

Abstract
There is an increasing level of interest in the use of black TiO2 prepared by thermal hydrogen treatments (H:TiO2) due to the potential to enhance both the photocatalytic and the light-harvesting properties of TiO2. Here, we examine oxygen-deficient H:TiO2 nanotube arrays that have previously achieved very high solar-to-hydrogen (STH) efficiencies due to incident photon-to-current efficiency (IPCE) values of >90% for photoelectrochemical water splitting at only 0.4 V vs RHE under UV illumination. Our transient absorption (TA) mechanistic study provides strong evidence that the improved electrical properties of oxygen-deficient TiO2 enables remarkably efficient spatial separation of electron–hole pairs on the submicrosecond time scale at moderate applied bias, and this coupled to effective suppression of microsecond to seconds charge carrier recombination is the primary factor behind the dramatically improved photoelectrochemical activity.
1. Introduction
Titanium dioxide has been studied as a photoanode material for water oxidation for over 40 years;1,2 however the large band gap energy of TiO2 (3.0 eV, rutile) limits the maximum theoretical STH efficiency to 2.2%,3 which is well below the anticipated required STH of 10% needed for commercial viability.4 A further complication has been that typical achieved STH efficiencies of TiO2 photoanodes have been well below the theoretical maximum, primarily because of the need for the application of large electrical biases to enable charge separation and minimize electron–hole recombination.5 Despite these drawbacks, TiO2 remains an important model for mechanistic studies; furthermore, new efforts toward both narrowing the band gap and minimizing recombination losses offer hope that the STH efficiency can be significantly increased.
Approaches to extend the photoactivity of TiO2 from UV to visible wavelengths have included elemental doping, dye-sensitization, and semiconductor sensitization,6−9 with the introduction of nonmetal dopants such as N, C, and S receiving particular attention.7,10−12 In 2011, Mao and co-workers reported a novel approach to obtaining TiO2 with an extended absorption profile.13 Hydrogen treatments of TiO2 nanoparticles at a moderate pressure (20 bar, 200 °C for 5 days) led to the formation of TiO2 with highly disordered surfaces and crystalline cores. The surface disorder leads to band gap narrowing down to 1.0 eV, with a large shift in the valence band edge taking place.14 The black TiO2 was found to exhibit remarkable photocatalytic activity for H2 evolution in the presence of a sacrificial electron donor, with a particularly high level of activity under UV illumination being a significant factor.13,15 DFT calculations indicate that the high photocatalytic efficiency can be attributed to the formation of localized midgap holes that are spatially separated from the conduction band electrons, limiting recombination losses.16
Li et al. have since explored the role of a lower pressure hydrogen treatment on TiO2 photoanodes for water oxidation, leading in 2011 to a new benchmark STH efficiency of 1.1% for a TiO2 photoanode.17 Rutile nanowires treated at 350 °C under a hydrogen atmosphere for 30 min (H:TiO2) became yellow and showed very high photocurrents under simulated solar irradiation. Despite significant improvements in the visible light absorption properties, only a small increase in the visible light activity was observed. Instead, a very large increase in the IPCE values under UV illumination (≥90% H:TiO2, ∼10% air annealed TiO2 (A:TiO2), λ = 300–375 nm) at relatively low applied biases was found to be the primary cause of the high STH.
In contrast to the materials prepared by Mao and co-workers,13 XPS measurements of the H:TiO2 nanowires showed no change in the valence band edge. Instead, an increase in the concentration of oxygen vacancies (Vo) lying 0.75 and 1.18 eV below the conduction band edge occurs (scheme 1), which is in line with previous studies on single crystal rutile TiO2 in which hydrogen treatment-induced Vo are known to lead to improved visible light absorption.18,19 In addition to changing the light-harvesting properties, it was proposed that the Vo act as electron donors, facilitating charge transport and separation, leading to the near unity IPCE values.17 The approach of introducing Vo into metal oxides to improve the electronic properties has since been further refined for TiO2 electrodes20 and extended to a range of other photoelectrodes for water splitting, including α-Fe2O3, WO3 and ZnO.18 Furthermore, it has been shown that a synergistic effect can be achieved by combining both a hydrogen treatment with N-doping of TiO2 nanowires21 or through visible light sensitization with gold nanoparticles22 to give improved photoactivity under visible light for the oxidation of water. In light of the remarkable photocatalytic activity of hydrogen-treated TiO2, it is important that an improved understanding of the underlying mechanisms occurring on the most active materials is obtained.15 Herein, we present a study on the factors controlling the very high STH efficiencies for the oxygen-deficient rutile TiO2 (H:TiO2) nanowire arrays previously reported by some of us.17
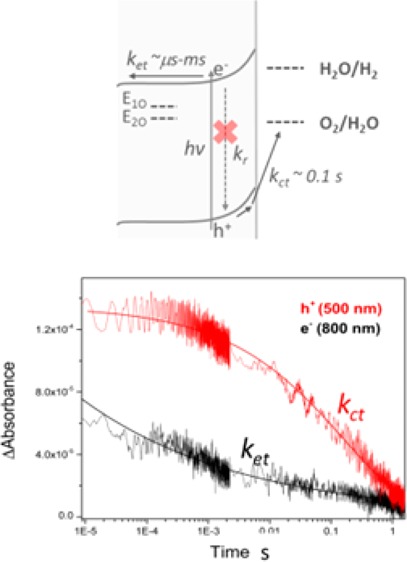
Scheme 1. Simplified Energy Diagram for a Cross Section of (a) A:TiO2 and (b) H:TiO2.

Under a positive applied bias at a distance away from the FTO interface showing the key kinetic processes occurring following absorption of UV light in which kct, kr, and ket correspond to the rates of charge transfer into solution, recombination, and electron transport and collection at the FTO Interface. E1O and E2O correspond to Vo at 0.75 and 1.2 eV below the conduction band edge. As described in the main text, A:TiO2 is anticipated to be fully depleted at even moderate applied biases.
Previous studies have proposed that the increased concentration of Vo decreases electron–hole recombination in hydrogen-treated TiO2 nanoparticles. Numerous mechanisms have been invoked, including electron trapping by O2 adsorbed at surface defects, electron trapping at Vo sites, and hole trapping at Ti3+ sites.23−25 In a photoelectrode under applied bias, the higher donor density of oxygen-deficient electrodes may also decrease recombination because the maximum voltage drop obtainable in the depletion layer of highly structured materials will be greater in materials with higher donor densities.18 Conversely, Vo centers have also been shown to act as recombination sites, lowering photocatalytic activity on several different types of TiO2.26−28 A high photocatalytic efficiency can also be achieved through enhancement of the rate of surface reactions, either via the presence of localized surface Vo, leading to enhanced dissociative adsorption of water,23 or through a change in the energy of the valence band edge.29 It is therefore apparent that the rates of recombination, electron transport, and charge transfer can all be altered to differing degrees in oxygen-deficient TiO2. To date, kinetic studies on oxygen-deficient TiO2 have concentrated on nanoparticulate powders and suspensions; however, when assessing the factors determining the high STH of H:TiO2 photoanodes, it is also important that we consider how the applied potential will modify the rates of recombination, transport, and charge transfer.
Transient absorption (TA) spectroscopy has been used to measure the change in concentration of photoelectrons and holes with time in complete photoelectrochemical (PEC) cells using a range of photoelectrodes, including TiO2,5 α-Fe2O3,30,31 WO3,32 and ZnO,33 providing important insights into the bias-dependent rates of charge carrier recombination, transport, and transfer. TiO2 has been studied with TA spectroscopy for over 25 years,34 and although slight differences in charge carrier spectra are observed, depending on the electrolyte, it is commonly accepted that on anatase TiO2, trapped photoholes absorb light at λ ∼450–550 nm, trapped photoelectrons absorb at λ∼800–900, nontrapped photoelectrons have an absorption profile that increases in intensity with wavelength (>900 nm),35−37 and trapping of holes and electrons is known to occur within 500 ps of the laser flash.38 Electron–hole recombination has been widely studied using TA spectroscopy across the picosecond-to-millisecond time scales, with kinetics being sensitive to the effective electron density,5,37 and recently, the required photohole lifetime for water oxidation, that is, the lifetime of hole transfer into solution during water oxidation, has also been measured and found to be ∼0.03–0.4 s, depending on the electrolyte pH.5,39 The potential of TA spectroscopy to provide insights into the role of trap states on recombination dynamics in hydrogen-treated metal oxide photoanodes has been demonstrated by a recent ultrafast study on hydrogen-treated ZnO;33 however, to the best of our knowledge, no previous studies have been reported for H:TiO2 photoanodes. In the following sections, we describe TA experiments on H:TiO2 photoanodes in a complete PEC cell, allowing us to elucidate the factors behind the very high IPCE values and (i) identify the critical role of hydrogen treatment on electron–hole recombination dynamics; (ii) demonstrate that photoholes generated in H:TiO2 require a lifetime for water oxidation similar to air-treated TiO2, in line with expectations for an unmodified valence band edge; and (iii) examine the factors behind the low level of visible light activity on H:TiO2.
2. Experimental Section
2.1. TiO2 Film Preparation
Rutile TiO2 nanoarrays with nanowire bundles of 100–200 nm diameter consisting of individual 10–20 nm diameter elements with typical lengths of 2–3 μm were prepared on fluorine-doped tin oxide (FTO) glass as previously reported.17 Samples were annealed in air at 550 °C for 3 h. Air-annealed samples (A:TiO2) were used without further modification. H:TiO2 samples were then annealed under hydrogen at 350 °C for 30 min.
2.2. Electrochemical Measurements
The working electrode was the TiO2 photoanode illuminated from the electrolyte–electrode (EE) side. The cell also contains a platinum gauze (99.9%) counter electrode and a 3 M KCl Ag/AgCl reference electrode (SSE, Bioanalytical Systems Ltd.), and all potentials are quoted versus this electrode unless otherwise stated. To prevent degradation of the reference electrode during long measurements, a double junction was employed, with the intermediate solution being 0.5 M NaClO4. The electrolyte in the main cell was 1 M NaOH (Aldrich) prepared with Milli-Q water (Millipore Corp, 18.2 MΩ cm at 25 °C). TiO2 samples were UV-cleaned (75 W Xe lamp) prior to experiments for a minimum of 20 min in the electrolyte. The electrolyte was then replaced following cleaning, and Argon gas was bubbled for at least 30 min through the electrolyte before experiments.
2.3. Transient Measurements
The TA apparatus has been described elsewhere.37 Briefly, the third harmonic of a Nd:YAG laser (Continuum, Surelite I-10, 355 nm, 4–6 ns pulse width) operating at 0.33 Hz was the UV excitation source. Visible light was obtained using an OPO (Continuum, Surelite OPO plus) pumped by 355 nm from the Nd:YAG. The repetition rate was chosen to ensure that all charge carriers had fully decayed prior to the next excitation event. The laser intensity employed was 70 μJ cm–2 at 355 nm and 250 μJ cm–2 at 575 nm, and this energy refers to that incident on the TiO2 sample with corrections for losses from the cell accounted for. A stabilized 75 W Xe lamp (OBB Corp.) coupled to a monochromator (OBB Corp., set to 4 nm resolution) acted as the probe light, and light transmitted through the photoanode was measured using a monochromator coupled to a Si photodiode and homemade amplification system. Typical experiments consisted of ∼300 laser shots per wavelength studied for data in the spectra and ∼600 laser shots for lower noise kinetic traces. All TA experiments were carried out on the complete PEC cells described in section 2.2 under potentiostatic control (Thomson, Ministat).
3. Results and Discussion
The photocurrent voltage plots for both a H:TiO2 and A:TiO2 photanodes in a 1 M NaOH electrolyte are shown in Figure 1. In agreement with the previous report, the H:TiO2 electrode is significantly more active for water oxidation under UV illumination, with both an earlier onset potential and a higher plateau photocurrent, and we observe an ∼10-fold enhancement of the photocurrent between A:TiO2 and H:TiO2 at −0.6 V vs Ag/AgCl.17
Figure 1.
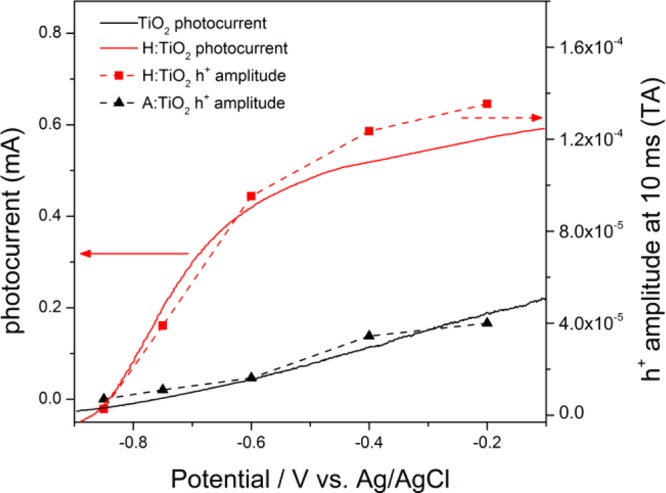
(Solid lines, left axis) Linear sweep voltammogram recorded at a scan rate of 50 mV s–1, sweeping from negative to positive potentials, of A:TiO2 (black) and H:TiO2 films (red) in 1 M NaOH(aq) under UV illumination (λ = 355 nm). (Dashed lines, right axis) Overlay of the relative photohole concentration 10 ms after pulsed laser (355 nm) excitation of A:TiO2 and H:TiO2 at the bias indicated measured by TA spectroscopy (500 nm probe).
TA Spectra of Rutile TiO2 Photoanodes
TA spectra of both H:TiO2 and A:TiO2 PEC cells under operating conditions at a range of applied potentials are shown in Figure 2. In light of the previous TA studies on anatase TiO2,36,38 we assign the TA signals between 425 and 550 nm on our rutile TiO2 samples to trapped photoholes. The TA spectrum of holes on anatase TiO2 has previously been assigned to transitions between surface and subsurface O– centers,40 and DFT calculations on rutile TiO2 have also indicated that hole trapping also occurs on similar oxygen lattice sites.41 The TA features from 750 to 900 nm in Figure 2 are assigned to trapped photoelectrons, also in agreement with the previous literature.37 It has been previously shown that TA measurements of the yield of long-lived photoholes, those which are sufficiently long-lived to participate in water oxidation, provide a quantitative measure of the level of charge separation.30,42 In Figure 1, we observe an excellent correlation between the amplitude of the TA signal measured at 500 nm, at 10 ms after the laser flash, and the measured photocurrent under continuous illumination. This further reinforces the assignment of the TA signals in the 425–550 nm region to trapped holes on rutile TiO2, and it also indicates that our transient measurements with a pulsed excitation source are a reasonable model for the same photoelectrochemical cell under continuous irradiation.
Figure 2.

TA spectra recorded after excitation of (a) A:TiO2 and (b) H:TiO2 in 1 M NaOH(aq) at the bias indicated (vs Ag/AgCl), 355 nm excitation, 70 μJ cm–2, 0.33 Hz laser repetition rate.
Figures 1 and 2 show that the yield of long-lived photoholes on H:TiO2 sharply increases between −0.85 and −0.6 V prior to leveling slightly at potentials greater than −0.6 V (figure 1), and it is clear that the overall level of electron–hole recombination in H:TiO2 is very sensitive to the applied electrical bias between −0.85 and −0.6 V. In contrast, on A:TiO2, we observe only a gradual increase in the yield of long-lived holes with applied bias across the whole potential window studied (−0.85 to −0.2 V vs Ag/AgCl), and a far greater electrical energy input is needed to drive charge separation, with the yield of long-lived holes on A:TiO2 at −0.2 V at 10 ms matching that achieved with 0.55 V less bias on H:TiO2. To rationalize the dramatic effect of the applied bias between −0.85 and −0.6 V on H:TiO2, we now examine the potential dependence of the photohole and electron kinetics in detail.
Role of Applied Bias on Charge Carrier Recombination Dynamics
At −0.85 V, minimal photocurrent is measured on both A:TiO2 and H:TiO2 under illumination from the Xe lamp (Figure 1). TA experiments at this potential show only weak and short-lived signals, with the decay kinetics of the photoelectrons and holes at −0.85 V being indistinguishable on the microseconds–seconds time scale, indicating a common decay mechanism, namely, recombination (Supporting Information Figure S1). The recombination kinetics at −0.85 V are well fitted to a power law decay function,43 and power law recombination kinetics have been reported for a number of different semiconductor materials with a high density of trap states in which nongeminate recombination occurs via multiple trapping–detrapping steps.44 As the applied bias is made more positive than −0.85 V, we note (i) a large increase in the electron–hole yield at the earliest times studied on H:TiO2 (1–10 μs, Figure 2a) and (ii) a decrease and decoupling of the rate of decay of the electron and hole TA signals on the microseconds–seconds time scales.
The increased yield of electrons and holes at 10 μs indicates retardation of fast (submicrosecond) recombination upon the application of the anodic potential. TA spectroscopy can be used to derive the concentration of photogenerated charge carriers in a material if the extinction coefficient is known, allowing us to estimate the relative charge carrier yields at 10 μs after laser excitation.42 Photoelectrons in single crystal rutile TiO2 have been reported to have ε = 600 M–1 cm–1 at 850 nm,45 and we estimate a maximum ΔA850 nm ∼1.2 × 10–4 when 100% of absorbed photons lead to the generation of trapped electrons. We therefore approximate that 30%, 55%, and 75% of photoelectrons remain in the H:TiO2 film at −0.85, −0.6, and −0.4 V, respectively, 10 μs after the laser flash.46
In contrast, using A:TiO2, only a minimal change in initial photoelectron yield is measured as the applied bias is varied (Figure 2b, 10 μs): between −0.85 and −0.6 V, the yield changes from 20% to 25%, and even at −0.4 V, a yield of only 30% achieved. It is noteworthy that the yield of the photogenerated charge carriers in H:TiO2 at 10 μs is much more sensitive to the applied bias than A:TiO2, and this effective initial fast charge separation is a significant factor behind the enhanced activity of the H:TiO2.
The decoupling of the electron and hole kinetics to give nonidentical decay traces on the microseconds–seconds time scales at potentials ≥−0.6 V on A:TiO2 and H:TiO2 is significant because it indicates that processes apart from direct electron–hole recombination are able to occur. In Figure 3a, the TA signal of the photoelectrons in H:TiO2 at −0.6 V decays by more than 50% between 10 μs and 1 ms, whereas the photohole (500 nm) concentration remains largely unchanged. The photoelectron decay is reasonably well fitted to either a power law or the tail of stretched exponential type function (τ ∼ 1.4 × 10–4 s), and because the lack of hole decay on this time scale precludes this being due to direct electron–hole recombination, we tentatively assign the electron kinetics in Figure 3a to electron transport to the external circuit. The kinetics of the hole signal on H:TiO2 in Figure 3a are fitted to a single stretched exponential function with a lifetime of 0.15 ± 0.03 s.43 It is expected that terminal charge transfer into solution will be the slowest photohole process measured, and we have previously found that water oxidation on anatase TiO2 photoelectrodes requires holes with a lifetime of between 0.03 and 0.4 s.39 Here, we also assign the slow decay of the 500 nm TA signal on H:TiO2 to hole transfer into solution. The decay of the photohole signal by this single pathway is striking because it indicates electron–hole recombination has been effectively blocked on H:TiO2 on the microseconds–seconds time scales at a relatively low applied potential (−0.6 V vs Ag/AgCl, +0.4 V vs RHE).
Figure 3.
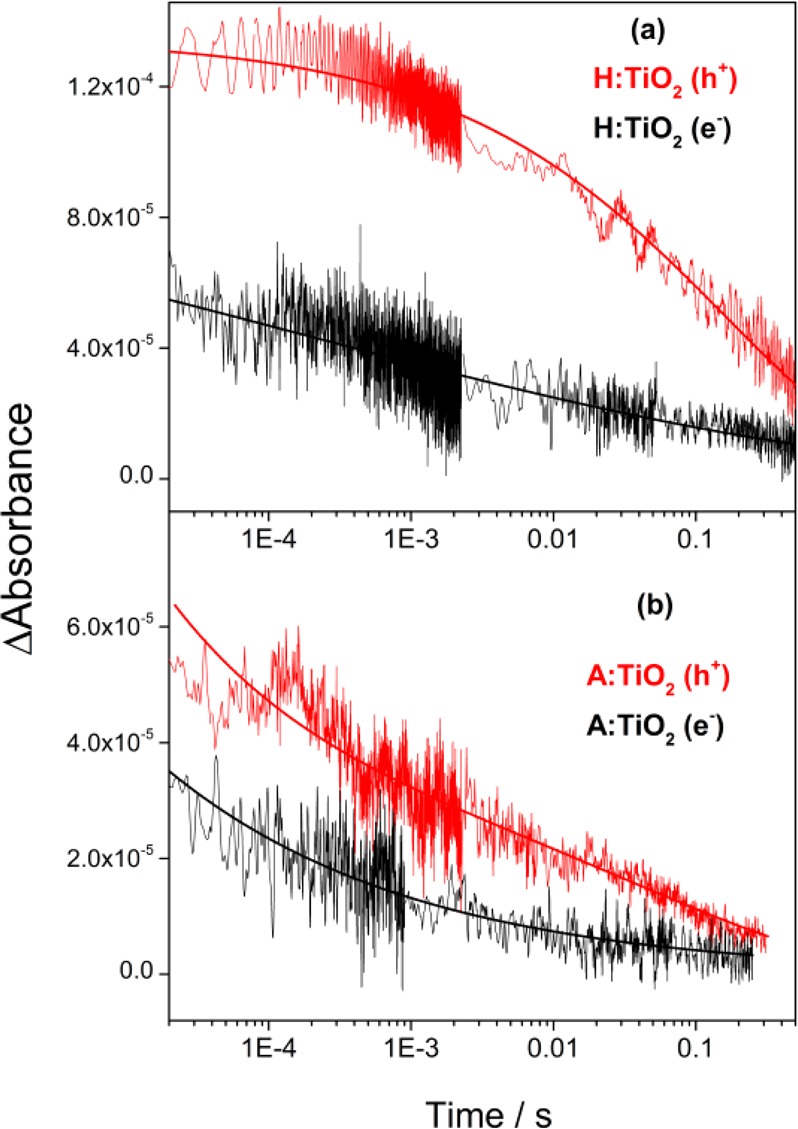
TA decay traces of photoholes (λ = 500 nm, red traces) and electrons (λ = 800 nm, black traces) recorded after excitation of (a) H:TiO2 and (b) A:TiO2 in 1 M NaOH(aq) at −0.6 V (vs Ag/AgCl), 355 nm excitation, 70 μJ cm–2, 0.33 Hz laser repetition rate. The functions employed to give the solid fit lines are described in the main text.43
In contrast, on A:TiO2, we observe high levels of electron–hole recombination on the microseconds–seconds time scale. At −0.6 V, the hole trace on A:TiO2 can be fitted to a combination of both a stretched exponential function and a power law type decay, indicating that at least two different kinetic pathways are operating (Figure 3b). The power law decay (b = 0.35) is assigned to electron–hole recombination, whereas the slow (∼0.1 s) exponential decay component is assigned to the hole transfer into solution. The small magnitude of the component assigned to hole transfer (A1 = 2.6 × 10–5) is due to kinetic competition with electron–hole recombination and is in line with the low (∼15%) IPCEs achieved previously.17 The photoelectron signal at −0.6 V on A:TiO2 is relatively weak, limiting our ability to accurately fit the data; however, it would be anticipated to be dominated by the electron–hole recombination, and the rate of decay of the 800 nm signal, which is similar to that of the hole at 500 nm, does indicate that this is the case.
Our TA experiments clearly demonstrate that initial charge separation (submicroseconds) and suppression of recombination on the microseconds–seconds time scale at −0.6 V is far more effective on H:TiO2 than A:TiO2. The large differences in the recombination kinetics with applied bias may be interpreted through a model in which effective spatial separation of charges occurs as a result of the presence of localized hole or electron traps following the thermal hydrogen treatment.16 Although we do not rule this out as a contributing mechanism, the general similarities of the electron and hole TA spectra on both A:TiO2 and H:TiO2 indicate that the nature of the trap states in both materials is similar. Alternatively, the differences in the role of the applied potential on the recombination kinetics can be interpreted within the context of the model developed by Gartner47 and Gerischer,48,49 in which electron–hole pair separation is driven by the presence of a depletion layer that drives holes toward the semiconductor–liquid junction (SCLJ) and electrons away from the interface.
In the band-bending model, the application of a positive bias increases the width and depth of the space–charge layer in the TiO2, leading to enhanced charge separation yields. The maximum possible radial field (depletion layer) depth depends upon the donor density, the dielectric constant, the radius of the nanowire, and the distance from the substrate contact.50,51 In highly nanostructured materials, it is common for the radial dimensions to be significantly smaller than the width of the space charge layer at a given applied potential, leading to complete depletion and limiting the degree of band bending achievable.52 Following a previous methodology, we estimate that for the air-annealed rutile TiO2 with 10–20 nm feature sizes studied here, the maximum voltage drop obtainable at axial distances more than a few tens of nanometers away from the substrate is ∼0.03–0.11 V (calculations are shown in the Supporting Information).50 In contrast, the numerous Vo in H:TiO2 act as electron donors, leading to a measured Nd ∼ 1022 cm–3, much greater than that of A:TiO2 (∼1018 cm–3).17 The higher Nd of H:TiO2 dramatically decreases the width of the space charge layer, making the individual nanowires more than thick enough to support a sufficiently large radial electric field for effective spatial charge separation, leading to both the high initial charge carrier yields and the suppression of slow (>μs) recombination (Scheme 1b).
In A:TiO2, the inability to maintain a significantly large radial electrical field leads to higher levels of recombination losses and lower IPCE values (Scheme 1a). We still do observe a clear, albeit weaker, bias dependence of the TA data for A:TiO2. This is interpreted through a previously invoked model for highly nanostructured photoanodes in which the primary effect of the application of a positive potential is to lower the Fermi level of the TiO2 and, hence, the background electron density, decreasing the rate of bimolecular electron–hole recombination.5,30 As the rate of recombination is decreased with the applied potential, electron transport and hole transfer into solution become viable pathways.
Hole Transfer Kinetics
To assess the possible contribution of modified hole transfer kinetics to the activity of H:TiO2, we have examined the slow hole kinetics of both H:TiO2 and A:TiO2 in detail (Figure 4). In the previous section, we assigned the slow decay at 500 nm on H:TiO2 at −0.6 V (τ = 0.15 ± 0.03 s) to the transfer of holes into solution. Analysis of the hole decay at −0.4 V on H:TiO2 also gives a very similar lifetime (τ = 0.13 ± 0.04 s), indicating that under the conditions employed here, the average rate of hole transfer is not very sensitive to the applied bias. An approximate lifetime of ∼0.1 s was also found for the hole transfer on A:TiO2 at −0.6 V vs Ag/AgCl; however, overlapping electron–hole recombination kinetics limited the accuracy of this lifetime measurement.
Figure 4.
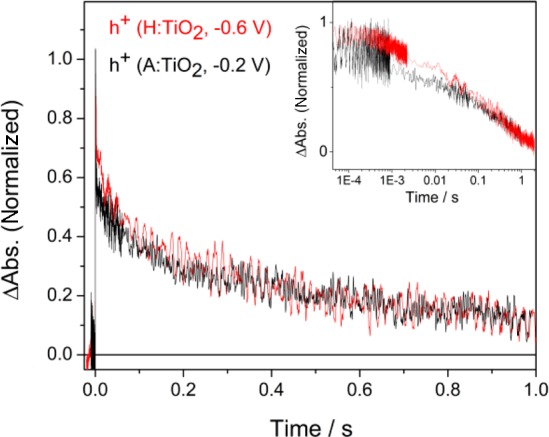
TA decay traces of photoholes (λ = 500 nm) on H:TiO2 at −0.6 V (red trace) and A:TiO2 at −0.2 V (black trace) following UV excitation (355 nm, 0.33 Hz). The inset shows the same data on a logarithmic time scale.
Application of a significantly more positive bias to A:TiO2 further lowers the level of electron–hole recombination, and at −0.2 V, we find that the hole transfer kinetics can be accurately fitted to a single stretched exponential with τ = 0.13 ± 0.02 s, which is the same, within the error, as that observed on H:TiO2 at −0.6 V. The insensitivity of the TA spectrum (Figure 2) and surface kinetics of the trapped photoholes to hydrogen treatment is in line with previous XPS studies, which showed no shift in the valence band edge in this particular form of H:TiO2. However, we do note that a limitation of our TA experiment is that we measure the average hole transfer rate over 300–600 shots, and changes induced in the rates of individual steps which occur on the H:TiO2 photoelectrodes will be hard to identify in these experiments. Nonetheless, our TA experiments do clearly show that the average rate of hole transfer into solution on H:TiO2 is not sufficiently different from that of A:TiO2 to account for the approximate 10-fold increase in IPCE under UV illumination. This further confirms that suppression of recombination and not enhanced surface reaction kinetics is the key factor for the improved photocatalytic activity of the H:TiO2.
Role of Visible Light with H:TiO2
Figure 5 shows TA spectra of H:TiO2 recorded following both the UV (355 nm) and visible light (575 nm, 2.2 eV) excitation at −0.6 V. In this experiment, the 575 nm laser light has been employed because this wavelength showed the highest IPCE in the visible region in the previous report.17 The intensity of the laser has been adjusted so that the number of photons absorbed is equivalent to that in the 355 nm experiment. Despite H:TiO2 absorbing light effectively at this wavelength (Supporting Information Figure S2), we observe no long-lived photohole or photoelectron signals following visible light excitation, in line with previously very low reported IPCE yields at λ > 400 nm, indicating that any electron–hole pairs that are generated are rapidly recombining. Oxygen vacancies in rutile TiO2 are localized at 0.75 and 1.2 eV below the conduction band edge,19 and charge carriers generated following photoexcitation to and from the E1O and E2O states (Scheme 1) have been proposed to be inactive as a result of their decreased energy and mobility. Here, our TA results also indicate that the low level of photocatalytic activity at 575 nm is due to an inability to generate suitably long-lived charge-separated states for water splitting.
Figure 5.
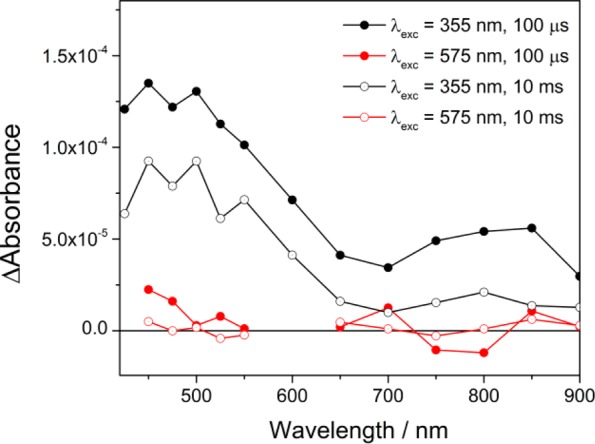
TA spectra recorded at the time scales indicated after UV (355 nm, 70 μJ cm–2, black traces) and visible light (575 nm, 250 μJ cm–2, red traces) excitation of H:TiO2 in 1 M NaOH(aq), at −0.6 V, 0.33 Hz laser repetition rate.
4. Conclusions
In light of the increased interest in hydrogen-treated TiO2 for a range of applications, including photocatalysis, DSSC, and supercapacitors,18 it is essential that an improved understanding of the fundamental mechanisms occurring is achieved. Here, our mechanistic study of the factors controlling the very high STH for the oxygen-deficient rutile TiO2 nanowire arrays provides strong evidence to support the hypothesis that the improved electrical properties of H:TiO2 enables efficient charge separation under an applied bias.17 The TA experiments demonstrate that near complete suppression of electron–hole recombination can be achieved at only −0.6 V vs Ag/AgCl on H:TiO2 following UV excitation. We also rule out a significant change in surface kinetics being an important factor behind the improved IPCE. Given that recent improvements in STH have also been achieved with a wider range of oxygen-deficient photoelectrodes, including α-Fe2O3 and WO3,18 it is apparent that the introduction of Vo for improved charge separation yields is a common design rule, making it probable that a similar mechanism of enhancement is also occurring in these materials, and further experiments to explore this hypothesis are now underway.
Acknowledgments
We thank Dr. C. J. Barnett for his help maintaining experimental equipment and Dr. Frank Jaeckel for helpful discussions. A.J.C. acknowledges funding from the EPSRC (EP/K006851/1).
Supporting Information Available
Details of calculations of the relative photoelectron yields and maximum voltage drops, UV/vis spectra of materials and recombination kinetics of charge carriers. This information is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Boddy P. J. Oxygen Evolution on Semiconducting TiO2. J. Electrochem. Soc. 1968, 115, 199. [Google Scholar]
- Fujishima A.; Honda K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37. [DOI] [PubMed] [Google Scholar]
- Murphy A. B.; Barnes P. R. F.; Randeniya L. K.; Plumb I. C.; Grey I. E.; Horne M. D.; Glasscock J. A. Efficiency of Solar Water Splitting Using Semiconductor Electrodes. Int. J. Hydrogen Energy 2006, 31, 1999. [Google Scholar]
- Basic Research Needs for the Hydrogen Economy; U.S. Department of Energy: Washington, DC, 2003.
- Cowan A. J.; Tang J.; Leng W.; Durrant J. R.; Klug D. R. Water Splitting by Nanocrystalline TiO2 in a Complete Photoelectrochemical Cell Exhibits Efficiencies Limited by Charge Recombination. J. Phys. Chem. C 2010, 114, 4208. [Google Scholar]
- Chen X.; Shen S.; Guo L.; Mao S. S. Semiconductor-Based Photocatalytic Hydrogen Generation. Chem. Rev. 2010, 110, 6503. [DOI] [PubMed] [Google Scholar]
- Ni M.; Leung M. K. H.; Leung D. Y. C.; Sumathy K. A Review and Recent Developments in Photocatalytic Water-Splitting Using TiO2 for Hydrogen Production. Renewable Sustainable Energy Rev. 2007, 11, 401. [Google Scholar]
- Mohamed A. E. R.; Rohani S. Modified TiO2 Nanotube Arrays (TNTAs): Progressive Strategies Towards Visible Light Responsive Photoanode, A Review. Energy Environ. Sci. 2011, 4, 1065. [Google Scholar]
- Youngblood W. J.; Lee S.-H. A.; Maeda K.; Mallouk T. E. Visible Light Water Splitting Using Dye-Sensitized Oxide Semiconductors. Acc. Chem. Res. 2009, 42, 1966. [DOI] [PubMed] [Google Scholar]
- Asahi R. Visible-Light Photocatalysis in Nitrogen-Doped Titanium Oxides. Science 2001, 293, 269. [DOI] [PubMed] [Google Scholar]
- Park J. H.; Kim S.; Bard A. J. Novel Carbon-Doped TiO2 Nanotube Arrays With High Aspect Ratios for Efficient Solar Water Splitting. Nano Lett. 2006, 6, 24. [DOI] [PubMed] [Google Scholar]
- Khan S. U. M.; Al-Shahry M.; Ingler W. B. Efficient Photochemical Water Splitting by a Chemically Modified n-TiO2. Science 2002, 297, 2243. [DOI] [PubMed] [Google Scholar]
- Chen X.; Liu L.; Yu P. Y.; Mao S. S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 2011, 331, 746. [DOI] [PubMed] [Google Scholar]
- Chen X.; Liu L.; Liu Z.; Marcus M. A.; Wang W.-C.; Oyler N. A.; Grass M. E.; Mao B.; Glans P.-A.; Yu P. Y.; et al. Properties of Disorder-Engineered Black Titanium Dioxide Nanoparticles through Hydrogenation. Sci. Rep. 2013, 3, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. H. A Highly Efficient Photocatalyst Hydrogenated Black TiO2 for the Photocatalytic Splitting of Water. Angew. Chem., Int. Ed. 2012, 51, 12410. [DOI] [PubMed] [Google Scholar]
- Liu L.; Yu P. Y.; Chen X.; Mao S. S.; Shen D. Z.. Hydrogenation and Disorder in Engineered Black TiO2. Phys. Rev. Lett. 2013, 111. [DOI] [PubMed] [Google Scholar]
- Wang G.; Wang H.; Ling Y.; Tang Y.; Yang X.; Fitzmorris R. C.; Wang C.; Zhang J. Z.; Li Y. Hydrogen-Treated TiO2 Nanowire Arrays for Photoelectrochemical Water Splitting. Nano Lett. 2011, 11, 3026. [DOI] [PubMed] [Google Scholar]
- Wang G.; Ling Y.; Li Y. Oxygen-Deficient Metal Oxide Nanostructures for Photoelectrochemical Water Oxidation and Other Applications. Nanoscale 2012, 4, 6682. [DOI] [PubMed] [Google Scholar]
- Cronemeyer D. C. Infrared Absorption of Reduced Rutile TiO2 Single Crystals. Phys. Rev. 1959, 113, 1222. [Google Scholar]
- Wang Z.; Yang C.; Lin T.; Yin H.; Chen P.; Wan D.; Xu F.; Huang F.; Lin J.; Xie X.; et al. Visible-Light Photocatalytic, Solar Thermal and Photoelectrochemical Properties of Aluminium-Reduced Black Titania. Energy Environ. Sci. 2013, 6, 3007. [Google Scholar]
- Hoang S.; Berglund S. P.; Hahn N. T.; Bard A. J.; Mullins C. B. Enhancing Visible Light Photo-oxidation of Water with TiO2 Nanowire Arrays via Cotreatment with H2 and NH3: Synergistic Effects between Ti3+ and N. J. Am. Chem. Soc. 2012, 134, 3659. [DOI] [PubMed] [Google Scholar]
- Pu Y.-C.; Wang G.; Chang K.-D.; Ling Y.; Lin Y.-K.; Fitzmorris B. C.; Liu C.-M.; Lu X.; Tong Y.; Zhang J. Z.; et al. Au Nanostructure-Decorated TiO2 Nanowires Exhibiting Photoactivity Across Entire UV-Visible Region for Photoelectrochemical Water Splitting. Nano Lett. 2013, 13, 3817. [DOI] [PubMed] [Google Scholar]
- Pan X.; Yang M.-Q.; Fu X.; Zhang N.; Xu Y.-J. Defective TiO2 with Oxygen Vacancies: Synthesis, Properties and Photocatalytic Applications. Nanoscale 2013, 5, 3601. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Zhang Y.; Jiang J.; Rong Y.; Wang Y.; Wu Y.; Pan C. Characterization of Oxygen Vacancy Associates within Hydrogenated TiO2: A Positron Annihilation Study. J. Phys. Chem. C 2012, 116, 22619. [Google Scholar]
- Rex R. E.; Knorr F. J.; McHale J. L. Comment on “Characterization of Oxygen Vacancy Associates within Hydrogenated TiO2: A Positron Annihilation Study”. J. Phys. Chem. C 2013, 117, 7949. [Google Scholar]
- Leshuk T.; Parviz R.; Everett P.; Krishnakumar H.; Varin R. A.; Gu F. Photocatalytic Activity of Hydrogenated TiO2. Appl. Mater. Interfaces 2013, 5, 1892. [DOI] [PubMed] [Google Scholar]
- Tachikawa T.; Takai Y.; Tojo S.; Fujitsuka M.; Irie H.; Hashimoto K.; Majima T. Visible Light-Induced Degradation of Ethylene Glycol on Nitrogen-Doped TiO2 Powders. J. Phys. Chem. B 2006, 110, 13158. [DOI] [PubMed] [Google Scholar]
- Irie H.; Watanabe Y.; Hashimoto K. Nitrogen-Concentration Dependence on Photocatalytic Activity of TiO2–xNx Powders. J. Phys. Chem. B 2003, 107, 5483. [Google Scholar]
- Naldoni A.; Allieta M.; Santangelo S.; Marelli M.; Fabbri F.; Cappelli S.; Bianchi C. L.; Psaro R.; Dal Santo V. Effect of Nature and Location of Defects on Bandgap Narrowing in Black TiO2 Nanoparticles. J. Am. Chem. Soc. 2012, 134, 7600. [DOI] [PubMed] [Google Scholar]
- Pendlebury S. R.; Cowan A. J.; Barroso M.; Sivula K.; Ye J.; Graetzel M.; Klug D. R.; Tang J.; Durrant J. R. Correlating Long-Lived Photogenerated Hole Populations with Photocurrent Densities in Hematite Water Oxidation Photoanodes. Energy Environ. Sci. 2012, 5, 6304. [Google Scholar]
- Huang Z.; Lin Y.; Xiang X.; Rodriguez-Cordoba W.; McDonald K. J.; Hagen K. S.; Choi K.-S.; Brunschwig B. S.; Musaev D. G.; Hill C. L.; et al. In Situ Probe of Photocarrier Dynamics in Water-Splitting Hematite (α-Fe2O3) Electrodes. Energy Environ. Sci. 2012, 5, 8923. [Google Scholar]
- Pesci F. M.; Cowan A. J.; Alexander B. D.; Durrant J. R.; Klug D. R. Charge Carrier Dynamics on Mesoporous WO3 during Water Splitting. Phys. Chem. Lett. 2011, 2, 1900. [Google Scholar]
- Cooper J. K.; Ling Y.; Longo C.; Li Y.; Zhang J. Z. Effects of Hydrogen Treatment and Air Annealing on Ultrafast Charge Carrier Dynamics in ZnO Nanowires Under in Situ Photoelectrochemical Conditions. J. Phys. Chem. C 2012, 116, 17360. [Google Scholar]
- Bahnemann D.; Henglein A.; Lilie J.; Spanhel L. Flash-Photolysis Observation of the Absorption Spectra of Trapped Holes and Electrons in Colloidal TiO2. J. Phys. Chem. 1984, 88, 709. [Google Scholar]
- Yoshihara T.; Katoh R.; Furube A.; Tamaki Y.; Murai M.; Hara K.; Murata S.; Arakawa H.; Tachiya M. Identification of Reactive Species in Photoexcited Nanocrystalline TiO2 Films by Wide-Wavelength-Range (400–2500 nm) Transient Absorption Spectroscopy. J. Phys. Chem. B 2004, 108, 3817. [Google Scholar]
- Yoshihara T.; Tamaki Y.; Furube A.; Murai M.; Hara K.; Katoh R. Effect of pH on Absorption Spectra of Photogenerated Holes in Nanocrystalline TiO2 Films. Chem. Phys. Lett. 2007, 438, 268. [Google Scholar]
- Tang J.; Durrant J. R.; Klug D. R. Mechanism of Photocatalytic Water Splitting in TiO2. Reaction of Water with Photoholes, Importance of Charge Carrier Dynamics, and Evidence for Four-Hole Chemistry. J. Am. Chem. Soc. 2008, 130, 13885. [DOI] [PubMed] [Google Scholar]
- Tamaki Y.; Furube A.; Murai M.; Hara K.; Katoh R.; Tachiya M. Dynamics of Efficient Electron–Hole Separation in TiO2 Nanoparticles Revealed by Femtosecond Transient Absorption Spectroscopy Under the Weak-Excitation Condition. Phys. Chem. Chem. Phys. 2007, 9, 1453. [DOI] [PubMed] [Google Scholar]
- Cowan A. J.; Barnett C. J.; Pendlebury S. R.; Barroso M.; Sivula K.; Graetzel M.; Durrant J. R.; Klug D. R. Activation Energies for the Rate-Limiting Step in Water Photooxidation by Nanostructured α-Fe2O3 and TiO2. J. Am. Chem. Soc. 2011, 133, 10134. [DOI] [PubMed] [Google Scholar]
- Zawadzki P. Absorption Spectra of Trapped Holes in Anatase TiO2. J. Phys. Chem. C 2013, 117, 8647. [Google Scholar]
- Zawadzki P.; Jacobsen K. W.; Rossmeisl J. Electronic Hole Localization in Rutile and Anatase TiO2 – Self-Interaction Correction in δ-SCF DFT. Chem. Phys. Lett. 2011, 506, 42. [Google Scholar]
- Cowan A. J.; Leng W.; Barnes P. R. F.; Klug D. R.; Durrant J. R. Charge Carrier Separation in Nanostructured TiO2 Photoelectrodes for Water Splitting. Phys. Chem. Chem. Phys. 2013, 15, 8772. [DOI] [PubMed] [Google Scholar]
- TA decay traces were fitted to stretched exponential functions (y = Ae(x/τ)^B + y0), power law type decays (y = Ae(−b) + y0) or combinations of the two processes.
- Nelson J.; Haque S. A.; Klug D. R.; Durrant J. R.. Trap-Limited Recombination in Dye-Sensitized Nanocrystalline Metal Oxide Electrodes. Phys. Rev. B 2001, 63. [Google Scholar]
- Katoh R.; Murai M.; Furube A. Electron-Hole Recombination in the Bulk of a Rutile TiO2 Single Crystal Studied by Sub-nanosecond Transient Absorption Spectroscopy. Chem. Phys. Lett. 2008, 461, 238. [Google Scholar]
- Details are given in the SI. The absolute yields do have potentially large errors due to the potential errors associated with the extinction coefficient employed; however, this does not affect our analysis of the trends in photoelectron yields.
- Gartner W. W. Depletion Layer Photoeffects in Semiconductors. Phys. Rev. 1959, 116, 84. [Google Scholar]
- Gerischer H. Electrochemical Behavior of Semiconductors Under Illumination. J. Electrochem. Soc. 1966, 113, 1174. [Google Scholar]
- Sodergren S.; Hagfeldt A.; Olsson J.; Lindquist S. E. Theroetical-Models for the Action Spectrum and the Current-Voltage Characteristics of Microporous Semiconductor Films in Photoelectrochemical Cells. J. Phys. Chem. 1994, 98, 5552. [Google Scholar]
- Bisquert J.; Garcia-Belmonte G.; Fabregat-Santiago F. Modelling the Electric Potential Distribution in the Dark in Nanoporous Semiconductor Electrodes. J. Solid State Electrochem. 1999, 3, 337. [Google Scholar]
- Barnes P. R. F.; Miettunen K.; Li X.; Anderson A. Y.; Bessho T.; Gratzel M.; O’Regan B. C. Interpretation of Optoelectronic Transient and Charge Extraction Measurements in Dye-Sensitized Solar Cells. Adv. Mater. 2013, 25, 1881. [DOI] [PubMed] [Google Scholar]
- Law M.; Greene L. E.; Johnson J. C.; Saykally R.; Yang P. D. Nanowire Dye-Sensitized Solar Cells. Nat. Mater. 2005, 4, 455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.