

Abstract
Spontaneous membrane-translocating peptides (SMTPs) have recently been shown to directly penetrate cell membranes. Adsorption of a SMTP, and some engineered extensions, at model silica surfaces is studied herein using fully atomistic molecular dynamics simulations in order to assess their potential to construct novel drug delivery systems. The simulations are designed to reproduce the electric fields above single, siloxide-rich charged surfaces, and the trajectories indicate that the main driving force for adsorption is electrostatic. An increase in the salt concentration slows down but does not prevent adsorption of the SMTP to the surface; it also does not result in peptide desorption, suggesting additional binding via hydrophobic forces. The results are used to design extensions to the peptide sequence which we find enhance adsorption but do not affect the adsorbed conformation. We also investigate the effect of surface hydroxylation on the peptide adsorption. In all cases, the final adsorbed conformations are with the peptide flattened to the surface with arginine residues, which are key to the peptide’s function, anchoring it to the surface so that they are not exposed to solution. This conformation could impact their role in membrane translocation and thus has important implications for the design of future drug delivery vehicles.
1. Introduction
Cell-penetrating peptides (CPPs) are an important class of peptides that can facilitate uptake of cargo ranging from small molecules to large proteins and nucleic acids into the cytoplasm of cells.1−6 This ability to deliver a drug payload into a cell offers significant therapeutic potential, as one in principle can target areas of therapeutic space that are difficult to access using small molecules.7 The mode of uptake that CPPs use to penetrate cell membranes can vary from an active endocytotic mechanism to direct translocation. The TAT peptide is one such CPP exemplar that is used extensively and is thought to facilitate the cell uptake of therapeutic cargo via a receptor-mediated endocytotic mechanism.8 As a result of this mode of uptake, the cell-penetrating activity of TAT peptides is generally confined to particular cell types and generally TAT peptides do not penetrate multicellular membrane barriers such as vascular epithelia and the blood brain barrier. A further limitation of this category is that cargo is typically trapped in endosomes, which can decrease the efficacy of large biologic therapeutics.
A recent study by Marks et al. reported a novel CPP that can directly penetrate cell membranes via direct membrane translocation.9 This spontaneous membrane penetrating peptide (SMPT) sequence [PLIYLRLLRGQFC-TAMRA] was found to penetrate synthetic membranes as well as the membranes of CHO cells, thus potentially providing a generic route to the delivery of therapeutic cargo to cells that circumvents the problems associated with CPPs functioning via active cell uptake mechanisms. It is intriguing to consider whether this SMPT could now be used to construct a novel drug delivery system employing silica nanoparticles decorated with SMPTs as platforms for drug delivery. For this, we need to understand in detail how the SMPT interacts with silica surfaces, and simulations provide the only means of obtaining the required insight.
Here we present a computational study aimed at understanding the non-covalent interactions between silica surfaces and a SMTP. A key question is the conformation the SMPT adopts upon adsorption, since conformation is a strong determinant of biological activity. Silica is a widely studied biomaterial and in its nanoparticulate form has potential as a drug delivery platform delivering coadsorbed therapeutic cargos into cells.10
We report molecular dynamics (MD) studies of the SMTP adsorption at three silica surface models: stoichiometric silica where the surface exposes 100% siloxide (≡SiO–) groups to solution; fully hydroxylated surface exposing OH groups; and a half-hydroxylated surface. These three cases represent a wide range of pH and counterion concentration. The simulation box is designed to create a suitable electric field above the charged surfaces with siloxide species, exploring the effect of electrostatics in the adsorption process, since experimental evidence shows that silica nanoparticles are charged at physiological pH.11 The native peptide as well as some engineered extensions were studied to elucidate the adsorption mechanism, and in particular what impact N- and C-terminal extensions can have on the adsorption and peptide conformation. Peptide engineering offers a great opportunity to improve CPP functions, for example, adding C-terminal cysteine to penetratin and its arginine enriched variant markedly enhances peptide affinity to DNA and the stability of the complex, which noticeably improves CPP function as a nucleic acid vector.12
The behavior of the arginine (residues 6 and 9) in the SMTP is of particular interest, since arginine is known to be crucial for CPP activity.4,13,14 It has been recently reported that arginine appears to govern protein and peptide adsorption on silica, anchoring biomolecules to charged surfaces where the driving force is electrostatic in nature.11,15−20 SMTP adsorption simulations can provide crucial insight into peptide conformation when interacting with the material surfaces and thus guide attempts to engineer effective drug delivery systems. In particular, we address the following questions:
-
1.
How does the SMTP interact with various silica surfaces?
-
2.
Does adsorption impede the availability of arginine for further membrane interactions?
-
3.
Can the peptide sequence be engineered to promote preferential conformation?
The simulations reflect experimental conditions close to the surface so that the nature of the peptide adsorption should give realistic guidance to the future design of drug delivery systems.
2. Methods
All simulations were performed with the NAMD 2.621 package using the Charmm27 force field, and analyzed using VMD.22 Since the 3D structure of SMTP has not yet been solved, the initial SMTP structures were created using one of 18 amino-acid sequences found by Marks et al., namely, Pro1Leu2Ile3Tyr4Leu5Arg6Leu7Leu8Arg9Gly10Gln11Phe12Cys13 (PLIYLRLLRGQFC)9 by appropriate mutation of residues forming a long and unstructured loop in hen egg white lysozyme (HEWL). The loop consists of residues 61–78, and the backbone coordinates of residues 65–78 were used. The peptide was placed in a rectangular box of water molecules (TIP3P) that extend 30 Å from any peptide atom. The net peptide charge was +2 e; therefore, the system was neutralized by adding NaCl salt with an ionic strength of 0.01 (mol/L), 0.05, and 0.7 M. The systems were subject to 1000 steps of water minimization only followed by 100 ps water equilibration at the target temperatures 293 K (room temperature), 310 K (body temperature), and 333 K (high temperature). By this, nine different systems were obtained.
The systems (water and peptide) were minimized for 10 000 steps, heated for 300 ps to the required temperature and equilibrated at constant temperature for 2.7 ns. The production MD simulations were pursued for 50 ns at the given temperature in the NVT ensemble. The integration step was 1 fs, and the SHAKE algorithm and PBC were used. The cutoff distance for van der Waals interactions was 12 Å, and the smooth particle mesh Ewald (SPME) summation23,24 was used for the Coulomb interactions. For ionizable residues, the most probable charge states at pH 7 were chosen. No additional restrictions on momentum in the simulations were used. The above protocol was used for the wild-type SMTP and engineered versions. Moreover, three trajectories at 293 K and various ionic strengths were produced using a 12 Å cutoff for electrostatic interactions instead of the SPME method, as a check on the model electrostatics.
The peptide structure after 50 ns dynamics in water only was used as a starting structure for adsorption simulations, i.e., in the presence of a silica surface. The initial peptide–surface orientation was random, while the initial distance was always 28 Å. The water box dimensions were 86 Å × 80 Å × 100 Å and simulations were run with three ionic strengths and at three temperatures using the protocol described above, producing 12 adsorption trajectories for the native peptide on a siloxide-rich surface (9 with SPME and 3 using a cutoff to calculate the electrostatic interactions). Additionally, for peptides adsorbed in low ionic strength (0.01 and 0.05 M), we have performed “washing” simulations by changing the ionic strength to 0.7 M.
For the surface model, a(101̅) slab of alpha-cristabolite with dimensions 86 Å × 80 Å × 13 Å was used following Patwardhan et al.11 The surface model has been carefully tested in ref (11), and it quantitatively agrees with experiment, reproducing well the density, vibration spectra, and surface and interface energies. Three variants of the surface were created: SiO2 surface with siloxide (≡SiO–) groups only on the top (denoted SiO2); fully hydroxylated SiO2 surface decorated by silanol (≡Si—OH) groups (denoted hSiO2); and half hydroxylated SiO2 surface with alternate ≡SiO– and ≡SiOH groups (denoted hhSiO2). The SiO2 slab model is neutral and stoichiometric, but the slab has been cut from a bulk crystal in such a way as to leave siloxide groups at the top of the slab and under-coordinated Si species at the bottom; the slab then has an intrinsic dipole moment across it. We model the silica as ions fixed in space, which is a common approximation in adsorption studies where surface relaxation is often found to make only small differences to adsorption energies. The 3D periodicity of the simulation box creates an electric field across the water/peptide space, mimicking the electric field above a single negatively charged silica surface with siloxide species11 (see Figure 1a). The hhSiO2 slab, with its alternate siloxide and silanol groups at both surfaces, has a weaker electric field across the water/peptide space (see Figure 1b). The hydroxylated slab (hSiO2, see Figure 1c) has OH groups on both its top and bottom surfaces, and thus by symmetry has no dipole moment across it and therefore creates no electric field across the water/peptide space. In the case of the surfaces producing the electric field across the simulation cell (surfaces SiO2 and hhSiO2, Figure 1a,b), the polarizing effect driving ions to the oppositely charged surface slabs is observed (data not shown). More precisely, in adsorption simulations on the SiO2 surface, sodium ions migrate toward the siloxide-rich surface (lower on Figure 1a), while chloride ions migrate toward the Si+-rich surface (upper on Figure 1a). Similarly, in the case of the hhSiO2 surface, Na+ ions migrate toward the negatively charged surface part (lower on Figure 1b), while Cl– ions migrate toward the positively charged one (upper on Figure 1b). The polarization effect is not visible in the case of hSiO2 which does not produce the electric field across the simulation cell; therefore, all ions are located randomly around the center of the cell.
Figure 1.

Illustration of the simulation boxes and crystal structures used in this study; silicon is yellow, oxygen red, and hydrogen gray. (a) The SiO2 surface: the alpha-cristobalite (101̅) surface is cut so that the upper surface is terminated with under-coordinated oxygen, inducing an electric field E across the water/peptide space due to the dipole moment of the crystal slab. (b) The hhSiO2 surface: alternate oxygens on the upper surface are converted to hydroxyl groups, and corresponding hydroxyl groups decorate the lower surface of the slab, reducing the magnitude of the electric field E across the box. (c) The hSiO2 surface: all oxygens on the upper surface converted to hydroxyls with corresponding hydroxyls on the lower surface, so that there is no electric field across the peptide/water space in the simulation box.
The three surface models allow us to assess the importance of the electric field above the surface in the peptide adsorption, as well as the competition between electrostatic interactions and hydrophobicity at the surface. We note that the Ewald summation has metallic boundary conditions with no jump in electrostatic potential across the box, so the magnitude of the electric field in the middle of the simulation box depends on the slab dipole moment and the overall box height.25 We measure the electric field in the empty SiO2 box to be 0.2 V/Å, corresponding to 0.16 charged silanol groups nm–2, which is comparable to estimates for large silica nanoparticles at pH 7.11 Thus, the surface models present realistic charge density as well as differing surface chemistry. Of course, in the presence of an ionic solution, the electric field is screened with Debye lengths of 30.4 and 13.6 Å for 0.01 and 0.05 M, respectively. We also use 0.7 M NaCl to investigate the impact of high counterion concentration at the charged surface.
In order to simulate the silica surfaces, we have parametrized the force field following the work of Patwardhan et al.11 by adjusting the parameters of the Charmm27 force field. The parameters we use are summarized in Table 1. Note that siloxide oxygen ions have the same charge as the bulk silica oxygen ions. The Si–O bonds and Si–O–Si bond angles were not included in the parametrization, since surface silicon and oxygen were fixed in all stages of our MD simulations. However, the hydrogen atoms were flexible so the O–H bond stretch and Si–O–H bond angle parameters were included.
Table 1. Charmm27 Force Field Parameters Used in Variants of the Alpha-Cristabolite (101̅) Slab.
| Charges | |
|---|---|
| atom | charge (e) |
| bulk silica | +1.10a |
| bulk oxygen | –0.55a |
| silanol oxygen | –0.692b |
| silanol hydrogen | +0.417b |
| van der Waals | ||
|---|---|---|
| atom | ε0 (kcal/mol) | 1/2R0 (Å) |
| bulk silicon | –0.50a | 2.00a |
| bulk oxygen | –0.25a | 1.75a |
| silanol oxygen | –0.1521b | 1.7682b |
| silanol hydrogen | –0.046b | 0.2245b |
| Geometry | ||
|---|---|---|
| spring constant | equilibrium value | |
| O–H | 495 kcal/mol·Å2 a | 0.945 Åa |
| Si–O–H | 50 kcal/mol·rad2 b | 115° a |
Parameters accord with those of the SiO2 surface from ref (11), adjusted to the Charmm27 force field.
Parameters combined with those of the TIP3P molecule in the Charmm27 force field, ensuring slab neutrality.
Our engineered peptides were created by inserting residues at the N-terminus, C-terminus, or both. We have constructed five alternatives by adding n Lys residues at the N-terminus of SMTP, denoted nLysSMTP respectively with n = 1, 2, 3, 4, and 5. These peptides were subject to the normal simulation protocol. Moreover, we added a 5x(Lys-Gly-Gly) sequence at the N-terminus of the peptide (denoted 5patchSMTP), at the C-terminus of the peptide (SMTP5patch), and at both ends (5patchSMTP5patch).
In most cases, the siloxide-rich SiO2 surface was employed in the adsorption simulations, due to the importance of the electric field above charged surfaces. Surface variants hSiO2 and hhSiO2 were used to simulate native SMTP adsorption at 310 K with 0.05 M ionic strength. These trajectories were repeated, so that we have four independent adsorption trajectories of the modified surface.
The total number of trajectories for peptide in water only was 23, while the total number of adsorption trajectories was 24 plus 3 “washing” trajectories; the length of each trajectory was 50 ns so that in total we have analyzed 2.5 μs of SMTP trajectories. Additionally, we have performed nine 50 ns trajectories for the system containing only the surface, water, and ions in various concentrations. Those trajectories performed at three different temperatures serve as references for the adsorption trajectories.
3. Results and Discussion
3.1. Simulations in Water Only
For the SMTP without extensions in water only (Figure 2), we have analyzed 12 trajectories, each 50 ns in duration, which is of comparable duration to other adsorption simulations.11 There are no indications that our 13-residue-long SMTP would adopt a folded structure in nature, and we find no evidence of folding in these trajectories; we have started from a random coil conformation instead of an extended chain conformation, so any folding (or at least the early stages of folding) should be visible if it was to occur. It is worth noting that involvement of CPP secondary structure in the intestinal absorption of conjugated cargo is still unclear.4
Figure 2.
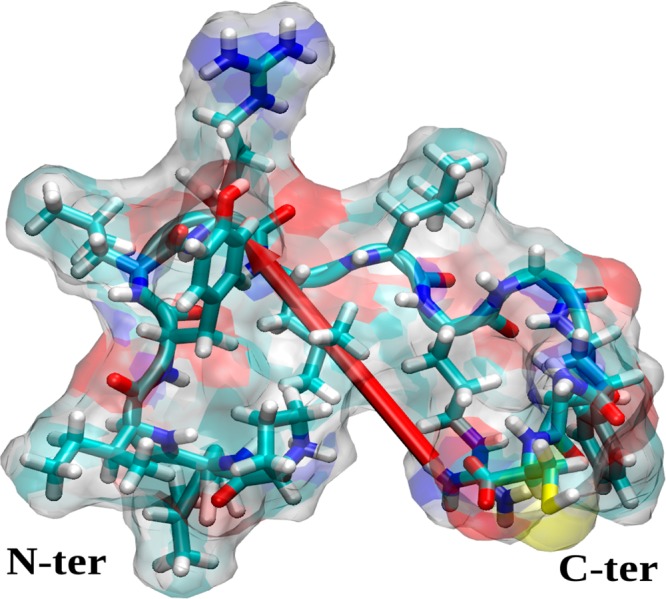
Initial structure of the SMTP peptide (PLIYLRLLRGQFC). The peptide surface is indicated as a ghost surface colored by name (C, cyan; H, white; N, blue; O, red; S, yellow), secondary structure is shown as a cartoon, and residues are shown by licorice. The red needle indicates the dipole moment of the peptide, and the peptide ends are annotated.
We have not observed folding in water; rather, the peptide samples the conformational space visiting the energy basins available. Figure 3 shows the final conformations of native SMTP at various temperatures and ionicities. In all trajectories, the peptide structure changed mostly during the minimization and heating period, while during the production trajectories the peptide remained relatively stable (data not shown). Secondary structure has not appeared in any case. The RMSD between the initial and final structures varied from trajectory to trajectory between 3.6 and 5.3 Å with the most frequent value ∼4.2 Å. Such high RMSD values were expected, since the trajectories started from trial initial conformations. The trajectories do not indicate any one particular low energy conformation for the native SMPT.
Figure 3.

Overlap of nine final structures of native SMTP in water only (water not shown) displayed by cartoon. The starting conformation indicated by green was always the same, and the final structures were observed after 50 ns of trajectories of various ionic strength: (a) 0.01 M; (b) 0.05 M; (c) 0.7 M. The colors code the temperature of the system in given ionic strength: 293 K, blue; 310 K, orange; 310 K, yellow. Peptide ends are annotated.
The addition of residues at the N-terminus (trajectories nLysSMTP with n = 1, 2, 3, 4, and 5 and 5patchSMTP; recall the patch is 5x(Lys-Gly-Gly)), the C-terminus (SMTP5patch), or both termini (5patchSMTP5patch) did not much change the fold pattern of the original part of the peptide. Analysis of RMSD plots (data not shown) revealed that, similarly to the native peptide, the biggest conformational changes appeared during the initial steps of the trajectories. During the production trajectories, the RMSD usually increased as well but not as substantially as in the initial stages. The common feature is again peptide flexibility and lack of secondary structure.
Interestingly, neither the ionic strength, the simulation temperature, nor the calculation method of the electrostatic interactions has been found to have any systematic effect on the overall dynamical behavior of isolated SMTP. The lack of a temperature-driven effect might be explained by the midrange temperatures used. The lack of an ionic-strength effect is probably due to the fact that hydrophobic interactions play a major role for peptide structure, and in this case, electrostatics have a negligible effect on peptide folding. This is supported by the observation that the peptide does not bind the ions present in the buffer.
3.2. Adsorption Simulations for Native Peptide at the SiO2 Surface
Adsorption simulations were performed at various temperatures (293, 310, and 333 K) and various ionic strengths (0.01, 0.05, and 0.7 M) using the SPME summation, yielding nine trajectories in total, each of 50 ns duration. Additionally, we have performed three trajectories with a 12 Å cutoff for electrostatic interactions at 293 K and various ionic strengths, also 50 ns long. Together, these allow us to explore the effects of temperature and salt concentration on the adsorption, as well as to provide different samples with which to observe general trends during SMTP adsorption on charged, siloxide-rich silica surfaces.
3.2.1. The Effect of the Salt Concentration, Temperature, and SPME Summation
The salt concentration had a strong effect on the adsorption kinetics but not on the adsorption mechanism. Using 0.01 and 0.05 M ionic strength, adsorption is very rapid and observed during the 2.7 ns equilibration period, so that the production trajectories started from an adsorbed state. Rapid adsorption was observed in all eight trajectories with these lower ionic strengths. The higher 0.7 M ionic strength slowed down the adsorption rate in two trajectories and prevented adsorption within the 50 ns time scale in one trajectory. During the two successful adsorption trajectories at 0.7 M, the peptide adsorbed after approximately 22.4 and 24.8 ns of the production trajectory (293 K without SPME and 310 K with SPME) employing the same adsorption mechanism as in lower ionic strengths. In one trajectory (333 K), after 20 ns, the peptide adsorbed to the periodic image of the surface. This trajectory was not analyzed further, since by the simulation slab’s construction (see Figure 1 and Methods above), the peptide adsorbed to a chemically different surface. In the final 0.7 M trajectory (293 K with SPME), the peptide did not adsorb within 50 ns.
In the system with 0.01 M ionic strength, four ions were present (1 Na+ and 3 Cl–), in the 0.05 M system 16 ions were present (7 Na+ and 9 Cl–), and the system with 0.7 M ionic strength contained 246 ions (122 Na+ and 124 Cl–). The lower adsorption success rate in the case of the 0.7 M trajectories indicates that the high ionic strength can strongly slow down adsorption and that electrostatic interactions play an important role in the SMTP adsorption, as observed in lysozyme adsorption at a charged ionic surface.15−19 Moreover, arginine residues seem to be crucial in anchoring the biomolecules to the surface, as previously reported for peptide adsorption on silica.11 As we will see below, hydrophobic as well as electrostatic forces play a role in the adsorption.
Similar to the simulations in water only, no effect of temperature on the adsorption mechanism or kinetics has been detected, and neither has a systematic effect of using SPME rather than a 12 Å cutoff to calculate electrostatic interactions been found.
3.2.2. Structural Changes Caused by the Adsorption
In the majority of the native SMTP adsorption trajectories on the siloxide-rich SiO2 surface, peptide folding was not observed. Only in 1 trajectory among the 12 studied was surface-induced peptide folding found. In this case, an α-helix comprising residues Tyr4 to Gln11 was created. Peptide folding to helix–coil–helix or an extended helix structure on the surface has been recently reported by Zhuang et al.26 Those computational studies suggested that the efficiency of surface induced protein folding depends on surface complementarity.
In the remaining adsorption trajectories, the peptide adopted various conformations on the surface, as illustrated in Figure 4. The difference, measured by RMSD, between final conformations (after 50 ns of the adsorption trajectories) varied from 3.0 to 6.1 Å, with an average value of 4.9 Å. The structural difference measured with respect to the initial (trial) peptide structure varied by the same amounts. Finally, the RMSD in each adsorption trajectory measured with respect to their starting structure (e.g., to the peptide structure found after 50 ns in water at the same temperature and ionic strength) varied from 3.2 to 5.3 Å, with average 4.3 Å. Together, these suggest that, even adsorbed, the peptide remains flexible and able to change its conformation. Nevertheless, the RMSD plotted as a function of time (Figure 5) reveals that the adsorption restricts the number of conformations available and elongates the time spent in each conformation. More precisely, when the peptide diffuses freely in water, the transitions between conformations are relatively smooth, while the adsorbed peptide rarely jumps between various conformations, the number of conformations adopted is reduced and the time spent in each conformation is enhanced (Figure 5a). Alternatively, adsorption restricts the number to just one stable conformation and the peptide does not change from it within 50 ns on the surface (Figure 5b). From these results, one can conclude that the surface (i) reduces the number of conformations available; (ii) enhances the time spent in each conformation; and (iii) makes transitions between alternative conformations more rapid.
Figure 4.

Overlap of nine final structures of native SMTP (shown by cartoon) adsorbed on the siloxide-rich SiO2 surface. The coloring scheme is the same as in Figure 3. The surface location is indicated by yellow (Si) and red (O) CPKs. Peptide ends are annotated.
Figure 5.
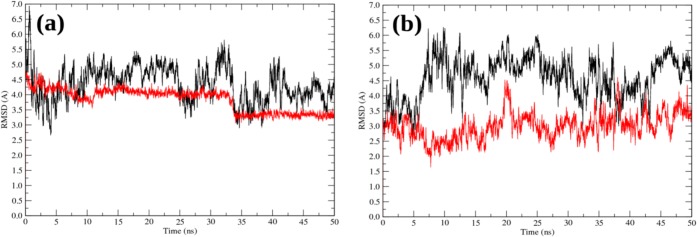
RMSD plots during 50 ns production trajectory in water (black) and the SiO2-surface adsorption trajectory (red) obtained for the native peptide under the same conditions: (a) 0.05 M ionic strength at 293 K; (b) 0.05 M ionic strength at 333 K. In both cases, the SPME method for electrostatic interactions was employed. The RMSD was calculated with respect to the starting structure for a given trajectory. The preparation period (minimization, heating, and equilibration) is omitted.
3.2.3. The Adsorption Mechanism
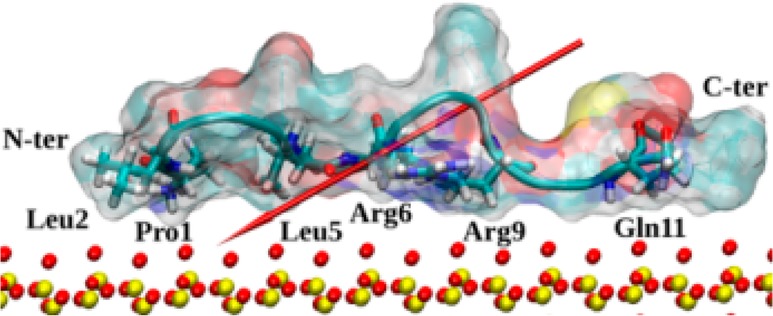
In all the trajectories, the initial orientation of the peptide with respect to the SiO2 surface was random, with an initial separation from the surface of 28 Å, while the distance to the lower surface of the image (see Figure 1) was 42 Å. This means that the peptide was placed approximately at the center of the box but closer to the surface than to the image. The size of the separation from the surface has not prevented rapid adsorption under low ionic strength. During minimization, the peptide rotated to direct its dipole moment toward the surface, aligning in the electric field across the box (see Methods above). Then, within the heating and equilibration period (3 ns in total), the peptide moved toward the surface and adsorbed at its N-terminus (Pro1 and Leu2). In the case of 0.05 M ionic strength trajectories, this happened after about 1.6–1.8 ns (depending on the trajectory) of heating and equilibration, while in the case of 0.01 M ionic strength trajectories it was visible after about 0.8–0.9 ns. Arg6 and Arg9 subsequently adsorbed, with the order varying with trajectory, and Gln11 and Leu5 adsorbed thereafter (a residue is considered as adsorbed if its distance to the surface is approximately constant at ∼4 Å). The side chains of the adsorbed arginine residues penetrate through the surface water and interact directly with the surface. Only arginine side chains can pass the water layer barrier and interact directly with the surface oxygen atoms. The number of direct contacts observed between arginine side chain hydrogen atoms and surface oxygen atoms is limited by the number of arginine residues. Water mediation of the SMTP–SiO2 interactions does not seem to be an important factor. The typical adsorbed peptide structure is shown in Figure 6.
Figure 6.
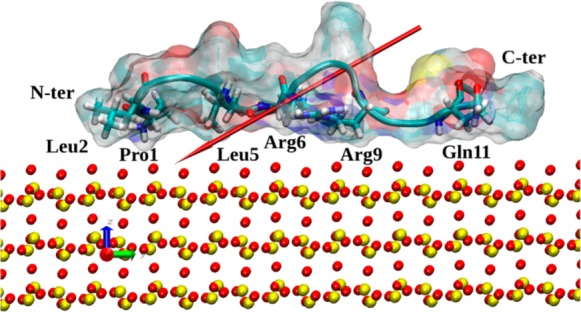
Typical structure of native SMTP adsorbed on the siloxide-rich SiO2 surface. The surface atoms are indicated as yellow (Si) and red (O) CPKs, and the peptide surface is shown as a ghost surface as in Figure 2. Secondary structure is shown as a cartoon, and residues strongly interacting with the surface (Pro1, Leu2, Leu5, Arg6, Arg9, and Gln11) are annotated and shown by licorice. Peptide ends are annotated as well, and the peptide dipole moment is indicated by the red arrow.
At the initial stages of the adsorption, when only the N-terminus is used, the protein “stands” on the surface for a short while and then bends or falls down to allow the arginine residues (Arg6 and Arg9) to adsorb. Thereafter, the immobilized peptide “lies” on the surface with its dipole moment oriented toward the surface. Peptide diffusion on the surface was not observed. In the case of the 0.7 M adsorption trajectories, the scenario was similar, the only difference being that the peptide was able to diffuse more freely in the bulk water during the preparation period and for about 20 ns of the production trajectory.
We therefore conclude that the steps for adsorption at the siloxide-rich surface are as follows: (1) redirection of the dipole moment toward the surface through peptide rotation and slight structural changes; (2) adsorption via N-terminus in a standing position, with the peptide long axis and the dipole moment perpendicular to the surface; (3) bending or falling down to the surface so that the peptide’s long axis is parallel to the surface; (4) Arg6 and Arg9 adsorption; (5) adsorption of Gln11 and Leu5. It is worth noting that after stage 2 the peptide is still mobile on the surface, while after stage 4 it is almost completely immobile. Adsorption energies and diffusion pathways will be probed in more detail using steered molecular dynamics27 in future work. Below, the impacts of various extensions are investigated to complete the picture for the adsorbed conformation.
3.2.4. “Washing” Simulations
To determine the influence of the salt concentration on the adsorption state, we have performed two additional trajectories for native SMTP previously adsorbed on the SiO2 surface at low (0.01 and 0.05 M) ionic strength and at 293 K. These trajectories use the same temperature, but now we introduce the high ionic strength 0.7 M. These reveal no impact of the salt on the adsorption state. As expected from our adsorption simulations conducted at high ionic strength, the peptide remained adsorbed in all cases. This suggests strong adsorption propensity of the peptides to the siloxide-rich SiO2 surface. It also suggests that hydrophobicity plays a role in the surface adsorption, so that replacement of the peptide by ions will not be energetically favorable. The role of hydrophobicity is more apparent below when we discuss the hSiO2 surface adsorption.
3.3. Adsorption Simulations for Engineered Peptide
Since it is known that the arginine residues are crucial for CPP membrane translocation13 and substrate binding,4 it would seem desirable to prevent Arg6 and Arg9 adsorption to keep the SMTP active on the surface. One possible solution might be to insert extra, positively charged sequences at one (or both) peptide ends so that they are available for surface adsorption. This might also prevent the arginine adsorption, due to the electrostatic repulsion between the inserted sequence(s) and the arginine residues; the peptide might then be protected against bending or falling onto the surface.
The addition of one up to five positively charged lysines at the N-terminus of the peptide did not much change the adsorption process. As found with the native SMTP, the peptide adsorbed rapidly during the preliminary steps of the simulations (the heating and the equilibration period). Despite the fact that initial peptide orientations with respect to the surface were random, the peptide rotated to direct the dipole moment toward the surface and then adsorbed to the surface by the inserted positively charged lysines. The peptide adsorbed by the first and second inserted Lys in a standing position with the dipole moment and the peptide long axis perpendicular to the surface. Nevertheless, the peptide was flexible enough to bend and allow both Arg6 and Arg9 to adsorb, a process unaffected by the electrostatic repulsion between the inserted lysines and the arginines. Gln11 was also involved in the adsorption.
These simulations clearly show that addition of hydrophilic chains at the N-terminus does not protect against arginine surface adsorption; all five steps (with some minor alterations) of the aforementioned adsorption process are still observed. Addition of the 5x(Lys-Gly-Gly) motif at the N-terminus (5patchSMTP) and both ends (5patchSMTP5patch) again does not change the general adsorption mechanism. The peptide spreads on the surface, which is understandable since there are no limitations coming from secondary structure because the peptide does not establish any, and the best way to minimize the energy is to increase the number of contacts with the surface. The addition of the 5Lys-Gly-Gly motif at the C-terminus (SMTP5patch) changed only the second step of the adsorption mechanism and affected the third step. The peptide first adsorbed by its C-terminus and the inserted sequence (cf. step 2), so that the peptide was already parallel to the surface and bending (step 3) is not possible. Nevertheless, the final situation remained unaffected. We therefore conjecture that attachment of a suitable cargo molecule to either SMTP termini should not change the adsorption affinity of this SMTP.
3.4. Adsorption at Hydroxylated and Half-Hydroxylated Surfaces
Adsorption simulations at hydroxylated (hSiO2) and half-hydroxylated (hhSiO2) surfaces were performed at 310 K with 0.05 M ionic strength. The simulation protocol was the same as that employed above with the SiO2 surface. RMSD analysis does not reveal new or unexpected features: adsorption restricts the peptide flexibility, limits the number of available conformations, and makes the transitions between conformations more rapid than otherwise observed in bulk water (data not shown). Peptide folding (defined as secondary structure creation) has not been detected on the hydroxylated surfaces during any of the eight 50 ns trajectories we have analyzed.
Compared to the SiO2 surface, the hydroxyl groups have a visible impact on the adsorption rate but not its mechanism. The adsorption rate was slowed down by a factor of 4–6. Nevertheless, the adsorption was rapid and most frequently appeared within the first 10 ns of the production trajectory. The surface chemistry does have an impact on the arginine side-chain orientation at the surface which in turn can potentially affect the adsorption strength. Final adsorbed structures on the SiO2, hSiO2, and hhSiO2 surfaces are shown in Figure 7. In the case of SiO2 and hSiO2 surfaces, the shortest surface–peptide distances come from the arginine side-chain ends (NH2 groups) which are oriented approximately parallel to the surface. With the hhSiO2 surface, arginines are still the closest residues to the surface, but here the side-chain part closer to the backbone is involved (around the Cα atom) in interactions with the surface and the NH2 side chain end of Arg6 is oriented away from the surface. Surface water is shown in Figure 7a to provide context for the distances shown, but for clarity, it is not shown in the other panels. The surface water prevents the peptide from coming close enough to the surface atoms to establish direct H-bonds with the surface. The anchoring arginine side chains lie parallel to the surface and due to the strong water–surface interactions, together with the water hydrogen bond network, they are not able to pass this barrier. Note that the shorter peptide–surface distances for the other surfaces shown in Figure 7 do not indicate that hydrogen bonds have been created; the shortest distances are now between arginine hydrogen and surface hydrogen atoms. Therefore, it seems that the surface decoration prevents the direct peptide–surface interactions.
Figure 7.

Typical structure of native SMTP adsorbed on the siloxide-rich SiO2 surface (a, b); on the fully hydroxylated hSiO2 surface (c, d); and on the half-hydroxylated hhSiO2 surface (e, f). The surface atoms are indicated as yellow (Si) and red (O) CPKs, the peptide is shown as a cartoon, and Arg6 and Arg9 are shown by licorice and annotated. The peptide ends and key distances to the surface are annotated. The distances are shown in Å. Water molecules are shown by the thin CPK model in part a only to keep the pictures clear.
Comparing the surface adsorbed states shown in Figure 7, we see that a worm-like configuration with no obvious secondary structure is common to all surfaces. That the adsorbed conformation at the fully hydroxylated surface hSiO2 is so similar to that at the siloxide-rich SiO2 surface is at first somewhat surprising. However, the arginine side chain is amphiphilic. Its positive guanidinium end group is attracted to the charged surface oxygen, while its aliphatic chain is hydrophobic. Therefore, we find that the arginine side chain plays a key role in immobilizing the peptide on both the charged, siloxide-rich SiO2 surface and the uncharged hydroxylated surface hSiO2. The absence of the electric field above the hydroxylated surface (see Methods above and Figure 1) slows down adsorption, and does not induce any peptide rotation to align its dipole moment, yet nevertheless yields a similar “flat” adsorbed conformation on the surface. The half-hydroxylated surface hhSiO2 presents the peptide with a mixture of adsorption sites and a weaker electric field above the surface, but again the final adsorption conformation is similar to those on the other surfaces. Experimentally, silica nanoparticles appear to present both hydroxyl and siloxide groups at their surface,11 so the hhSiO2 model used here is perhaps more representative of nature.
Summary and Conclusions
The conjugation of peptides to silica nanoparticles offers a possible route to designing new drug delivery systems and thus has a great potential for future therapeutics.10 In this work, we have presented the first atomistic simulation study of how a SMTP peptide9 adsorbs to silica surfaces. We employed simulation boxes designed for siloxide-rich surfaces with an electric field across the water/peptide space. We have also used hydroxylated silica surfaces which have no such electric field, and a model which presents a mixture of sites and a weaker electric field to mimic silica nanoparticles.11
The key result from our simulations is that the adsorbed SMTP flattens onto the surface whichever surface we use. Significantly, the polar residues in the peptide, Arg6 and Arg9, interact strongly with surfaces due to their amphiphilic side chains. This agrees with other recent studies which identify arginine as a key residue for immobilizing proteins and peptides at various surfaces. Therefore, adsorbed SMTP is not exposed to solution, and its arginines are not readily available to interact with other materials. Our study suggests that future utility of an SMTP-decorated silica nanoparticle requires passivation of the surface followed by SMTP conjugation, in order to eliminate adverse interactions between the silica and peptide. These interactions include both electrostatic and hydrophobic forces between the Arg residues and the nanoparticle surface.
We have also explored the idea of engineering chain segments to promote adsorption to charged siloxide-rich surfaces. Using combinations of lysines, we can readily enhance adsorption through the number of contacts between the peptide and the surface. However, we were not able to successfully engineer a chain which keeps the SMTP arginines away from the surface through electrostatic repulsion alone, due to the flexible nature of the peptide. Nevertheless, the results do suggest that it might be possible to attach selected cargo molecules at the SMTP ends without impacting the propensity to adsorb to silica.
Our studies provide vital insights into the interactions occurring at the peptide–material interface and should guide future efforts toward effective surface functionalization. In particular, our simulations indicate that further work should focus on reducing the surface–arginine interactions, since it is believed that these residues play vital roles in the cell membrane penetration.
Acknowledgments
This work was supported by a University of Strathclyde Pump-priming grant funded by the UK’s Engineering and Physical Sciences Research Council (EP/J013765/1). Results were obtained using the Faculty of Engineering High Performance Computer at the University of Strathclyde.
Glossary
Abbreviations
- SMTP
Spontaneous membrane-translocating peptide
- CPP
cell-penetrating peptide
- MD
molecular dynamics
- HEWL
hen egg white lysozyme
- SPME
smooth particle mesh Ewald
- SiO2
SiO2 surface with siloxide (≡SiO–) groups only on the top
- hSiO2
fully hydroxylated SiO2 surface decorated by silanol (≡Si—OH) groups
- hSiO2
half hydroxylated SiO2 surface with alternate ≡SiO– and ≡SiOH groups
- nLysSMTP
SMTP with n Lys residues added at the N-terminus of the peptide with n = 1, 2, 3, 4, and 5
- 5patchSMTP
SMTP with added 5x(Lys-Gly-Gly) sequence at the N-terminus of the peptide
- SMTP5patch
SMTP with added 5x(Lys-Gly-Gly) sequence at the C-terminus of the peptide
- 5patchSMTP5patch
SMTP with added 5x(Lys-Gly-Gly) sequence at both ends of the peptide
- RMSD
root-mean-square distance
Author Contributions
K.K.-O. performed all the simulations and analyses, and led the writing of the manuscript. S.V.P. advised on the experimental relevance of the simulations with particular regard to material systems, and contributed to the interpretation of results and the manuscript development. G.B. advised on the experimental relevance of the simulations with particular regard to the CPPs, and contributed to the interpretation of results and the manuscript development. P.A.M. led the project, supervised the simulations and analyses, and oversaw the interpretation of results and the manuscript development.
The authors declare no competing financial interest.
References
- Heitz F.; Morris M. C.; Divita G. Twenty Years of Cell-Penetrating Peptides: From Molecular Mechanisms to Therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra M.; Prakash S. Targeted Drug Delivery Across Blood-Brain-Barrier Using Cell Penetrating Peptides Tagged Nanoparticles. Curr. Nanosci. 2011, 7, 81–93. [Google Scholar]
- Mussbach F.; Franke M.; Zoch A.; Schaefer B.; Reissmann S. Transduction of Peptides and Proteins into Live Cells by Cell Penetrating Peptides. J. Cell. Biochem. 2011, 112, 3824–3833. [DOI] [PubMed] [Google Scholar]
- Khafagy E. S.; Morishita M. Oral Biodrug Delivery Using Cell-Penetrating Peptide. Adv. Drug Delivery Rev. 2012, 64, 531–539. [DOI] [PubMed] [Google Scholar]
- Milletti F. Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discovery Today 2012, 17, 850–860. [DOI] [PubMed] [Google Scholar]
- Mo R. H.; Zaro J. L.; Shen W. C. Comparison of Cationic and Amphipathic Cell Penetrating Peptides for siRNA Delivery and Efficacy. Mol. Pharmaceutics 2012, 9, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. The Druggable Genome. Nat. Rev. Drug Discovery 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Brooks H.; Prevot P.; Lebleu B.; Chernomordik L. V. Cellular Uptake of Unconjugated TAT Peptide Involves Clathrin-dependent Endocytosis and Heparan Sulfate Receptors. J. Biol. Chem. 2005, 280, 15300–15306. [DOI] [PubMed] [Google Scholar]
- Marks J. R.; Placone J.; Hristova K.; Wimley W. C. Spontaneous Membrane-Translocating Peptides by Orthogonal High-Throughput Screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L.; He Q.; Liu J.; Chen Y.; Ma M.; Zhang L.; Shi J. Nuclear-Targeted Drug Delivery of TAT Peptide-Conjugated Monodisperse Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2012, 134, 5722–5725. [DOI] [PubMed] [Google Scholar]
- Patwardhan S. V; Emami F. S.; Berry R. J.; Jones S. E.; Naik R. R.; Deschaume O.; Heinz H.; Perry C. C. Chemistry of Aqueous Silica Nanoparticle Surfaces and the Mechanism of Selective Peptide Adsorption. J. Am. Chem. Soc. 2012, 134, 6244–6256. [DOI] [PubMed] [Google Scholar]
- Amand H. L.; Norden B.; Fant K. Functionalization with C-terminal Cysteine Enhances Transfection Efficiency of Cell Penetrating Peptides Through Dimer Formation. Biochem. Biophys. Res. Commun. 2012, 418, 469–474. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Lai G. H.; Schmidt N. W.; Sun V. Z.; Rodriguez A. R.; Tong R.; Tang L.; Cheng J.; Deming T. J.; Kamei D. T.; Wong G. C. L. Translocation of HIV TAT Peptide and Analogues Induced by Multiplexed Membrane and Cytoskeletal Interactions. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16883–16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal A. M.; Khanduri R.; Naik R. J.; Ganguli M. Structural Rearrangements and Chemical Modifications in Known Cell Penetrating Peptide Strongly Enhance DNA Delivery Efficiency. J. Controlled Release 2012, 157, 260–271. [DOI] [PubMed] [Google Scholar]
- Mulheran P. A; Kubiak K. Protein Adsorption Mechanisms on Solid Surfaces: Lysozyme-on-mica. Mol. Simul. 2009, 35, 561–566. [Google Scholar]
- Kubiak K.; Mulheran P. A. Molecular Dynamics Simulations of Hen Egg White Lysozyme Adsorption at a Charged Solid Surface. J. Phys. Chem. B 2009, 113, 12189–12200. [DOI] [PubMed] [Google Scholar]
- Kubiak – Ossowska K.; Mulheran P. A. What Governs Protein Adsorption and Immobilization at a Charged solid Surface?. Langmuir 2010, 26, 7690–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubiak – Ossowska K.; Mulheran P. A. Mechanism of Hen Egg White Lysozyme Adsorption on a Charged Solid Surface. Langmuir 2010, 26, 15954–15965. [DOI] [PubMed] [Google Scholar]
- Kubiak – Ossowska K.; Mulheran P. A. Multiprotein Interactions during Surface Adsorption: a Molecular Dynamics Study of Lysozyme Aggregation at a Charged Solid Surface. J. Phys. Chem. B 2011, 115, 8891–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Pandey R. B.; Berry R., J; Farmer B., L.; Naik R., R.; Heinz H. Adsorption Mechanism of Single Amino Acid and Surfactant Molecules to Au {111} Surfaces in Aqueous Solution: Design Rules for Metal-Binding Molecules. Soft Matter 2011, 7, 2113–2120. [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot Ch.; Skeel R. D.; Kale L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pederson L. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Kastenholz M. A.; Hünenberger P. H. Influience of Artificial Periodicity and Ionic Strenght in Molecular Dynamics Simulations of Charged Biomolecules Employing Lattice-Sum Methods. J. Phys. Chem. B 2004, 108, 774–788. [Google Scholar]
- Frenkel D.; Smit B.. Understanding Molecular Simulation; Academic Press: London, 2002. [Google Scholar]
- Zhuang Z.; Jewett A., I.; Kuttimalai S.; Bellesia G.; Gnanakaran S.; Shea J. E. Assisted Peptide Folding by Surface Pattern Recognition. Biophys. J. 2011, 100, 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubiak – Ossowska K.; Mulheran P. A. Protein Diffusion and Long-Term Adsorption States at Charged Solid Surfaces. Langmuir 2012, 28, 15577–15585. [DOI] [PubMed] [Google Scholar]