG (tanespimycin) and 17-DMAG (alvespimycin) have demonstrated anti-tumorigenic and -angiogenic properties both in vitro and in vivo. However, GA was never brought to the clinic due to its poor pharmacological properties and hepatoxicity in animal models [181,182]. 17-AAG was the first-in-class Hsp90 inhibitor to enter Phase I trials, where it showed promise; however, it showed poor results in Phase II trials, most likely due to its poor bioavailability and solubility [183-186]. 17-DMAG, an orally available agent, has shown promise in the clinic, with success in Phase I trials, but needs further evaluation [187,188].
Conclusion
Hypoxic conditions in the cancer microenvironment lead to increased resistance to both chemotherapy and radiotherapy. In most cases, the HIF pathway is the primary pathway responsible for this more malignant phenotype and its activation leads to increased cancer metastasis and poor patient prognosis. There has been much effort in this area to develop small-molecule inhibitors of the HIF pathway (mostly focused on HIF-1) as well as upstream and downstream effects. There have been no approved drugs that directly inhibit the HIF pathway, but there have been a few that indirectly affect the HIF pathway, as well as many more that have failed to demonstrate therapeutic efficacy in clinical trials for cancer patients. Some of these failures can be accredited to a lack of specificity and/or redundancy in the complexity of tumor signaling/metabolism that can overcome the inhibition effects. Another contributing factor to the failure of these compounds can be attributed to the lack of patient selection in clinical trials. Although many clinical trials evaluate the efficacy of anticancer therapeutics and examine their effects on HIF levels, patients are not selected based on their HIF-expression levels. If patients do not have elevated levels of HIF, therapeutics that target the HIF pathway may be less effective. More work needs to be done to identify novel, potent and more specific inhibitors targeting clearly defined points in the HIF pathway. Such new agents should be used in combination therapy and will hopefully overcome resistance that may develop during the initial treatment.
Future perspective
Recently, the HIF pathway was touted as “technically undruggable or at the very least as extremely challenging to target by medicinal chemists using small molecules” [189]. This attitude can be attributed not only to the difficulty of targeting such a complex pathway, but also to the challenges of targeting protein–protein interactions [190]. Small molecules typically inhibit protein function by binding with high affinity and specificity to hydrophobic pockets on or near the protein’s surface. When trying to disrupt interactions between proteins, these binding sites may no longer be accessible and the interactions between proteins are so multi farious that one small molecule may not be able to interrupt the key interactions. There have been advances in this field of targeting protein–protein interaction, such as stapled peptide inhibitors of transcription factors [191] and small-molecule inhibitors of the MDM2-p53 interaction as anticancer therapeutics [192]. Some interesting features of the small molecules identified are that they tend to be large, lipophilic, and rigid structures with complex 3D shape that form few hydrogen bonds [193,194]. These structural differences vary from the typical Lipinksi rule of five for drug-like properties [195] and could portend a new paradigm for drug-like qualities for small molecules that inhibit protein–protein interactions.
Interest in small-molecule inhibitors of the HIF pathway has steadily increased over the past 15 years. The number of patent applications, publications, citations and clinical trials has risen since the late 1990s and early 2000s (Figure 3). Although there have been few drugs that have made it all the way through clinical trials, the field is ripe and interest is increasing. It still remains to be seen whether inhibitors capable of distinguishing HIF-1 and HIF-2 complexes can be developed. In addition, new therapeutics are being developed against downstream HIF targets, such as MCT (91) and CA-IX (196). Inhibitors of post-translational modifications of HIF-1α have also shown some promise. Kaempferol was recently shown to inactivate the p44/42 MAPK phosphorylation of HIF-1α, causing its mislocalization into the cytoplasm in hepatocellular carcinoma [197]. Results from high-throughput screenings and natural product discovery [198] have also yielded new hits and lead compounds, but even more new methods need to be identified. Recently, novel HIF inhibitors from frankincense were identified through a new method using a molecularly imprinted polymer [199]. Acriflavine was identified as a HIF-dimerization inhibitor from a screening of drugs that had previously made it to Phase II clinical trials [200]. In addition, targeting the HIF-2α/-1β dimerization has been suggested as a valid target for anticancer therapeutics [201]. The well-known HIF/p300 interaction inhibitor chetomin [202] and its family, the epidithiodiketopiperazines, act via a mechanism of action involving disruption of the zinc-binding sites in the CH1 domain of p300 [203]. In addition, artificial α-helices have been demonstrated to interfere with the HIF/p300 interaction [204]. Oncolytic viruses dependent upon HIF expression for their replication have also been developed and showed strong antitumor effects [205-207]. Recently, a group of collaborators, including the authors, reported a new class of HIF-1 inhibitors, that appears to target the interaction between HIF-1 and p300 and, therefore, HIF-mediated transcription [208-214]. The lead arylsulfonamide compound (KCN1) showed potent anticancer activity in several cancer models [215,216]. Moreover, these inhibitors do not show intrinsic cytotoxicity and thus are promising compounds for further clinical development.
Figure 3. Trends of hypoxia inducible factor inhibitors from 2000–2012.

(A) Number of patent issued by year. (B) Number of publications by year. (C) Number of clinical trials by year. (D) Number of citations by year.
Executive summary.
Hypoxia in cancer
-
■
Hypoxia is an important element in the tumor microenvironment.
-
■
Hypoxia leads to a more malignant phenotype that is: resistant to radiation and chemotherapy; more invasive; genetically unstable; resistant to apoptosis; greater metastatic potential.
The hypoxia inducible factor pathway
-
■The hypoxia inducible factor (HIF) pathway is a critical target for anticancer therapeutics.
-
■The HIF complex acts as a transcription factor for many genes that increase tumor survival, including pro-angiogenic and pro-glycolytic genes.
-
■Increased HIF-1α levels in tumors lead to decreased patient survival rate.
-
■
-
■Distinct roles of HIF-α subunits.
-
■HIF-1α, HIF-2α, and HIF-3α all have differing roles and effects on cancer progression.
-
■
-
■Distinct functions of HIF-1α and HIF-2α in certain types of cancer can be explained by differential direct and indirect transcriptional effects.
-
■HIF-1 and HIF-2 activate different sets of transcriptional targets; HIF3 is an inhibitor.
-
■>100 different genes have been identified as HIF targets, with a variety of functions such as angiogenesis, erythropoiesis, increased glucose metabolism, immune evasion, immortalization, genetic instability, increased invasion and metastasis, pH regulation, cancer stem cell maintenance, and proliferation.
-
■
Molecular targets of HIF-1 inhibitors
-
■
HIF inhibitors target the HIF pathway by a variety of mostly indirect mechanisms: HIF-1α protein synthesis, HIF-1α protein stabilization, HIF-1α–HIF-1β dimerization, HIF-1 dimer DNA binding and interactions with other proteins.
HIF-1 inhibitors in the clinic: hits & misses
-
■
Many anticancer agents with indirect HIF-1 inhibition have made it to clinical trials and a few have been US FDA-approved for cancer treatment, such as: camptothecins: camptothecin, topotecan, irinotecan; bortezomib, romidepsin, temsirolimus, perifosine, 2-Methoxyestradiol, echinomycin; ansamycins: Geldanamycin, 17-AAG, 17-DMAG.
Conclusion
-
■
More work needs to be done to identify novel, potent and more specific inhibitors targeting clearly defined points in the HIF pathway.
Future perspective
-
■
Interest in the HIF field is increasing and looking to new mechanisms for inhibition will be key to this field.
Key Terms
- Cancer stem-like cells
Undifferentiated cells with the capacity to self-renew and reconstitute tumors in vivo that are phenotypically similar to the parental tumor.
- Hypoxia-inducible factor
Transcription factors regulated by the presence or absence of oxygen.
- Hypoxia
Reduction of oxygen supply to tissues.
- Angiogenesis
Process of new blood vessel formation from existing vasculature.
- Drug-like
Following Lipinski’s rule of five, no more than five hydrogen bond donors, no more than ten hydrogen bond acceptors, molecular weight of less than 500, logP of less than 5.
Footnotes
Financial & competing interests disclosure
EG Van Meir received financial support from the NIH (Grant R01CA116804), the St. Baldrick, the MaxCure, Samuel Waxman Cancer Research and V foundations. SK Burroughs gratefully acknowledges the fellowships received from the Georgia State University Center for Diagnostics and Therapeutics and Molecular Basis of Disease Program. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
■■ of considerable interest
- 1.Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26(2):225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 2.Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001;61(24):8903–8908. [PubMed] [Google Scholar]
- 3.Matsumoto S. Imaging cycling tumor hypoxia. Cancer Res. 2010;70(24):10019–10023. doi: 10.1158/0008-5472.CAN-10-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belozerov VE, Van Meir EG. Inhibition of hypoxia-inducible factor-1 signaling. Curr. Opin. Investig. Drugs. 2006;7(12):1067–1076. [PubMed] [Google Scholar]
- 5.Wilson W, Hay M. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer. 2011;11(6):393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 6.Brown JM, Wilson WR. Exploiting tumor hypoxia in cancer treatment. Nat. Rev. Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 7■.Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumors. Nature. 1996;379(6560):88–91. doi: 10.1038/379088a0. First study to show that hypoxia selects for tumor cells with mutations in the p53 gene that prevent hypoxia-induced apoptosis.
- 8.Subarsky P, Hill RP. The hypoxic tumor microenvironment and metastatic progression. Clin. Exp. Metastasis. 2003;20(3):237–250. doi: 10.1023/a:1022939318102. [DOI] [PubMed] [Google Scholar]
- 9.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995;270(3):1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 10■.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. Reports the original cloning of hypoxia inducible factor-1α.
- 11.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22(16):4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003;278(33):30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 13.Maxwell PH, Wiesener MS, Chang GW, et al. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 14■.Ivan M, Kondo K, Yang HF, et al. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. doi: 10.1126/science.1059817. Along with [13], describes the discovery of VHL as the substrate-recognition unit of the E3 ubiquitin ligase responsible for oxygen-dependent hypoxia inducible factor-α degradation.
- 15.Jeong JW, Bae MK, Ahn MY, et al. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell. 2002;111(5):709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 16.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci. STKE. 2005;2005(306):re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 17.Kimura H, Weisz A, Ogura T, et al. Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J. Biol. Chem. 2001;276(3):2292–2298. doi: 10.1074/jbc.M008398200. [DOI] [PubMed] [Google Scholar]
- 18.Fath DM, Kong X, Liang D, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-α. J. Biol. Chem. 2006;281(19):13612–13619. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α. Cancer Res. 2006;66(7):3688–3698. doi: 10.1158/0008-5472.CAN-05-4564. [DOI] [PubMed] [Google Scholar]
- 20.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Kaelin WG., Jr ROS: really involved in oxygen sensing. Cell Metab. 2005;1(6):357–358. doi: 10.1016/j.cmet.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Czibik G. Complex role of the HIF system in cardiovascular biology. J. Mol. Med. 2010;88(11):1101–1111. doi: 10.1007/s00109-010-0646-x. [DOI] [PubMed] [Google Scholar]
- 23.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1 alpha. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):U739–U752. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26■.Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):U485–U144. doi: 10.1038/nature10898. Report showing that 2-hydroxyglutarate produced in IDH mutant tumor cells activates prolyl hydroxylase and destabilizes hypoxia inducible factor.
- 27.Bardos J, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim. Biophys. Acta. 2005;25(2):107–120. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Déry M-A, Michaud MD, Richard DE. Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell. Biol. 2005;37(3):535–540. doi: 10.1016/j.biocel.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Flügel D, Görlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1α and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biochem. 2007;27(9):3253–3265. doi: 10.1128/MCB.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mottet D, Dumont V, Deccache Y, et al. Regulation of hypoxia-inducible factor-1α protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3β pathway in HepG2 cells. J. Biol. Chem. 2003;278(33):31277–31285. doi: 10.1074/jbc.M300763200. [DOI] [PubMed] [Google Scholar]
- 31.Flügel D, Görlach A, Kietzmann T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood. 2012;119(5):1292–1301. doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu D, Yao Y, Lu L, Costa M, Dai W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1α. J. Biol. Chem. 2010;285(50):38944–38950. doi: 10.1074/jbc.M110.160325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell. 2010;40(4):509–520. doi: 10.1016/j.molcel.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minet E, Arnould T, Michel G, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468(1):53–58. doi: 10.1016/s0014-5793(00)01181-9. [DOI] [PubMed] [Google Scholar]
- 35.Gradin K, Takasaki C, Fujii-Kuriyama Y, Sogawa K. The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J. Biol. Chem. 2002;277(26):23508–23514. doi: 10.1074/jbc.M201307200. [DOI] [PubMed] [Google Scholar]
- 36.Mottet D, Ruys SPD, Demazy C, Raes M, Michiels C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int J. Cancer. 2005;117(5):764–774. doi: 10.1002/ijc.21268. [DOI] [PubMed] [Google Scholar]
- 37.Richard DE, Berra E, Gothié E, Roux D, Pouysségur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999;274(46):32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 38.Mylonis I, Chachami G, Samiotaki M, et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1α. J. Biol. Chem. 2006;281(44):33095–33106. doi: 10.1074/jbc.M605058200. [DOI] [PubMed] [Google Scholar]
- 39.Kalousi A, Mylonis I, Politou AS, Chachami G, Paraskeva E, Simos G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010;123(17):2976–2986. doi: 10.1242/jcs.068122. [DOI] [PubMed] [Google Scholar]
- 40.Van De Sluis B, Mao X, Zhai Y, et al. COMMD1 disrupts HIF-1alpha/beta dimerization and inhibits human tumor cell invasion. J. Clin. Invest. 2010;120(6):2119–2130. doi: 10.1172/JCI40583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho H, Ahn D-R, Park H, Yang EG. Modulation of p300 binding by posttranslational modifications of the C-terminal activation domain of hypoxia-inducible factor-1α. FEBS Lett. 2007;581(8):1542–1548. doi: 10.1016/j.febslet.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 42.Huang LE, Bunn HF. Hypoxia-inducible factor and its biomedical relevance. J. Biol. Chem. 2003;278(22):19575–19578. doi: 10.1074/jbc.R200030200. [DOI] [PubMed] [Google Scholar]
- 43.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biochem. 2003;23(24):9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raval RR, Lau KW, Tran MGB, et al. Contrasting properties of HIF-1 and HIF-2 in Von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005;25(13):5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat. Rev. Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14(6):435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu J, Wang B, Xu Y, et al. Epigenetic regulation of HIF-1 alpha in renal cancer cells involves HIF-1 alpha/2 alpha binding to a reverse hypoxia-response element. Oncogene. 2012;31(8):1065–1072. doi: 10.1038/onc.2011.305. [DOI] [PubMed] [Google Scholar]
- 48.Giatromanolaki A, Koukourakis MI, Sivridis E, et al. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumors and survival. Br. J. Cancer. 2001;85(6):881–890. doi: 10.1054/bjoc.2001.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franovic A, Holterman CE, Payette J, Lee S. Human cancers converge at the HIF-2α oncogenic axis. Proc. Natl Acad. Sci. USA. 2009;106(50):21306–21311. doi: 10.1073/pnas.0906432106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biswas S, Troy H, Leek R, et al. Effects of HIF-1alpha and HIF-2alpha on growth and metabolism of clear-cell renal cancer 786-0 xenografts. J. Oncol. 2010;2010:757908. doi: 10.1155/2010/757908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (HIF)-1alpha versus HIF-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63(19):6130–6134. [PubMed] [Google Scholar]
- 52.Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66(11):5641–5647. doi: 10.1158/0008-5472.CAN-05-3345. [DOI] [PubMed] [Google Scholar]
- 53.Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1 alpha. Mol. Cell. 2010;38(6):864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 54.Dioum EM, Chen R, Alexander MS, et al. Regulation of hypoxia-inducible factor 2 alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324(5932):1289–1293. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 55.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2 alpha promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell. 2007;11(4):335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moeller BJ, Dreher MR, Rabbani ZN, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8(2):99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 57.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1 alpha. Nature. 1998;392(6674):405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 58.Bertout JA, Majmundar AJ, Gordan JD, et al. HIF2 alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl Acad. Sci. USA. 2009;106(34):14391–14396. doi: 10.1073/pnas.0907357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hadjipanayis CG, Van Meir EG. Tumor initiating cells in malignant gliomas: biology and implications for therapy. J. Mol. Med. 2009;87(4):363–374. doi: 10.1007/s00109-009-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer. 2010;102(5):789–795. doi: 10.1038/sj.bjc.6605551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soeda A, Park M, Lee D, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1 alpha. Oncogene. 2009;28(45):3949–3959. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]
- 62.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15(6):501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pietras A, Hansford LM, Johnsson AS, et al. HIF-2 alpha maintains an undifferentiated state in neural crest-like human neuroblastoma tumor-initiating cells. Proc. Natl Acad. Sci. USA. 2009;106(39):16805–16810. doi: 10.1073/pnas.0904606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gustafsson MV, Zheng XW, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005;9(5):617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 65.Covello KL, Kehler J, Yu HW, et al. HIF-2 alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20(5):557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moreno-Manzano V, Rodriguez-Jimenez FJ, Acena-Bonilla JL, et al. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. J. Biol Chem. 2010;285(2):1333–1342. doi: 10.1074/jbc.M109.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17(1):126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 69.Ortiz-Barahona A, Villar D, Pescador N, Amigo J, del Peso L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010;38(7):2332–2345. doi: 10.1093/nar/gkp1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37(14):4587–4602. doi: 10.1093/nar/gkp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanimoto K, Tsuchihara K, Kanai A, et al. Genome-wide identification and annotation of HIF-1α binding sites in two cell lines using massively parallel sequencing. HUGO J. 2010;4(1-4):35–48. doi: 10.1007/s11568-011-9150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117(23):e207–e217. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mole DR, Blancher C, Copley RR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009;284(25):16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Simon MP, Tournaire R, Pouyssegur J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell Physiol. 2008;217(3):809–818. doi: 10.1002/jcp.21558. [DOI] [PubMed] [Google Scholar]
- 75.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3(4):347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 76.Lofstedt T, Jogi A, Sigvardsson M, et al. Induction of ID2 expression by hypoxia-inducible factor-1: a role in dedifferentiation of hypoxic neuroblastoma cells. J. Biol. Chem. 2004;279(38):39223–39231. doi: 10.1074/jbc.M402904200. [DOI] [PubMed] [Google Scholar]
- 77.Grosfeld A, André J, Hauguel-De Mouzon S, Berra E, Pouysségur J, Guerre-Millo M. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. J. Biol. Chem. 2002;277(45):42953–42957. doi: 10.1074/jbc.M206775200. [DOI] [PubMed] [Google Scholar]
- 78.Melillo G, Musso T, Sica A, Taylor LS, Cox GW, Varesio L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 1995;182(6):1683–1693. doi: 10.1084/jem.182.6.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang Y, Hickey RP, Yeh JL, et al. Cardiac myocyte-specific HIF-1α deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004;18(10):1138–1140. doi: 10.1096/fj.04-1510fje. [DOI] [PubMed] [Google Scholar]
- 80.Han Z-B, Ren H, Zhao H, et al. Hypoxia-inducible factor (HIF)-1α directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF) Carcinogenesis. 2008;29(10):1853–1861. doi: 10.1093/carcin/bgn066. [DOI] [PubMed] [Google Scholar]
- 81.Ceradini DJ, Kulkarni AR, Tepper OM, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004;10(8):858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 82.Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J. Biol. Chem. 1995;270(22):13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- 83.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62(12):3387–3394. [PubMed] [Google Scholar]
- 84.Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl Acad. Sci. USA. 1991;88(13):5680–5684. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miyazaki K, Kawamoto T, Tanimoto K, Nishiyama M, Honda H, Kato Y. Identification of functional hypoxia response elements in the promoter region of the DEC1 and DEC2 genes. J. Biol. Chem. 2002;277(49):47014–47021. doi: 10.1074/jbc.M204938200. [DOI] [PubMed] [Google Scholar]
- 86.Koshiji M, To KKW, Hammer S, et al. HIF-1α induces genetic instability by transcriptionally downregulating MutSα expression. Mol. Cell. 2005;17(6):793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 87.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1: interaction between H-ras and hypoxia. J. Biol. Chem. 2001;276(12):9519–9525. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 88.Yoon DY, Buchler P, Saarikoski ST, Hines OJ, Reber HA, Hankinson O. Identification of genes differentially induced by hypoxia in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2001;288(4):882–886. doi: 10.1006/bbrc.2001.5867. [DOI] [PubMed] [Google Scholar]
- 89.Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001;276(46):43407–43412. doi: 10.1074/jbc.M108181200. [DOI] [PubMed] [Google Scholar]
- 90.Semenza GL, Jiang B-H, Leung SW, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996;271(51):32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 91.Sonveaux P, Copetti T, De Saedeleer CJ, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE. 2012;7(3):e33418. doi: 10.1371/journal.pone.0033418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994;269(38):23757–23763. [PubMed] [Google Scholar]
- 93.Nishi H, Nakada T, Kyo S, Inoue M, Shay JW, Isaka K. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT) Mol. Cell. Biol. 2004;24(13):6076–6083. doi: 10.1128/MCB.24.13.6076-6083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Synnestvedt K, Furuta GT, Comerford KM, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Invest. 2002;110(7):993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang Y, Hickey RP, Yeh JL, et al. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004;18(10):1138–1140. doi: 10.1096/fj.04-1510fje. [DOI] [PubMed] [Google Scholar]
- 96.Krishnamachary B, Berg-Dixon S, Kelly B, et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003;63(5):1138–1143. [PubMed] [Google Scholar]
- 97.Ben-Yosef Y, Lahat N, Shapiro S, Bitterman H, Miller A. Regulation of endothelial matrix metalloproteinase-2 by hypoxia/reoxygenation. Circ. Res. 2002;90(7):784–791. doi: 10.1161/01.res.0000015588.70132.dc. [DOI] [PubMed] [Google Scholar]
- 98.Petrella BL, Lohi J, Brinckerhoff CE. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2 alpha in von Hippel-Lindau renal cell carcinoma. Oncogene. 2005;24(6):1043–1052. doi: 10.1038/sj.onc.1208305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumor suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 100.Erler JT, Bennewith KL, Nicolau M, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 101.Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008;7(14):2090–2096. doi: 10.4161/cc.7.14.6324. [DOI] [PubMed] [Google Scholar]
- 102.Wykoff CC, Beasley NJP, Watson PH, et al. Hypoxia-inducible expression of tumorassociated carbonic anhydrases. Cancer Res. 2000;60(24):7075–7083. [PubMed] [Google Scholar]
- 103.Koshiji M, Kageyama Y, Pete EA, Horikawa Barrett JC, Huang LE. HIF-1[alpha] induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23(9):1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 1999;59(16):3915–3918. [PubMed] [Google Scholar]
- 105.Krishnamurthy P, Ross DD, Nakanishi T, et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004;279(23):24218–24225. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 106.Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008;283(52):36542–36552. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mathieu J, Zhang Z, Zhou W, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71(13):4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2009;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005;23(5):1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 110.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438(7070):937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 111.Liao D, Hypoxia Johnson R. A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26(2):281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 112.Jensen R, Ragel B, Whang K, Gillespie D. Inhibition of hypoxia inducible factor-1α(HIF-1α) decreases vascular endothelial growth factor (VEGF) secretion and tumor growth in malignant gliomas. J. Neurooncol. 2006;78(3):233–247. doi: 10.1007/s11060-005-9103-z. [DOI] [PubMed] [Google Scholar]
- 113.Kim J-W, Gao P, Dang C. Effects of hypoxia on tumor metabolism. Cancer Metastasis Rev. 2007;26(2):291–298. doi: 10.1007/s10555-007-9060-4. [DOI] [PubMed] [Google Scholar]
- 114.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 115.Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumor hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J. Cell. Mol. Med. 2010;14(4):771–794. doi: 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Potter C, Harris AL. Hypoxia inducible carbonic anhydrase IX, marker of tumor hypoxia, survival pathway and therapy target. Cell Cycle. 2004;3(3):159–162. [PubMed] [Google Scholar]
- 117.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5(5):429–441. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 118.Moeller B, Richardson R, Dewhirst M. Hypoxia and radiotherapy: opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev. 2007;26(2):241–248. doi: 10.1007/s10555-007-9056-0. [DOI] [PubMed] [Google Scholar]
- 119.Chandel NS, Mcclintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia. J. Biol. Chem. 2000;275(33):25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 120.Jung SN, Yang WK, Kim J, et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis. 2008;29(4):713–721. doi: 10.1093/carcin/bgn032. [DOI] [PubMed] [Google Scholar]
- 121.Brown LM, Cowen RL, Debray C, et al. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol. Pharmacol. 2006;69(2):411–418. doi: 10.1124/mol.105.015743. [DOI] [PubMed] [Google Scholar]
- 122.Yang DI, Chen SD, Yang YT, Ju TC, Xu JM, Hsu CY. Carbamoylating chemoresistance induced by cobalt pretreatment in C6 glioma cells: putative roles of hypoxia-inducible factor-1. Br. J. Pharmacol. 2004;141(6):988–996. doi: 10.1038/sj.bjp.0705687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu L, Ning X, Sun L, et al. Hypoxia-inducible factor-1α contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008;99(1):121–128. doi: 10.1111/j.1349-7006.2007.00643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sasabe E, Zhou X, Li D, Oku N, Yamamoto T, Osaki T. The involvement of hypoxia-inducible factor-1α in the susceptibility to γ-rays and chemotherapeutic drugs of oral squamous cell carcinoma cells. Int. J. Cancer. 2007;120(2):268–277. doi: 10.1002/ijc.22294. [DOI] [PubMed] [Google Scholar]
- 125.Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updat. 2011;14(3):191–201. doi: 10.1016/j.drup.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 126.Comerford KM, Wallace TJ, Karhausen JR, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62(12):3387–3394. [PubMed] [Google Scholar]
- 127.Seagroves TN. Pharmaceutical Perspectives of Cancer Therapeutics. Springer Science and Business Media, LLC; NY, USA: 2009. The complexity of the HIF-1-dependent hypoxic response in breast cancer presents multiple avenues for therapeutic intervention; p. 540. [Google Scholar]
- 128.Tan Z, Wu Y, Zhang H, Xiao X. CoCl2-induced chemotherapy resistance in SW480 cells and its mechanism. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2006;31(3):345–349. [PubMed] [Google Scholar]
- 129.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012;33(4):207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xia Y, Choi H-K, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 2012;49:24–40. doi: 10.1016/j.ejmech.2012.01.033. [DOI] [PubMed] [Google Scholar]
- 131.Kola L, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug. Discov. 2004;3(8):1474–1776. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 132.Adams DJ. The valley of death in anticancer drug development: a reassessment. Trends Pharmacol. Sci. 2012;33(4):173–180. doi: 10.1016/j.tips.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Atkuri KR, Herzenberg LA, Herzenberg LA. Culturing at atmospheric oxygen levels impacts lymphocyte function. Proc. Natl Acad. Sci. USA. 2005;102(10):3756–3759. doi: 10.1073/pnas.0409910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ivanovic Z. Hypoxia or in situ normoxia: the stem cell paradigm. J. Cell. Physiol. 2009;219(2):271–275. doi: 10.1002/jcp.21690. [DOI] [PubMed] [Google Scholar]
- 135.Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56(6):1194–1198. [PubMed] [Google Scholar]
- 136.Gerweck LE, Kozin SV, Stocks SJ. The pH partition theory predicts the accumulation and toxicity of doxorubicin in normal and low-pH-adapted cells. Br. J. Cancer. 1999;79(5-6):838–842. doi: 10.1038/sj.bjc.6690134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wall ME, Wani MC. Camptothecin and taxol: discovery to clinic-thirteenth Bruce F. Cain memorial award lecture. Cancer Res. 1995;55(4):753–760. [PubMed] [Google Scholar]
- 138.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985;260(27):14873–14878. [PubMed] [Google Scholar]
- 139.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl Acad. Sci. USA. 2002;99(24):15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1. Cancer Res. 2004;64(4):1475–1482. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 141.Baranello L, Bertozzi D, Fogli MV, Pommier Y, Capranico G. DNA topoisomerase I inhibition by camptothecin induces escape of RNA polymerase II from promoter-proximal pause site, antisense transcription and histone acetylation at the human HIF-1α gene locus. Nucleic Acids Res. 2010;38(1):159–171. doi: 10.1093/nar/gkp817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rothenberg ML. Irinotecan (CPT-11): recent developments and future directions – colorectal cancer and beyond. Oncologist. 2001;6(1):66–80. doi: 10.1634/theoncologist.6-1-66. [DOI] [PubMed] [Google Scholar]
- 143.Leblanc R, Catley LP, Hideshima T, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62(17):4996–5000. [PubMed] [Google Scholar]
- 144.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–3076. [PubMed] [Google Scholar]
- 145.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011;10(11) doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shin DH, Chun Y-S, Lee DS, Huang LE, Park JW. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood. 2008;111(6):3131–3136. doi: 10.1182/blood-2007-11-120576. [DOI] [PubMed] [Google Scholar]
- 147.Kaluz S, Kaluzova M, Stanbridge EJ. Proteasomal inhibition attenuates transcriptional activity of hypoxia-inducible factor 1 (HIF-1) via specific effect on the HIF-1α C-terminal activation domain. Mol. Cell. Biol. 2006;26(15):5895–5907. doi: 10.1128/MCB.00552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Befani C, Vlachostergios P, Hatzidaki E, et al. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 2012;90(1):45–54. doi: 10.1007/s00109-011-0805-8. [DOI] [PubMed] [Google Scholar]
- 149.Bross PF, Kane R, Farrell AT, et al. Approval summary for Bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004;10(12):3954–3964. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]
- 150.Ueda H, Nakajima H, Hori Y, et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968 I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 1994;47(3):301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 151.Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 1998;241(1):126–133. doi: 10.1006/excr.1998.4027. [DOI] [PubMed] [Google Scholar]
- 152.Shigematsu N, Ueda H, Takase S, Tanaka H, Yamamoto K, Tada T. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No.968 II. Structure determination. J. Antibiot. 1994;47(3):311–314. doi: 10.7164/antibiotics.47.311. [DOI] [PubMed] [Google Scholar]
- 153.Ueda H, Manda T, Matsumoto S, et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No.968 III. Antitumor activities on experimental tumors in mice. J. Antibiot. 1994;47(3):315–323. doi: 10.7164/antibiotics.47.315. [DOI] [PubMed] [Google Scholar]
- 154.Fath DM, Kong X, Liang D, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-1α. J. Biol. Chem. 2006;281(19):13612–13619. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Qian DZ, Kachhap SK, Collis SJ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1α. Cancer Res. 2006;66(17):8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 156.Rini BI. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin. Cancer Res. 2008;14(5):1286–1290. doi: 10.1158/1078-0432.CCR-07-4719. [DOI] [PubMed] [Google Scholar]
- 157.Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006;12(1):122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 158.Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002;22(20):7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60(6):1541–1545. [PubMed] [Google Scholar]
- 160.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003;2(11):1093–1103. [PubMed] [Google Scholar]
- 161.Ghobrial IM, Leleu X, Rubin N, et al. Phase II trial of the novel oral Akt inhibitor perifosine in relapsed and/or refractory Waldenstrom macroglobulinemia (WM) J. Clin. Oncol. 2008;26(Suppl. 15):8546. [Google Scholar]
- 162.Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy. 2003;23(2):165–172. doi: 10.1592/phco.23.2.165.32088. [DOI] [PubMed] [Google Scholar]
- 163.Mabjeesh NJ, Escuin D, Lavallee TM, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3(4):363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 164.Carbonaro M, O’Brate A, Giannakakou P. Microtubule disruption targets HIF-1α mRNA to cytoplasmic P-bodies for translational repression. J. Cell Biol. 2011;192(1):83–99. doi: 10.1083/jcb.201004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dahut WL, Lakhani NJ, Gulley JL, et al. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol. Ther. 2006;5(1):22–27. doi: 10.4161/cbt.5.1.2349. [DOI] [PubMed] [Google Scholar]
- 166.Harrison MR, Hahn N, Pili R, et al. Phase II study of 2-methoxyestradiol (2ME2) NanoCrystal Dispersion (NCD) in patients with taxane-refractory, metastatic hormone-refractory prostate cancer (HRPC) J. Clin. Oncol. 2008;26(Suppl. 15):5173. doi: 10.1007/s10637-010-9455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bruce J, Eickhoff J, Pili R, et al. A Phase II study of 2-methoxyestradiol nanocrystal colloidal dispersion alone and in combination with sunitinib malate in patients with metastatic renal cell carcinoma progressing on sunitinib malate. Invest. New Drugs. 2012;30(2):794–802. doi: 10.1007/s10637-010-9618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Formica JV, Waring MJ. Effect of phosphate and amino acids on echinomycin biosynthesis by Streptomyces echinatus. Antimicrob. Agents Chemother. 1983;24(5):735–741. doi: 10.1128/aac.24.5.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Van Dyke M, Dervan P. Echinomycin binding sites on DNA. Science. 1984;225(4667):1122–1127. doi: 10.1126/science.6089341. [DOI] [PubMed] [Google Scholar]
- 170.Nickols NG, Jacobs CS, Farkas ME, et al. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem. Biol. 2007;2(8):561–571. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kong D, Park EJ, Stephen AG, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65(19):9047–9055. doi: 10.1158/0008-5472.CAN-05-1235. [DOI] [PubMed] [Google Scholar]
- 172.Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J. Cell. Mol. Med. 2009;13(9a):2780–2786. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Deboer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. J. Antibiot. 1970;23(9):442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 174.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl Acad. Sci. USA. 2000;97(20):10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl Acad. Sci. USA. 1994;91(18):8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dehner A, Furrer J, Richter K, Schuster I, Buchner J, Kessler H. NMR chemical shift perturbation study of the N-terminal domain of Hsp90 upon binding of ADP, AMP-PNP, geldanamycin, and radicicol. ChemBioChem. 2003;4(9):870–877. doi: 10.1002/cbic.200300658. [DOI] [PubMed] [Google Scholar]
- 177.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010;11(7):515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 178.Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol. Cancer Ther. 2004;3(8):1021–1030. [PubMed] [Google Scholar]
- 179.Minet E, Mottet D, Michel G, et al. Hypoxia-induced activation of HIF-1: role of HIF-1alpha-Hsp90 interaction. FEBS Lett. 1999;460(2):251–256. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 180.Mabjeesh NJ, Post DE, Willard MT, Kaur B, et al. Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002;62(9):2478–2482. [PubMed] [Google Scholar]
- 181.Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995;36(4):305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- 182.Neckers LM. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb. Exp. Pharmacol. 2006;172:259–277. doi: 10.1007/3-540-29717-0_11. [DOI] [PubMed] [Google Scholar]
- 183.Richardson PG, Chanan-Khan AA, Alsina M, et al. Tanespimycin monotherapy in relapsed multiple myeloma: results of a Phase I dose-escalation study. Br. J. Haematol. 2010;150(4):438–445. doi: 10.1111/j.1365-2141.2010.08265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Solit DB, Osman I, Polsky D, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin. Cancer Res. 2008;14(24):8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ronnen EA, Kondagunta GV, Ishill N, et al. A Phase II trial of 17-(allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest. New Drugs. 2006;24(6):543–546. doi: 10.1007/s10637-006-9208-z. [DOI] [PubMed] [Google Scholar]
- 186.Heath EI, Hillman DW, Vaishampayan U, et al. A Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 2008;14(23):7940–7946. doi: 10.1158/1078-0432.CCR-08-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pacey S, Wilson RH, Walton M, et al. A Phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin. Cancer Res. 2011;17(6):1561–1570. doi: 10.1158/1078-0432.CCR-10-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kaufmann SH, Karp JE, Litzow MR, et al. Phase I and pharmacological study of cytarabine and tanespimycin in relapsed and refractory acute leukemia. Haematologica. 2011;96(11):1619–1626. doi: 10.3324/haematol.2011.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol. 2012;6(2):155–176. doi: 10.1016/j.molonc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Valkov E, Sharpe T, Marsh M, Greive S, Hyvönen M. Targeting protein-protein interactions and fragment-based drug discovery. In: Davies TG, Hyvönen M, editors. Fragment-Based Drug Discovery and X-Ray Crystallography. Springer; Berlin/Heidelberg, Germany: 2012. pp. 145–179. [DOI] [PubMed] [Google Scholar]
- 191.Verdine GL, Hilinski GJ. All-hydrocarbon stapled peptides as synthetic cell-accessible mini-proteins. Drug Discov. Today Technol. 2012;9(1):e41–e47. doi: 10.1016/j.ddtec.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 192.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008;14(17):5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Higueruelo AP, Schreyer A, Bickerton GRJ, Pitt WR, Groom CR, Blundell TL. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: the TIMBAL database. Chem. Biol. Drug Des. 2009;74(5):457–467. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- 194.Fry DC. Drug-like inhibitors of protein-protein interactions: a structural examination of effective protein mimicry. Curr. Protein Pept. Sci. 2008;9(3):240–247. doi: 10.2174/138920308784533989. [DOI] [PubMed] [Google Scholar]
- 195.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 196.Monti S M, Supuran C T, De Simone G. Carbonic anhydrase IX as a target for designing novel anticancer drugs. Curr. Med. Chem. 2012;19(6):821–830. doi: 10.2174/092986712799034851. [DOI] [PubMed] [Google Scholar]
- 197.Mylonis I, Lakka A, Tsakalof A, Simos G. The dietary flavonoid kaempferol effectively inhibits HIF-1 activity and hepatoma cancer cell viability under hypoxic conditions. Biochem. Biophys. Res. Commun. 2010;398(1):74–78. doi: 10.1016/j.bbrc.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 198.Nagle D-G, Zhou Y-D. Natural product-based inhibitors of hypoxia-inducible factor-1 (HIF-1) Curr. Drug Targets. 2006;7(3):355–369. doi: 10.2174/138945006776054979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lakka A, Mylonis I, Bonanou S, Simos G, Tsakalof A. Isolation of hypoxia-inducible factor 1 (HIF-1) inhibitors from frankincense using a molecularly imprinted polymer. Invest. New Drugs. 2011;29(5):1081–1089. doi: 10.1007/s10637-010-9440-4. [DOI] [PubMed] [Google Scholar]
- 200.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl Acad. Sci. USA. 2009;106(42):17910–17915. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 201.Scheuermann TH, Tomchick DR, Machius M, Guo Y, Bruick RK, Gardner KH. Artificial ligand binding within the HIF2a PAS-B domain of the HIF2 transcription factor. Proc. Natl Acad. Sci. USA. 2009;106(2):450–455. doi: 10.1073/pnas.0808092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kung AL, Zabludoff SD, France DS, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6(1):33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 203.Cook KM, Hilton ST, MecinoviĆ J, Motherwell WB, Figg WD, Schofield CJ. Epidithiodiketopiperazines block the interaction between hypoxia-inducible factor-1α (HIF-1α) and p300 by a zinc ejection mechanism. J. Biol. Chem. 2009;284(39):26831–26838. doi: 10.1074/jbc.M109.009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. Inhibition of hypoxia inducible factor 1-transcription coactivator interaction by a hydrogen bond surrogate α-helix. J. Am. Chem. Soc. 2009;132(3):941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22(14):2065–2072. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 206.Post DE, Devi NS, Li ZC, et al. Cancer therapy with a replicating oncolytic adenovirus targeting the hypoxic microenvironment of tumors. Clin. Cancer Res. 2004;10(24):8603–8612. doi: 10.1158/1078-0432.CCR-04-1432. [DOI] [PubMed] [Google Scholar]
- 207.Post DE, Sandberg EM, Kyle MM, et al. Targeted cancer gene therapy using a hypoxia inducible factor-dependent oncolytic adenovirus armed with interleukin-4. Cancer Res. 2007;67(14):6872–6881. doi: 10.1158/0008-5472.CAN-06-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tan C, de Noronha RG, Roecker AJ, et al. Identification of a novel small-molecule inhibitor of the hypoxia-inducible factor 1 pathway. Cancer Res. 2005;65(2):605–612. [PubMed] [Google Scholar]
- 209.Narita T, Yin S, Gelin CF, et al. Identification of a Novel Small Molecule HIF-1 alpha Translation Inhibitor. Clin. Cancer Res. 2009;15(19):6128–6136. doi: 10.1158/1078-0432.CCR-08-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tan C, de Noronha RG, Devi NS, et al. Sulfonamides as a new scaffold for hypoxia inducible factor pathway inhibitors. Bioorg. Med. Chem. Lett. 2011;21(18):5528–5532. doi: 10.1016/j.bmcl.2011.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Mooring SR, Jin H, Devi NS, et al. Design and synthesis of novel small-molecule inhibitors of the hypoxia inducible factor pathway. J. Med. Chem. 2011;54(24):8471–8489. doi: 10.1021/jm201018g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mun J, Jabbar AA, Devi NS, Liu Y, Van Meir EG, Goodman MM. Structure-activity relationship of 2,2-dimethyl-2H-chromene based arylsulfonamide analogs of 3,4-dimethoxy-N-[(2,2-dimethyl-2H-chromen-6-yl)methyl]-N-phenylbenzenesulfonamide, a novel small molecule hypoxia inducible factor-1 (HIF-1) pathway inhibitor and anti-cancer agent. Bioorg. Med. Chem. 2012;20(14):4590–4597. doi: 10.1016/j.bmc.2012.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mun J, Jabbar AA, Devi NS, et al. Design and in vitro activities of N-alkyl-N-[(8-R-2,2-dimethyl-2H-chromen-6-yl)methyl] heteroarylsulfonamides, novel, small-molecule hypoxia inducible factor-1 pathway inhibitors and anticancer agents. J. Med. Chem. 2012;55(15):6738–6750. doi: 10.1021/jm300752n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shi Q, Yin S, Kaluz S, et al. Binding model for the interaction of anticancer arylsulfonamides with the p300 transcription cofactor. ACS Med. Chem. Lett. 2012;3(8):620–625. doi: 10.1021/ml300042k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yin S, Kaluz S, Devi NS, et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1 alpha interaction with cofactors p300/CBP. Clin. Cancer Res. 2012;18(24):6623–6633. doi: 10.1158/1078-0432.CCR-12-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang W, Ao L, Rayburn ER, et al. KCN1, a novel synthetic sulfonamide anticancer agent: in vitro and in vivo anti-pancreatic cancer activities and preclinical pharmacology. PLoS ONE. 2012;7(9):e44883. doi: 10.1371/journal.pone.0044883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 301.American Cancer Society [Accessed 11 April 2012];Cancer facts and figures 2011. www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-figures-2011.
- 302.World Cancer Research Fund International [Accessed 1 April 2012];General world cancer statistics: GLOBOCAN 2008 database. www.wcrf.org/cancer_statistics/world_cancer_statistics.php.
- 303. [Accessed 30 April 2012];US FDA approves topotecan hydrochloride (hycamtin) in combination with cisplatin for the treatment of Stage IVB recurrent or persistent carcinoma of the cervix. www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm095638.htm.
- 304. [Accessed 4 May 2012];GlaxoSmith Kline receives approval for hycamtin (topotecan) capsules for the treatment of relapsed small cell lung cancer. www.drugs.com/newdrugs/gsk-receives-approval-hycamtin-topotecan-capsules-relapsed-small-cell-lung-cancer-671.html.
- 305.National Cancer Institute [Accessed 1 June 2011];Romidepsin. www.cancer.gov/cancertopics/druginfo/romidepsin.
- 306. [Accessed 4 May 2012];US FDA approves new drug for advanced kidney cancer. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108924.htm.
- 307.Genetic Engineering & Biotechnology News [Accessed 11 May 2012];Aeterna Zentaris regains North American rights to Akt inhibitor from Keryx. www.genengnews.com/gen-news-highlights/aeterna-zentaris-regains-north-american-rights-to-akt-inhibitor-from-keryx/81246731/
- 308. [Accessed 26 February 2013];Patent Database Search Results: ISD 1/1/2000->12/31/2012 AND HIF AND cancer AND inhibitor in US Patent Collection. http://patft.uspto.gov/netacgi/nph-Parser?Sect1=PTO2&Sect2=HITOFF&u=%2Fnetahtml%2FPTO%2Fsearch-adv.htm&r=0&f=S&l=50&d=PTXT&RS=%28%28%28ISD%2F20120101-%3E20121231+AND+HIF%29+AND+Cancer%29+AND+inhibitor%29&Refine=Refine+Search&Refine=Refine+Search&Query=ISD%2F1%2F1%2F2000-%3E12%2F31%2F2012+AND+HIF+AND+Cancer+AND+inhibitor.
- 309. [Accessed 26 February 2013];Web of Knowledge – All Database Results: Topic=(HIF AND CANCER AND INHIBITOR) Timespan=2000-2012. http://apps.webofknowledge.com/summary.do?SID=1F83DLk7akON2aaKN9E&product=UA&qid=18&search_mode=GeneralSearch.
- 310. [Accessed 26 February 2013];Search of: HIF and cancer – List Results – ClinicalTrials.gov. ClinicalTrials.gov www.clinicaltrials.gov/ct2/results?term=hif+and+cancer+&Search=Search.