

Abstract
Hypoxia is linked to epithelial mesenchymal transition (EMT) and tumor progression in numerous carcinomas. Responses to hypoxia are thought to operate via hypoxia-inducible factors (HIFs), but the importance of co-factors that regulate HIF signaling within tumors is not well understood. Here we elucidate a signaling pathway that physically and functionally couples tyrosine phosphorylation of β-catenin to hypoxia-inducible factor-1α (HIF1α) signaling and HIF1α-mediated tumor EMT. Primary human lung adenocarcinomas accumulate pY654-β-catenin and HIF1α. All pY654-β-catenin, and only the tyrosine phosphorylated form, was found complexed with HIF1α and active Src, both within human tumors and in lung tumor cell lines exposed to hypoxia. Phosphorylation of Y654, generated by hypoxia mediated, reactive oxygen species (ROS)-dependent Src kinase activation, was required for β-catenin to interact with HIF1α and Src, to promote HIF1α transcriptional activity, and for hypoxia-induced EMT. Mice bearing hypoxic pancreatic islet adenomas, generated by treatment with anti-vascular endothelial growth factor antibodies, accumulate HIF1α/pY654-β-catenin complexes and develop an invasive phenotype. Concurrent administration of the ROS inhibitor N-acetylcysteine abrogated β-catenin/HIF pathway activity and restored adenoma architecture. Collectively, the findings implicate accumulation of pY654-β-catenin specifically complexed to HIF1α and Src kinase as critically involved in HIF1α signaling and tumor invasion. The findings also suggest that targeting ROS-dependent aspects of the pY654-β-catenin/ HIF1α pathway may attenuate untoward biological effects of anti-angiogenic agents and tumor hypoxia.
Keywords: hypoxia, signaling, transcription, tumor, epithelial
Introduction
The process of tumor metastasis is thought to involve EMT (1, 2). EMT involves transcriptional reprogramming whereby epithelial tumor cells lose cell polarity and cell junction proteins (such as E-cadherin) and at the same time acquire signal transduction activities associated with mesenchymal cells and mesenchymal cell markers, e.g. fibronectin (Fn), collagen I, and metalloproteinases. This reprogramming facilitates migration, survival, and ultimately metastasis at distal sites (3, 4). Both hypoxia and overexpression of hypoxia inducible factor (HIF)-1α and/or HIF-2α have been shown to promote EMT and metastatic phenotypes (5, 6). Indeed the presence of tumor hypoxia has long been associated with poor cancer outcome (7). This problem has received additional attention because of the recently observed untoward effects of anti-angiogenic therapies on tumor invasion, likely operating at least in part through the generation of hypoxia (8, 9). Mechanisms possibly connecting hypoxia with EMT include intracellular reactive oxygen species (ROS)-dependent HIF accumulation (10), Snail translocation (11) and HIF1α-dependent accumulation of the transcription factors Snail, Twist, ZEB1 and ZEB2, key regulators of EMT (12). HIF1α is reported to directly bind the Snail and Twist promoter (13, 14). Although HIF1α accumulation is a fundamental regulator of the cellular response to hypoxia, HIF1α transcriptional activity is dependent not only on binding to its canonical DNA hypoxia response element (HRE) but also on a complex array of co-factors that dictate which genes are preferentially activated in different cells exposed to hypoxia (15). One such co-factor previously reported to bind HIF1α and promote its transcriptional activity is β-catenin (16).
Signaling through Wnt/β-catenin has been implicated in EMT in breast cancer cells via upregulation of the Wnt target gene Axin2 followed by stabilization of nuclear Snail (17, 18). In other cells Wnt is reported to mainly influence tumor cell proliferation via induction of c-myc and cyclin D1 (19). Indeed the principal mechanism underlying the strong association between stabilizing mutations in β-catenin and tumor development is thought to be β-catenin driven-tumor cell proliferation (20). In addition, a number of tyrosine phosphorylations of β-catenin have been reported and these appear to function not simply by promoting canonical Wnt target genes but instead by modifying the repertoire of β-catenin binding partners. For example, Y654-β-catenin phosphorylation disrupts the association between β-catenin and E-cadherin, favoring its transcriptional activity (21). We have previously reported that pY654-β-catenin is found in complexes with p-Smad2 following transforming growth factor (TGF) β1 signaling and such complexes strongly correlate with TGFβ1-induced EMT in kidney and lung alveolar epithelial cells both ex vivo and in vivo (22, 23). Accumulation of pY654-β-catenin following TGFβ1 stimulation had little or no contribution to canonical Wnt pathway signaling (24). Whether hypoxia-induced EMT in tumor cells either generates or requires pY654-β-catenin and whether tyrosine phosphorylation of Y654 regulates β-catenin association with HIF1α is currently unknown.
Activation of several oncogenic tyrosine kinases including Src family kinases, epidermal growth factor receptor (EGFR) (25) and hepatocyte growth factor receptor c-Met (26) have been reported to lead to β-catenin phosphorylation. However, only Src kinase(s) has been shown to directly phosphorylate Y654. Though mechanisms remain undefined, over-expression of activated Src kinase has been found to promote HIF1α accumulation and its transcriptional activity (27, 28), raising the possibility of an intrinsic linkage between Src kinase activity and HIF1α signaling. Elevated Src activation has been reported in hypoxic regions of tumor xenografts, but whether Src kinase activity is promoted by hypoxia in human cancer is not reported (29). Hence it is also unknown whether hypoxia-regulated tyrosine kinases contribute to the phosphorylation of β-catenin and hypoxia-induced EMT in human carcinomas. This is an important point because tumor invasion and metastasis may depend as much or more on activation of pro-invasive pathways within developed tumors as on driver mutations implicated in tumor initiation(30, 31). In this study we explore the importance of pY654-β-catenin accumulation in tumor responses to hypoxia.
Results
pY654-β-catenin accumulates and associates with HIF1α and Src in human lung adenocarcinomas
To test for the presence of pY654-β-catenin in human lung cancers, protein extracts of flash frozen tumor tissues and matched contiguous normal lung tissues surgically dissected from lung adenocarcinoma patients were analyzed by immunoblotting. pY654-β-catenin was easily detected in all the tumor tissues but not in the contiguous normal tissues (Figure 1a, top) although total β-catenin varied among the samples (Figure 1a, bottom). As observed in prior studies of fibrosis (23), p-Smad2 co-precipitated with pY654-β-catenin in all of the tumor specimens (Figure 1a). Several tyrosine kinases that are known to promote Y654-β-catenin phosphorylation including activated Src and EGFR (25), as well as activated and total c-Met, were also upregulated in the tumor tissues compared to the matched normal controls (Figure 1b and 1c). Further, both the EMT markers Snail1 and Twist as well as the hypoxia marker HIF1α accumulated in these lung tumors (Figure 1b). To initially assess whether there is a functional link between active Src, pY654-β-catenin, and EMT markers, slices of fresh adenocarcinomas were cultured in vitro with or without Src kinase inhibitors and then immunoblotted. The presence of pY654-β-catenin, active Src, and Snail1/Twist in lysates from cultured fresh tumor slices (Figure S1) were all abrogated by the Src inhibitors PP2 and SU6656, suggesting that Src family kinases play an important role in phosphorylating β-catenin and the subsequent EMT.
Figure 1.

Multiple activated tyrosine kinases in human lung adenocarcinomas associated with formation of pY654-β-catenin complexes with HIF1α and p-Smad2. (a) Human lung tumors (T) and contiguous normal tissues (N) were lysed and the lysates immunoprecipitated for pY654-β-catenin and blotted for β-catenin and p-Smad2. Equal amount of protein from both normal and tumor lysates were mixed together as input for mouse IgG control. (b–d) The above lysates were blotted for EMT-related proteins listed in (b), immunoprecipitated for p-c-Met and blotted for total c-Met (c), and sequentially immunoprecipitated for pY654-β-catenin and total β-catenin and blotted for β-catenin, HIF1α, and Src (d). (e) Lysates were sequentially immunoprecipitated for two rounds of HIF1α and one round of pY654-β-catenin and blotted for β-catenin, HIF1α, and Src.
Because hypoxia-induced HIF1α has been shown to promote EMT and bind β-catenin (16, 32), we tested whether the tyrosine phosphorylated form of β-catenin preferentially interacts with HIF1α. Indeed pY654-β-catenin immunoprecipitation co-precipitated HIF1α and Src whereas sequential total β-catenin precipitates did not contain detectable HIF1α or Src (Figure 1d), implying association with HIF1α and Src depends on pY654. Consistent with this conclusion, immuno-depletion of HIF1α removed virtually all pY654-β-catenin from tumor lysates (Figure 1e). To explore further the requirement for pY654 in the interaction of β-catenin with HIF1α and its functional importance in tumor EMT we turned to model systems.
Hypoxia induces pY654-β-catenin association with HIF1α/Src and increases EMT markers in lung adenocarcinoma cells in a Src-dependent manner
We used human lung adenocarcinoma cell lines H358 and A549 as ex vivo models to test the role of HIF1α/pY654-β-catenin in hypoxia-stimulated EMT. After 2 or 12 hours incubation under hypoxic conditions (1% O2), there was a robust increase of pY654-β-catenin and low levels of pY654-β-catenin/p-Smad2 complexes, reflecting the low basal levels of p-Smad2 in H358 cells that is not altered by hypoxia (Figure 2a and S2a).
Figure 2.

Hypoxia induces Src kinase-dependent pY654-β-catenin/HIF1α complexes and EMT in human lung adenocarcinoma cells. (a) pY654-β-catenin immunoprecipitation of H358 cells incubated in normoxia (21% O2) (N) or hypoxia (1% O2) (H) for 2 h or 12 h. (b) Immunoblots for HIF1α, pY416-Src, total Src, and Snail1 of H358 cells under normoxia or hypoxia for 24 h ± Src inhibitor. TGFβ1 stimulation-positive control; β-actin blot-loading control. (c, d) Sequential immunoprecipitation for pY654-β-catenin and total β-catenin (c) or two rounds of HIF1α and one round of pY654-β-catenin (d) of H358 cells under normoxia or hypoxia or hypoxia with Src inhibitor SU6656 (5 µM) (H+SU) for 4 h. (e) Myc immunoprecipitation of H358 cells expressing Myc-tagged wt (W) or Y654F mutant (F) β-catenin under normoxia or hypoxia for 4 h. (f) Fibronectin (orange) and E-cadherin (green) staining of H358 cells under normoxia or hypoxia for 56 h ± Src inhibitor. Scale bar, 100 µm.
Exposure of H358 cells to hypoxia increased HIF1α protein level (Figure 2b and S2b, right) and Src activity (Figure 2c). HIF1α and Src were detected in pY654-β-catenin IP from cells exposed to hypoxia but not from cells cultured under normoxia or from hypoxic cells treated with Src inhibitor SU6656 (Figure 2b and S2b, left). Again, there was no HIF1α and Src discernible in the subsequent total β-catenin immunoprecipitates (Figure 2b and S2b, middle) and HIF1α depletion removed all detectable pY654-β-catenin (Figure 2d and S2c), similar to observations in human lung adenocarcinomas.
To further test whether pY654 is required for HIF1α and Src association, we stably expressed Myc-tagged wt (W) or a non-phosphorylated Y654F mutant (F) β-catenin in H358 cells. Myc immunoprecipitates of these cells under hypoxic conditions confirmed that only the wt but not the Y654F β-catenin associates with HIF1α and Src (Figure 2e). In additional experiments we observed that over-expression of either wt or Y654F β-catenin in 293 fibroblastic cells resulted in comparable HIF1α/β-catenin interactions, consistent with direct in vitro binding observed in a prior report (16). But at endogenous levels of HIF1α and β-catenin, at least in epithelial cells where the bulk of β-catenin is bound to E-cadherin, the only species of β-catenin found in association with HIF1α is pY654-β-catenin. Generation of this β-catenin species requires active Src kinase and presumably Y654 subsequently acts as a binding site for Src SH2 domain, though we have not specifically addressed this point.
Active Src kinase(s) was also found to be required for hypoxia-induced EMT. Under normoxia, lung cancer cells strongly expressed the epithelial marker E-cadherin at cell:cell contacts and had little mesenchymal marker Fn staining, whereas cells cultured under hypoxic conditions for 56 hours underwent clear morphological changes (Figure 2f and S2d, top). This phenotype was reversed by SU6656 (Figure 2f and S2d, bottom). Immunoblotting showed both hypoxia-induced Src activation and Snail1 expression were inhibited by SU6656 (Figure 2c). While hypoxia-induced upregulation of HIF1α does not require Src activity (Figure 2c), knockdown of HIF1α completely blocked hypoxia-induced EMT in H358 cells (Figure S3), indicating a critical role for both Src and HIF1α.
pY654-β-catenin and HIF1α act as a functional unit
To address the functional importance of pY654-β-catenin in hypoxia-induced EMT we utilized a T antigen-immortalized alveolar epithelial cell line homozygous for a floxed β-catenin allele, termed AECT. When plated onto Fn for 3 days AECTs activate latent TGFβ1 and undergo EMT as indicated by the induction of p-Smad2, loss of intact adherens junctions, and induction of mesenchymal genes (24). To test whether TGFβ1 signaling is required for hypoxia-induced EMT we treated cells with ALK5 inhibitor SB431542. As expected, when these cells were exposed to hypoxia there was induction of HIF1α (Figure 3a) and pY654-β-catenin that was not blocked by ALK5 inhibitor (Figure S4a). All pY654-β-catenin was again associated with HIF1α and this association was Src kinase dependent (Figure 3a). Cells null for β-catenin were generated upon adenovirus-Cre exposure (Ad-Cre). Under hypoxia, AECTs treated with Ad-Cre maintained an epithelial phenotype with strong cell border staining of E-cadherin (green) whereas AECTs treated with control adenovirus-GFP (Ad-GFP) completely lost cell:cell contact and border staining of E-cadherin (green) and β-catenin (red) (Figure 3b). The deletion of β-catenin also blocked hypoxia-induced EMT biomarkers collagen I (Figure 3c), Twist (Figure 3d), as well as the invasive metalloproteinase, MMP-2 (33), which was Src kinase dependent (Figure 3e). Depletion of either β-catenin (Figure 3f) or HIF1α (Figure 3g) blocked collagen I and Snail 1 mRNA induction by hypoxia, indicating both β-catenin and HIF1α are functionally important for hypoxia-induced EMT.
Figure 3.

Hypoxia-induced EMT requires both HIF1α and β-catenin expression. (a) HIF1α and pY654-β-catenin sequential immunoprecipitation of AECTs under normoxia or hypoxia ± Src inhibitor for 4 h. (b) Immortalized AECTs with floxed β-catenin were infected with Ad-Cre or Ad-GFP and cultured in hypoxia for 56 h. Cells were stained for E-cadherin (green) and β-catenin (orange). Scale bar, 50 µm. (c–e) AECTs were infected with Ad-Cre or Ad-GFP and incubated in hypoxia for 56 h ± ALK5 inhibitor SB431542 or Src inhibitor SU6656 (5 µM) before immunoblotting for proteins as indicated in panels (c) and (d). Conditioned medium was concentrated 10× for zymography with recombinant MMP-2 as positive control (e). (f, g) AECTs infected with Ad-Cre or Ad-GFP (f) or transfected with HIF1α or non-targeting control siRNA (g) were incubated in normoxia or hypoxia for 24 h before qRT-PCR analysis. Collagen I, Snail1, β-catenin, HIF1α mRNA levels were normalized to β-actin mRNA level. * indicates p<0.05 by t-test.
We next reconstituted the β-catenin null AECTs with wild-type or specific point mutants of β-catenin to specifically assess the role of Y654. A Y654E mutation mimics its phosphorylated form, while a Y654F mutation mimics the non-phosphorylated form of β-catenin (21). We verified that the Y654E mutant β-catenin was recognized by the pY654-β-catenin antibody (Figure S4b). Interestingly, Y654E mutant cells already show a mesenchymal phenotype under normoxia as judged by disrupted border staining of E-cadherin (green) and strong staining of EMT marker, α-SMA (orange) while Y654F mutant cells maintain an epithelial phenotype (Figure 4a). When AECTs with β-catenin deletion (Cre), wt β-catenin (W), and Y654E (E) or Y654F (F) mutant β-catenin were exposed to hypoxia, Y654E mutant cells showed high levels of collagen I, 〈-SMA, Twist (Figure 4b) as well as MMP-2 (Figure 4c) under both normoxia and hypoxia. While these markers were significantly induced by hypoxia in WT cells, Y654F mutant cells failed to respond to hypoxia, similar to the AECTs with ®-catenin deletion (Figure 4b–4d). Y654E-®-catenin proved to be most efficient in EMT induction among different ®-catenin forms, as illustrated by the EMT marker/®-catenin protein expression ratio quantified and pooled from 3 independent experiments (Figure 4d).
Figure 4.

Y654-β-catenin phosphorylation is required for hypoxia-induced EMT and β-catenin promotion of HIF1α transcriptional activity. (a) E-cadherin (green) and α-SMA (orange) staining of AECTs expressing Y654E or Y654F-β-catenin. Scale bar, 100 µm. (b) Immunoblots for collagen I, α-SMA, Twist, and HIF1α of AECTs with β-catenin deletion (Cre), cells expressing wt (W) and Y654E (E) or Y654F (F) mutant in normoxia or hypoxia for 56 h. (c) Conditioned medium from the cells above were concentrated for zymography with recombinant MMP-2 as control. (d) EMT markers from B and C were quantified using Image J and the mean value normalized to β-catenin level from 3 independent experiments were shown. ns, not significant. * indicates p<0.05 by t-test. (e) HRE reporter activity of 293 cells expressing W, E or F mutant β-catenin under normoxia or hypoxia for 16 hours. Renilla activity was used as internal control. Mean value of hypoxia/normoxia ratio from 4 independent experiments was shown. * indicates p<0.05 by t-test. 293 cell lysates were also blotted for Myc-tagged β-catenin and β-actin.
To examine whether hypoxia-induced HIF1α transcriptional activity is regulated by the phosphorylation status of Y654, we transfected a HRE reporter construct into 293 cells expressing either wt (W), Y654E (E) or Y654F (F) mutant ®-catenin (Figure 4e, left). Under hypoxia, non-transfected cells expressing only endogenous ®-catenin had ~10-fold increase in luciferase activity. The ratio of hypoxia/normoxia for these cells was assigned a value of 1. Either wt or Y654E mutant cells showed 3–4-fold higher HRE activity over non-transfected cells whereas the HRE activity of Y654F mutant cells (Figure 4e, right) were no different than non-transfected controls. These findings demonstrate that Y654 phosphorylation directly promotes HIF1α transcriptional activity, consistent with our findings of ®-catenin/HIF1α-dependent induction of mRNA for EMT genes (Figure 3). Interestingly, in addition to EMT genes, a number of other classical HIF1α responsive genes (Figure S5a) were also found to be sensitive to Src activity (Figure S5b) and β-catenin expression (Figure S5c). Taken together, these data indicate that tumor cell responses to hypoxia that depend on upregulated HIF1α activity also require association of HIF1α with pY654-®-catenin for a full transcriptional response.
ROS activity is required for hypoxia-induced tyrosine kinase activation, β-catenin phosphorylation, and subsequent EMT
In some cell systems hypoxia is reported to generate ROS that can then lead to Src activation and EMT (32, 34). Indeed hypoxia significantly increased ROS activity in H358 cells (Figure 5a) and A549 cells (Figure S6a) as measured by fluorescence response of 3’-(p-aminophenyl) fluorescein (APF) (35). Both ROS inhibitors EUK-134 and/or N-acetyl-cysteine (NAC) inhibited hypoxia-induced Src activation and pY654-β-catenin formation in H358 cells (Figure 5b) and A549 cells (Figure S6b). In contrast, Src activation and pY654-β-catenin formation initiated by TGFβ1 signaling (24) were unaffected by ROS inhibitors (Figure 5b and S6b). Notably, HIF1α expression levels were suppressed by ROS inhibitor(s) in H358 cells (Figure 5b) but not in A549 cells (Figure S6b) indicating that pY654-β-catenin/HIF1α complexes rather than HIF1α levels alone track with EMT and its suppression by ROS inhibitors.
Figure 5.

Hypoxia-induced pY654-β-catenin formation and EMT are dependent on ROS. (a) H358 cells were cultured in normoxia or hypoxia for 2 h ± ROS scavenger NAC (10 mM). APF signal (green) indicates ROS activity and the fluorescence intensity was quantified by Image J. * indicates p< 0.05 by t-test. Scale bar, 50 µm. (b) H358 cells were cultured in normoxia or hypoxia for 4 h or treated with TGFβ1 (4 ng/ml) for 2 h ± EUK-134 or NAC. The lysates were immunoprecipitated for pY654-β-catenin and the lysates were blotted for β-catenin, pY416-Src, Src, HIF1α, and β-actin. (c) Immumoblots for Snail1 in H358 cells under normoxia or hypoxia for 24 h ± NAC. The numbers indicate relative Snail1 level normalized to β-actin. (d) H358 cells were incubated in normoxia or hypoxia for 24 h ± EUK-134 or NAC. The conditioned media were concentrated for zymography with recombinant MMP-9 as positive control.
Hypoxia-induced activation of EGFR and c-Met was blocked by ROS inhibitors (Figure S7a and S7b) as well as Src inhibition (Figure S7c and S7d), implying that the activation of these receptor tyrosine kinases is triggered by hypoxia-increased ROS activity, but is also downstream of Src activation. Longer exposure of H358 cells to hypoxia confirmed that hypoxia-induced Snail1 (Figure 5c), the invasive marker MMP-9 (Figure 5d), and wound closure (Figure S8) were all dependent on ROS activity. Collectively, these findings indicate that hypoxia-induced ROS leads to activation of Src kinase(s) and then EGFR and c-Met activation that in turn further promotes Src activation and pY654-β-catenin accumulation. These co-factors then cooperate with HIF1α to drive an invasive EMT program. We next asked whether these ex vivo observations operate in vivo.
Anti-VEGF neutralizing antibodies induce tumor hypoxia, pY654-β-catenin/HIF1α/Src accumulation, and EMT in vivo
In a mouse model of pancreatic islet carcinogenesis (RIP-Tag2), Casanovas and colleagues reported that VEGF blockade in late-stage tumors resulted in hypoxia-mediated induction of VEGF-independent proangiogenic factors (36). Sennino and colleagues recently reported that anti-VEGF antibodies in this model led to both tumor EMT and marked tumor invasiveness, including metastasis (37). We therefore addressed our ex vivo findings in the RIP-Tag2 model. When 14-week old RIP-Tag2 mice were treated with a goat anti-VEGF antibody or control IgG daily for a week, there was marked pY654-β-catenin as well as HIF1α accumulation with the anti-VEGF antibody but not the controls (Figure 6a–6c). We also noted that p-Smad2 was associated with pY654-β-catenin in the tumor lysates, similar to human lung tumors (Figure 1), but anti-VEGF antibodies had no discernible effect on p-Smad2 levels (Figure 6a). All or virtually all pY654-β-catenin induced by anti-VEGF in these tumors was complexed with HIF1α and Src (Figure 6b). Western blot analysis confirmed upregulation of active Src, N-cadherin, vimentin, Snail1, and Twist by anti-VEGF antibody treatment (Figure 6c), indicating activation of an EMT program. Thus there is a strong correlation between anti-VEGF induced-hypoxia, pY654-β-catenin/HIF1α generation, and EMT reprogramming in the RIP-Tag2 tumor model.
Figure 6.

pY654-β-catenin/HIF1α/Src complexes and EMT markers are increased in anti-VEGF antibody treated pancreatic tumors. (a) RIP-Tag2 tumor lysates from anti-VEGF- or control IgG-treated mice were immunoprecipitated for pY654-β-catenin and blotted for β-catenin and p-Smad2. (b) The above lysates were sequentially immunoprecipitated for two rounds of HIF1α and one round of pY654-β-catenin and blotted for β-catenin, HIF1α, and Src. (c) The above lysates were blotted for pY416-Src, Src, E-cadherin, N-cadherin, vimentin, Snail1, Twist, HIF1α, and β-actin.
Finally we asked whether anti-VEGF induced pY654-β-catenin/HIF1α/Src activation and EMT in this model require ROS. Fourteen-week old RIP-Tag2 mice were concurrently treated with goat anti-VEGF antibody and the antioxidant NAC or control buffer daily for 4–5 days. Co-immunostaining of E-cadherin (red) and the RIP-Tag2 tumor cell marker insulin (green) revealed marked diminution of E-cadherin staining by antibody treatment and the antibody effect was reversed by NAC treatment (Figure 7a). Consistent with abrogation of the EMT program, pancreatic tumors in mice concurrently given NAC had sharp borders (Figure 7a and S9), typical of the adenomas prior to acquiring an invasive phenotype. Concurrent immunoblotting indicated that overall protein levels of E-cadherin did not decrease in the tumor specimens (Figure 6c) only the intensity of staining which reflects the loss of integrity of adherens junctions (37). Consistent with this finding, the margins of the tumors were more irregular and interlaced with pancreatic tissue in anti-VEGF treated mice (Figure 7a). We cannot exclude the possibility that some of the total E-cadherin protein in tumors of the anti-VEGF treated mice derives from normal pancreatic tissue infiltrated by the tumors. In contrast, the anti-VEGF antibody-enhanced pY654-β-catenin/HIF1α, EMT markers, Src activity and phospho-c-Met (Figure 7b–7e) were all reversed by NAC treatment. Reduced HIF1α is consistent with its reported stabilization by ROS (10).
Figure 7.

pY654-β-catenin/HIF1α and p-c-Met accumulation, EMT markers, and tumor invasiveness induced by anti-VEGF are blocked by ROS inhibition. (a) Confocal images of anti-VEGF treated-RIP-Tag2 tumors ± NAC for 5 days stained for insulin (tumor cells, green) and E-cadherin (acinar cells and tumor cells, red). Scale bar, 200 µm. (b) Combination (anti-VEGF+NAC) or anti-VEGF alone (Anti-VEGF)-treated RIP-Tag2 tumor lysates were immunoprecipitated for pY654-β-catenin and blotted for β-catenin. (c–e) The above lysates were blotted for E-cadherin, N-cadherin, vimentin, Snail1, Twist, HIF1α, and β-actin (c) or pY416-Src, Src, pY845-EGFR, and EGFR (d) or immunoprecipitated with p-c-Met and blotted for c-Met (e).
Discussion
In this study we identify a transcriptional complex that coordinates HIF1α and β-catenin signaling as a function of hypoxia-induced ROS and Src kinase activation. The data reported here indicate that activity of the HIF1α/pY654-β-catenin complex, over either element alone, promotes tumor cell acquisition of a mesenchymal phenotype both in vitro and in vivo. These findings provide a conceptual paradigm that extends prior studies of HIF1α, β-catenin, and Src kinase in tumor invasion and metastasis by linking the function of each element, and the complex as a whole, to the presence of hypoxia-induced ROS (Figure 8). Whereas β-catenin transcriptional activity as a function of Wnt-induced or mutation-based cytoplasmic stabilization mainly promotes epithelial proliferation (20), tyrosine phosphorylation of β-catenin switches β-catenin to an interaction with HIF1α that promotes mesenchymal transition. Interestingly, the interaction of pY654-β-catenin with HIF1α is required for a full transcriptional response of multiple HIF1α responsive genes to hypoxia (Figure S5). The levels of pY654-β-catenin, and therefore HIF1α/pY654-β-catenin complexes, could be expected to rise if Wnt signaling is superimposed on hypoxia(24), though hypoxia per se has little or no impact on Wnt activity in human lung adenocarcinoma cell lines (Figure S10a). The failure of β-catenin complexed with HIF1α to signal via the classical Wnt pathway has been previously noted (16), though the requirement and consequences of tyrosine phosphorylation were not evaluated. Overall, β-catenin appears to be at the center of intersecting signaling pathways that collectively determine the response of tumor cells to a hypoxic microenvironment. These findings provide further mechanistic insight into the established association between hypoxia and tumor invasion and highlight a pathway of acquisition of tumor invasiveness that, while involving activation of tyrosine kinases, operates through common features of a solid tumor without invoking acquisition of new driver mutations. This conclusion is consistent with the lack of evidence for metastasis as representing simply a genetic evolution of the primary tumor (31, 38).
Figure 8.
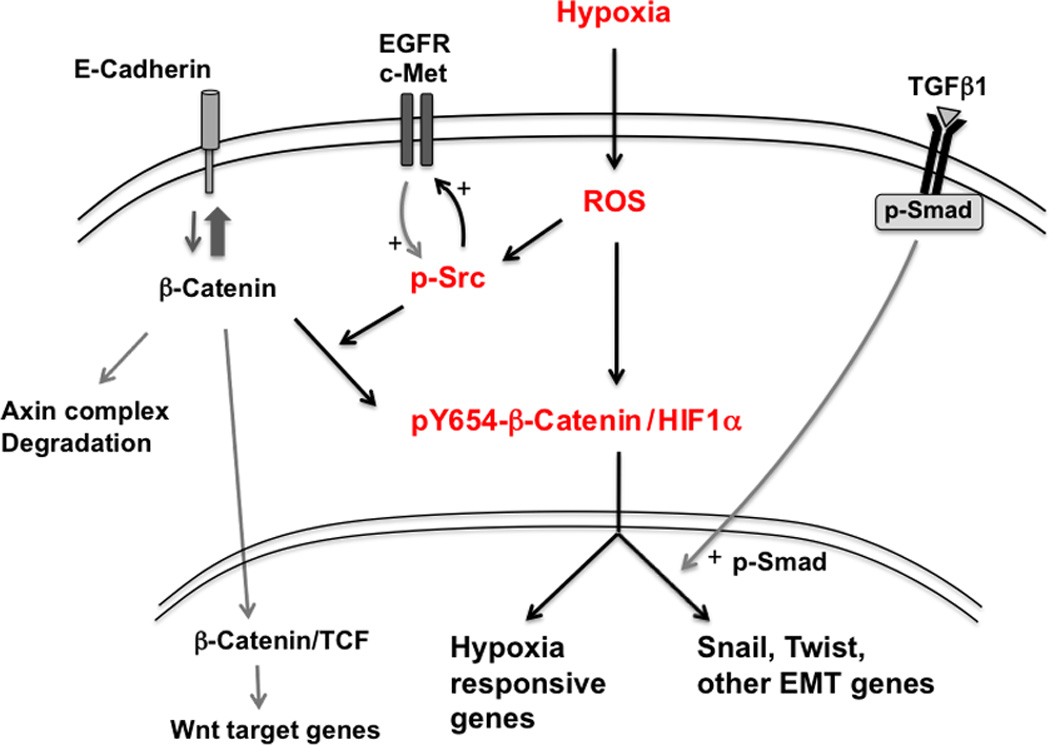
Schematic diagram illustrating central role for ROS and Src in hypoxia-induced pY654-β-catenin/HIF1α formation and tumor EMT. Hypoxia exposure generates reactive oxygen species (ROS) and stabilizes HIF1α (10). Increased ROS activity results in the activation of Src family kinase(s) (p-Src) (45) that then promotes activation of tyrosine kinases such as EGFR and c-Met, further promoting p-Src. Active Src phosphorylates β-catenin at Y654, favoring β-catenin association with HIF1α over β-catenin degradation, binding to E-cadherin, or association with TCFs in the Wnt pathway. pY654-β-catenin/HIF1α complexes promote transcription of EMT genes as well as other hypoxia responsive genes. Although not shown, Src is also in complexes of pY654-β-catenin/HIF1α. TGFβ1 signaling is not enhanced by hypoxia but remains active within the tumor microenvironment and further promotes hypoxia-induced EMT.
Prior studies demonstrate that HIF1α can directly promote EMT, at least in part through binding HIF response elements in the Twist and Snail1 promoters (14). Expression of HIF2α promotes lung tumorigenesis and EMT in a mutant K-Ras mouse model (39) and HIF1α expression levels correlate with tumor progression in several human cancers, including NSCLC (6). As reported here, HIF1α accumulation is prominent both in primary human lung adenocarcinomas and in experimentally induced tumor hypoxia (Figure 1 and 6), co-existing with markers of EMT including up-regulation of Twist and Snail1. However our analysis of hypoxia- and HIF1α-dependent EMT in three independent cell systems indicate the promoting effects of HIF1α on EMT require association with pY654-β-catenin. Expression of a phosphorylation mimic of β-catenin enhanced HIF1α promoter activity 3–4-fold over that of native β-catenin whereas a non-phosphorylatible form of β-catenin was without effect. In vivo, accumulation of HIF1α and pY654-β-catenin developed in parallel in mice bearing pancreatic tumors and exposed to anti-VEGF antibodies. Accumulation of both proteins, along with evidence of EMT, abated in response to ROS inhibitors. Collectively our findings invite the view that at least in epithelial cells HIF1α and pY654-β-catenin act as a functional signaling unit. If so, this concept implies determinants of Src kinase activity are also intrinsically linked to HIF1α signaling.
Of particular interest is the activation state of c-Met in the RIP-Tag2 tumors from mice treated with anti-VEGF antibodies. Activation of c-Met was recently reported to be crucial to anti-VEGF antibody-induced EMT and metastasis in the RIP-Tag2 tumor model (37). In our studies c-Met activation was ROS and Src kinase dependent both ex vivo and in vivo (Figure 5 and 7). An alternative mechanism for anti-VEGF mediated c-Met activation was recently reported in neuroblastoma cells. VEGF-dependent accumulation of the tyrosine phosphatase PTP1B in complexes of c-Met and VEGF receptors acts as a brake on c-Met activation. Anti-VEGF antibodies enhanced c-Met activation by attenuating phosphatase activity near the c-Met receptors (40). These mechanisms may not be mutually exclusive as the active site cysteines in a number of tyrosine phosphatases, including PTP1B, are known to be targeted by ROS (41). We speculate that hypoxia induced ROS may effect c-Met activation both by activation of Src leading to ligand independent activation of c-Met and ROS-dependent PTP1B phosphatase inhibition, further supporting c-Met activation.
Prior studies have identified Wnt-independent signaling events operating through tyrosine phosphorylation of β-catenin. For example, EGFR-dependent Src kinase activation leads to pY333-β-catenin that promotes its association with the glycolytic enzyme, pyruvate kinase M2. This β-catenin complex was shown to translocate to the nucleus and promote proliferation through a classical Wnt target gene, cyclin D1 (42). Whether Y654-β-catenin is phosphorylated in this signaling cascade is unclear. We have previously reported TGFβ1-dependent Src kinase activation leading to pY654-β-catenin and its association with p-Smad2. Formation of pY654-β-catenin/p-Smad2 complexes promoted EMT of kidney and lung alveolar epithelial cells (22, 23). Activation of Src kinase through hypoxia and TGFβ1 signaling is distinct because the former is critically dependent on ROS whereas TGFβ1-mediated Src activation is not affected by ROS inhibitors (Figure 5b). In vivo, as judged by co-immunoprecipitation experiments in extracts of human lung adenocarcinomas (Figure 1), both p-Smad2 and HIF1α are found in association with pY654-β-catenin, indicating both pathways likely contribute to EMT within lung cancers. This conclusion is supported by experiments in vitro with H358 cells that indicate although ROS-dependent pY654-β-catenin formation is critical to an EMT response under hypoxic conditions, activation of TGFβ1 signaling further promotes the response (Figure S10b).
Hypoxia and overexpression of HIF-1α have been associated with radiation therapy and chemotherapy resistance, an increased risk of invasion and metastasis, and a poor clinical prognosis of solid tumors (7). Similarly, a hypoxia-induced invasive phenotype and increased metastasis has been reported to result from anti-angiogenic treatment in preclinical models (8, 9) (5). The administration of anti-VEGF antibodies to mice bearing RIP-Tag2 pancreatic tumors was recently reported to result in both hypoxia-induced tumor EMT and enhanced hepatic metastases (37). Here we demonstrate activation of the HIF1α/pY654-β-catenin pathway in this model, the presence of EMT, and its dependence on ROS generation. This problem is also potentially a complication of anti-angiogenic therapy in humans and may contribute to the marginal survival benefits reported for such therapy in colon, breast, and lung cancer treatments (43). Neuroblastomas appear particular prone to acquire invasive properties in the presence of anti-VEGF induced tumor hypoxia (44). Indeed in preliminary studies we have observed marked activation of pY654-β-catenin in brain biopsies of patients progressing on Avastin when compared with the initial tumors (44) (Figure S11). Our data thus provide new information that is potentially clinically relevant. The finding that hypoxia-induced invasiveness in vivo can be markedly attenuated by concurrent administration of the anti-oxidant N-acetylcysteine (Figure 7) raises the possibility that blockade of hypoxia induced ROS accumulation, likely from mitochondrial sources (34) or administration of this or other anti-oxidants could attenuate the invasive switch that is apparently experienced by some patients receiving anti-angiogenic adjunctive therapy.
Materials and methods
Cells and cell culture
Human lung adenocarcinoma cell line H358 and human embryonic kidney 293 cells were purchased from ATCC (Manassas, VA) and grown in RPMI1640 medium or DMEM supplemented with L-glutamine, 10% FBS (Hyclone). Stable cell line of H358 that express wt or Y654F β-catenin were established by infection of lentivirus and selected by puromycin. T antigen immortalized mouse lung alveolar epithelial cells (AECTs) were generated as previously described (24) and were maintained on Matrigel (BD Biosciences) in small airway growth medium (SAGM, Lonza, Wakersville, MD) supplemented with 5% FBS and keratinocyte growth factor (KGF). For experiments, cells were incubated with Adenovirus-Cre (50 pfu/cell) to delete β-catenin or with control Adenovirus-GFP. AECTs expressing various forms of β-catenin were generated as previously described (24).
Tissue samples
Fresh or frozen tumor and adjacent normal tissues were obtained from patients with lung adenocarcinomas who were undergoing surgical resection of the primary tumor. The study was approved by the University of California San Francisco, Institutional Review Board (IRB#: 10-00959).
Animals and treatment
Tumor-bearing RIP-Tag2 transgenic mice (C57BL/6 background) (14 weeks old) were treated for 1 week with normal goat IgG or function-blocking goat anti-mouse VEGF antibody (150 µg in 50 µL sterile PBS) injected ip three times. Some mice were concurrently treated with NAC (1g/kg/day by gavage) for 4–5 days. Body weight and survival were recorded during the treatment period. All animal procedures were approved by the Institutional Animal Care and Use Committee of UCSF.
Additional methods
Detailed reagents list and all other experimental procedures are available in Supplementary Materials and Methods.
Supplementary Material
Acknowledgements
The authors thank Roshni Ray, Mazen Sidani, Toshina Ishiguro-Oonuma, Casey W. Williamson, Thomas Kim, Yonghyun Kim, Yang Gao, and Ronald Tsang for technical assistance; and Drs. Miguel Ramalho Santos and Martin Brown for generous gifts of reagents.
Grant Support: This work was supported by NIH grants to HL-44712 and CA-125564 (HAC).
Footnotes
Conflicts of Interest
No conflicts of interest.
References
- 1.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009 Apr;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 2.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004 Jun 25;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009 Nov 25;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression? Nat Clin Pract Oncol. 2008 May;5(5):280–290. doi: 10.1038/ncponc1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010 Dec 15;16(24):5928–5935. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hung JJ, Yang MH, Hsu HS, Hsu WH, Liu JS, Wu KJ. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and Snail expression in resectable non-small cell lung cancer. Thorax. 2009 Dec;64(12):1082–1089. doi: 10.1136/thx.2009.115691. [DOI] [PubMed] [Google Scholar]
- 7.Ruan K, Song G, Ouyang G. Role of hypoxia in the hallmarks of human cancer. J Cell Biochem. 2009 Aug 15;107(6):1053–1062. doi: 10.1002/jcb.22214. [DOI] [PubMed] [Google Scholar]
- 8.De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol. 2011 Jul;8(7):393–404. doi: 10.1038/nrclinonc.2011.83. [DOI] [PubMed] [Google Scholar]
- 9.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009 Mar 3;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008 Apr;15(4):660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 11.Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008 Dec;29(12):2267–2278. doi: 10.1093/carcin/bgn216. [DOI] [PubMed] [Google Scholar]
- 12.Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006 Mar 1;66(5):2725–2731. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 13.Luo D, Wang J, Li J, Post M. Mouse snail is a target gene for HIF. Mol Cancer Res. 2011 Feb;9(2):234–245. doi: 10.1158/1541-7786.MCR-10-0214. [DOI] [PubMed] [Google Scholar]
- 14.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008 Mar;10(3):295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 15.Kaluz S, Kaluzova M, Stanbridge EJ. Regulation of gene expression by hypoxia: integration of the HIF-transduced hypoxic signal at the hypoxia-responsive element. Clin Chim Acta. 2008 Sep;395(1–2):6–13. doi: 10.1016/j.cca.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007 Feb;9(2):210–217. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 17.Stemmer V, de Craene B, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene. 2008 Aug 28;27(37):5075–5080. doi: 10.1038/onc.2008.140. [DOI] [PubMed] [Google Scholar]
- 18.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006 Dec;8(12):1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 19.Morin PJ. beta-catenin signaling and cancer. Bioessays. 1999 Dec;21(12):1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 20.Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999 Feb;9(1):15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 21.Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999 Dec 17;274(51):36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 22.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009 Jan;119(1):213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim Y, Kugler MC, Wei Y, Kim KK, Li X, Brumwell AN, et al. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol. 2009 Jan 26;184(2):309–322. doi: 10.1083/jcb.200806067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ulsamer A, Wei Y, Kim KK, Tan K, Wheeler S, Xi Y, et al. Axin pathway activity regulates in vivo pY654-beta-catenin accumulation and pulmonary fibrosis. J Biol Chem. 2012 Feb 10;287(7):5164–5172. doi: 10.1074/jbc.M111.322123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005 Oct;17(5):459–465. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Zeng G, Apte U, Micsenyi A, Bell A, Monga SP. Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-beta-catenin interactions. Exp Cell Res. 2006 Nov 1;312(18):3620–3630. doi: 10.1016/j.yexcr.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem. 2002 Nov 8;277(45):42919–42925. doi: 10.1074/jbc.M206141200. [DOI] [PubMed] [Google Scholar]
- 28.Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997 Dec 1;57(23):5328–5335. [PubMed] [Google Scholar]
- 29.Pham NA, Magalhaes JM, Do T, Schwock J, Dhani N, Cao PJ, et al. Activation of Src and Src-associated signaling pathways in relation to hypoxia in human cancer xenograft models. Int J Cancer. 2009 Jan 15;124(2):280–286. doi: 10.1002/ijc.23912. [DOI] [PubMed] [Google Scholar]
- 30.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003 Apr;3(4):347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 31.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010 Oct 28;467(7319):1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009 Dec;297(6):L1120–L1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Littlepage LE, Sternlicht MD, Rougier N, Phillips J, Gallo E, Yu Y, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010 Mar 15;70(6):2224–2234. doi: 10.1158/0008-5472.CAN-09-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Res. 2007 Aug 1;67(15):7368–7377. doi: 10.1158/0008-5472.CAN-07-0515. [DOI] [PubMed] [Google Scholar]
- 35.Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem. 2003 Jan 31;278(5):3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 36.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005 Oct;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012 Mar;2(3):270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008 Mar 18;105(11):4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim WY, Perera S, Zhou B, Carretero J, Yeh JJ, Heathcote SA, et al. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest. 2009 Aug;119(8):2160–2170. doi: 10.1172/JCI38443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell. 2012 Jul 10;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, et al. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. FEBS J. 2008 Jan;275(1):69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- 42.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011 Dec 1;480(7375):118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grepin R, Pages G. Molecular mechanisms of resistance to tumour anti-angiogenic strategies. J Oncol. 2010;2010:835680. doi: 10.1155/2010/835680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A. 2011 Mar 1;108(9):3749–3754. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao W, Irby R, Coppola D, Fu L, Wloch M, Turner J, et al. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene. 1997 Dec 18;15(25):3083–3090. doi: 10.1038/sj.onc.1201496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.