

Abstract
Two recent epidemiological investigations in children exposed to valproic acid (VPA) treatment in utero have reported a significant risk associated with neurodevelopmental disorders and autism spectrum disorder (ASD) in particular. Parallel to this work, there is a growing body of animal research literature using VPA as an animal model of ASD. In this focused review we first summarize the epidemiological evidence linking VPA to ASD and then comment on two important neurobiological findings linking VPA to ASD clinicopathology, namely, accelerated or early brain overgrowth and hyperexcitable networks. Improving our understanding of how the drug VPA can alter early development of neurological systems will ultimately improve our understanding of ASD.
1. Introduction
The core clinicopathology of autism spectrum disorder (ASD) is currently thought to be characterized by accelerated or early brain tissue overgrowth in regions involved in emotional, social, and communication functions [1–6]. Although much of the earlier evidence regarding accelerated or early brain overgrowth was based on head circumference measurements which may not be as robust as previously thought [7] (but see [8]), more recent longitudinal brain imaging studies have provided further evidence of this abnormal pattern of brain growth in ASD [3, 6]. In fact, abnormal pathological overgrowth in ASD may even persist into adulthood in certain brain regions [9].
Exposure to exogenous chemicals during pregnancy can interfere with cortical development and the maturation of offspring. One such exogenous chemical is the short chain fatty acid valproic acid (VPA). VPA is a drug used in humans primarily for epilepsy and seizure control, although it has been used in many nonepileptic conditions as well [10]. Based on several case studies and small-scale population-based studies in humans [11–16], in addition to mounting experimental evidence in animals [17–32], VPA has known teratogenicity and has long been suspected as a risk factor for ASD. This year, however, both a prospective study and a large-scale population-based study were published providing the most substantial evidence to date linking prenatal VPA exposure to an increased risk of ASD [33, 34]. Thus, in this paper we will review epidemiological evidence linking VPA to ASD and will discuss two promising leads in the study of ASD in light of human clinical findings and the valproic acid animal model of ASD.
2. Valproic Acid and ASD: Epidemiological Evidence
Previous links between the antiepileptic drug VPA and ASD have been found and explored in several case studies and retrospective studies. VPA has long been known to cause physical malformations and developmental disabilities, features of the clinical entity termed fetal valproate syndrome (FVS). Laegreid and colleagues in a 1993 study examined seven children with FVS, two of which were also exposed to benzodiazepines. Two of the seven cases showed autistic traits, with 2 more showing marked developmental delays [16]. The next year Christianson et al. published a case study describing two sibling pairs, all having FVS to various degrees, with one having been diagnosed with infantile autism [11]. A second case of a boy with FVS and ASD was reported by Williams et al. in 1997, followed up with 5 more cases described by the same authors in 2001 [12, 13]. Several larger retrospective studies have also been published providing further support that in utero VPA exposure and fetal valproate syndrome may be linked to an increased risk of ASD, where, for example, it has been reported that the rate of ASD in the children of VPA-treated mothers may be roughly eight times larger than that of the general population [14, 15, 35]. Nevertheless, many of these studies had mentioned the need for future study to confirm the association between VPA and ASD.
A recently published prospective cohort study by Bromley et al. [33] explored the relationship between prenatal exposure to anti-epileptic drugs (including VPA) and the risk of neurodevelopmental disorders such as ASD, ADHD, and Dyspraxia. They performed an 11-year study, observing the physical and cognitive development of 415 children born to mothers with epilepsy and nonepileptic controls, with a final outcome of neurodevelopmental diagnosis by 6 years of age. Their results showed a significant increase in the risk of neurodevelopmental disorders in those women taking VPA during pregnancy, with autism being the most frequent diagnosis. The researchers also suspected a dose-dependent mechanism of VPA action but were not able to show it with significance in the study due to low numbers of mothers taking VPA. Although this study had a fairly small cohort and a relatively early endpoint (the average age of diagnosis of ASD in the UK is 5–11 years of age), it was the first prospective study to explore the relationship of VPA to ASD [33].
A much larger and detailed population-based study by Christensen et al. [34] that focused more on ASD than other neurodevelopmental disorders was also published this year. The large sample (655,615 children) and thorough control of the study provide the strongest evidence to date on the relationship between VPA and ASD. Data were from children born in Denmark from 1996 through 2006 over 14 years until the end of 2010. In their analysis they controlled for other known risk factors of ASD and potential confounders such as parental psychiatric disease, parental age at conception, congenital malformations, and maternal epilepsy. They divided the outcomes as Autism Spectrum Disorder (ASD) or childhood autism (the most severe diagnosis, simply referred to as “autism”) and reported absolute risks over the 14 years and hazard ratios (HR) for each. The absolute risks for all children studied for ASD and autism were 1.53% and 0.48%, respectively, whereas for those exposed to VPA in utero, the risks were 4.42% (HR 2.9) and 2.5% (HR 5.2). In fact, even when looking at all stratifications and controls, it was concluded that there was a significantly increased risk of children developing either ASD or autism in women taking VPA during pregnancy [34].
Accumulating epidemiological studies have shown an increased risk of children developing a neurodevelopmental disorder, and ASD in particular, if their mothers take VPA during pregnancy. These findings should encourage a discussion on the risks and benefits of the treatment during pregnancy and provide an opportunity to explore how this chemical can alter early developing biological systems that may relate to ASD in animal models. This may facilitate the discovery of other VPA-like substances that may subsequently prove to be environmental or nutritional risk factors for the development of ASD, and these could further elucidate the role of exogenous chemicals on ASD pathology. The authors of the two recent papers discussed above also noted a need to define the mechanism behind the increased risk of ASD with VPA exposure. Several questions arise as to the relationship between environmental exposure to chemicals such as VPA in the manifestation of ASD [33, 34]. Christensen et al. suggested several mechanisms by which VPA may increase the risk of ASD that need further study, including interference in neurotransmitter function, neuronal apoptosis or plasticity, histone deacetylase inhibition, and disruption of folic acid metabolism [34]. Studying the effects of VPA on biological tissue will provide many avenues to explore the possible mechanism(s) of ASD.
3. Valproic Acid Rodent Model of ASD
The model of VPA exposure proposed by Rodier et al. in 1996 is one of the most frequently studied animal models of ASD [36, 37] (also see [38] for review). As reviewed by Dufour-Rainfray et al., this model exhibits many of the structural and behavioural features that can be observed in ASD patients [39]. For example, rats exposed to VPA in utero can exhibit physical malformations (e.g., ear malformations) which have been compared to similar abnormalities that have been observed in some autistic patients (e.g., posterior rotation of the outer ear) [39]. Furthermore, several independent laboratories have also shown that prenatal VPA exposure can lead to behavioral abnormalities that are strikingly similar to those observed in autistic patients; including decreased social interactions and sensitivity to pain, increased sensitivity to nonpainful stimuli, repetitive/stereotypic-like activity, increased anxiety, abnormally high and longer lasting fear memories which are over-generalized and harder to extinguish, and changes in specific types of pup ultrasonic vocalizations [17–28, 30, 31, 39]. At the cellular level, recent morphological data has also suggested that a decrease in spine density in forebrain structures may be a substrate for distal hypo-connectivity, while the increase in dendritic length might support the enhancement of local hyperconnectivity [40]. As pointed out by the authors, this notion is consistent with the ‘‘intense world” theory of ASD which postulates a main neuropathology characterized by hyperfunctioning of local neural microcircuits [40–42].
An animal model, to be considered as a relevant model of a psychiatric condition described in humans, should fit several criteria usually described as construct, face, and predictive validity [43]. Although the effects of VPA have been tested in rodents for many years, only relatively recently it has been used to model ASD in rodents for studying ASD-like behavioural features and potential treatment interventions [1, 26]. Roullet et al. have noted that the VPA model exhibits both construct and face validity [38], and recent work has now provided evidence that it may also exhibit predictive validity [26, 44]. For example, Schneider et al. previously reported that environmental enrichment including multisensory stimulation reversed almost all behavioral alterations observed in pups exposed to VPA in utero [26], while Woo and Leon have recently shown in a randomized controlled trial that environmental enrichment in the form of multiple sensorimotor stimuli was also effective in ameliorating some of the symptoms in autistic children [44].
It has been suggested that methodological issues may have limited the effectiveness of utilizing the prenatal VPA rodent model to study ASD [45]. Interestingly, similar to the prenatal model, many ASD-like behavioural and pathological features have also been observed with VPA exposure during the early postnatal period (equivalent to the third trimester and perinatal period in humans) as previously shown by Yochum et al. [46–49]. This suggests a mechanism that is effective over different developmental time points and may offer an additional approach for studying ASD in rodents. Indeed, the postnatal VPA model may partly be explained by the clinical observation that the maternal serum VPA free-fraction increases in the third trimester and is highest at birth [50]. This is consistent with the idea that bioavailability may play an important role in VPA teratogenicity and that pre- and early postnatal VPA exposure may be quite useful in studying the effects of VPA to help elucidate neuropathological mechanisms of ASD [47]. For instance, VPA exposure during prenatal and early postnatal periods in rodents can lead to accelerated or early brain overgrowth that is reminiscent of that observed in humans. Prenatal VPA exposure on the one hand has been shown to induce a more generalized pathology and can induce macrocephaly in rat brain [51], while early postnatal VPA exposure, on the other hand, has been suggested to induce a more regionally selective pattern of aberrant overgrowth that may be particularly prominent in the temporal association cortex [49]. Thus, while it has been reported that the period of exposure to VPA that is likely to result in ASD is the first trimester of pregnancy, the possibility that ASD can also result from later effects should not be excluded [39].
4. Valproic Acid and Epigenetic Regulation
Dysregulated biochemical pathways can have a profound impact on key cellular processes that may ultimately alter neuronal and network developmental trajectories. We know that the coordinated execution of gene programs involved in neural network maturation is highly regulated by epigenetic processes and that many biochemical pathways can converge to influence these processes [52–55] (Figure 1). Some of the mechanisms that may be involved include histone acetylation, histone methylation, and possibly DNA methylation.
Figure 1.
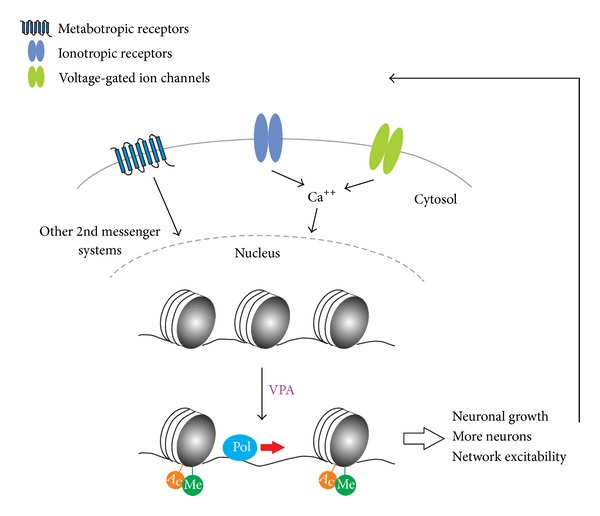
A schematic of some key molecular and cellular changes associated with VPA related to ASD neuropathology. Common clinicopathology associated with ASD is accelerated or early brain overgrowth and increased network excitability. Early or accelerated overgrowth may result from more synapses, increased denritic growth, and/or increased number of cells, while increased network excitability may result from hyperconnectivity and/or hyperplasticity of microcircuits, presumably driven by synaptic mechanisms (i.e., enhanced paired-pulse facilitation and long-term potentiation) that have been shown in rat brain both in vitro and in vivo following prenatal VPA exposure [56, 57]. All of these aspects are also likely regulated by histone acetylation (orange circles “Ac”) and/or histone methylation (green circles “Me”) and possibly DNA methylation (DNA methylation not shown). VPA has been shown to increase histone acetylation and histone methylation that can promote gene activation (symbolized by blue circle “Pol”; RNA polymerase). While VPA may disrupt the balance between excitatory and inhibitory neuronal activities through histone acetylation modulation [58], the role of histone methylation on VPA-related ASD neuropathology is much less clear. Increased connectivity and network excitability may further influence this process by activating metabotropic, ionotropic, and voltage-gated ion channels and subsequent intracellular signaling cascades.
Valproic acid is a commonly used antiepileptic drug [39]. Classically, the mechanism of action of VPA has focused on increases in brain concentrations of gamma-aminobutyric acid (GABA), the major inhibitory neurotransmitter [39], but has also included modulation of voltage-gated sodium channels and glutamatergic signaling [59]. However, it was previously shown in an animal model of seizure that VPA treatment did not significantly affect seizure strength or frequency for the first several days following induction but did, interestingly, protect the animals from seizure-induced cognitive impairment [60]. The authors concluded that their results could be explained, at least partially, by histone deacetylase (HDAC) inhibition [60]. In fact, several studies have since been published supporting the notion that VPA may block HDAC activity in neurons [52, 58, 61].
Histone deacetylases (HDACs) play an important role in regulating gene transcription and phenotype differentiation [53–55]. For example, MacDonald and Roskams reported specific expression patterns of HDAC1 and HDAC2 (both class I HDACs) in the murine brain at multiple developmental ages. HDAC1 is expressed in neural stem cells/progenitors and glia, while HDAC2 is upregulated in postmitotic neuroblasts and neurons but not in fully differentiated glia [62]. HDAC modulation in different cell types and at different maturational time points may therefore lead to dramatically different outcomes and may help to explain why HDAC inhibition in adulthood can improve ASD-like symptoms in rodents exposed to VPA in utero [63].
VPA is a nonselective HDAC inhibitor. Class I HDACs, which include HDAC1 and HDAC2, are expressed in the CNS, and VPA has the net effect of inhibiting their activity via different mechanisms. VPA inhibits HDAC1 through interaction with the enzyme's catalytic site, while VPA induces proteasomal degradation of HDAC2 [64, 65]. This is important as inhibition of class I HDACs can lead to increases in synapse numbers and a robust facilitation of excitatory synapse maturation [52]. Furthermore, VPA can also lead to increased neurite growth and promote neural proliferation [51, 66], both of which are thought to contribute to the accelerated or early overgrowth observed in ASD [1].
ASD structural pathology is regionally selective, exhibiting changes that are prominent in the prefrontal and temporal association cortices [1–3] that can even persist into adulthood [9]. These areas have been found to exhibit a slow and protracted endogenous pattern of development under normal conditions, remaining relatively immature well into postnatal life [67–72]. This pattern of maturation appears to be needed for appropriate cognitive development and allowing for the maturation and stabilization of lower-order networks with which to build upon [73–75]. However, the protracted maturation pattern of these prefrontal and temporal areas may confer an enhanced vulnerability to HDAC inhibition compared to networks that have already attained full maturity [1]. This vulnerability may lead to prominent increases in the structural assembly of synapses and growth of neural networks, both of which are thought to contribute to the underlying clinicopathology of ASD [1, 76].
Histone methylation is another important epigenetic mechanism related to histone acetylation that can regulate gene transcription, developmental gene programs, and cellular phenotype [55, 77, 78]. Much like that of histone acetylation, it has been proposed that histone methylation may be reversible, dynamic, and also important in the teratogenicity of VPA [78]. For example, Tung and Winn reported that not only did in utero exposure to VPA lead to HDAC inhibition and increased histone acetylation of mouse fetal tissue, but it was also associated with changes in histone methylation [78]. In fact, this study found significant increases in di- and trimethylated histone 3-K4 and significant decreases in monomethylated histone 3-K9, which may represent sites involved in gene activation and repression, respectively [77]. Indeed, these epigenetic changes are likely involved in the multiple genes that can be influenced by early VPA exposure [58].
Adding another layer of complexity to the epigenetic control of gene regulation is DNA methylation, where methyl groups covalently bound to the 5′ position of cytosine project into the major groove of DNA, thus inhibiting the binding of transcription factors [78]. While there is interplay between DNA methylation and histone acetylation [79], somewhat surprisingly, the significance of DNA methylation as a mechanism of VPA-induced teratogenesis remains debatable [78]. Thus, the role VPA plays on DNA methylation and the interplay between DNA methylation and histone acetylation will be an important area for future research.
5. Valproic Acid, Overconnectivity, and Hyperexcitable Networks
Clinically apparent seizures are not uncommon in ASD [80]. While there has been a long standing association between ASD and seizure, even children that do not present with clinical seizure can exhibit subclinical seizure and epileptiform electroencephalographic (EEG) abnormalities [80, 81]. Thus, it is postulated that accelerated or early brain overgrowth is accompanied by pathological changes in network excitability.
The rodent VPA model of ASD allows for intrusive electrophysiological study of living tissue unavailable in humans to uncover the nature and potential causes of these suspected electrophysiological abnormalities. One prevailing theory of ASD, the “intense world” theory [41, 42], has been refined through the study of the VPA rat model and postulates that several areas of the brain including the prefrontal cortex and amygdala display hyperreactivity of the microcircuitry of pyramidal cells compared to controls [22, 23, 41, 42, 56, 57]. The amygdala bares particular relevance, for example, as it has been functionally related to ASD due to its role in socioemotional behaviour and can also exhibit enlargement and hyperactivation in autistic children [82, 83]. Markram's group has shown enhanced fear memories, enhanced fear generalization, and impaired fear extinction in prenatally exposed VPA treated rats, along with the other ASD-like behavioural phenotypes [23]. As well, electrophysiological data from lateral amygdaloid nucleus slices showed increased reactivity to stimulation, increased long-term potentiation, and impaired inhibition [23]. A similar result was found by Lin et al. as they also reported hyperexcitability and enhanced LTP in amygdala lateral nucleus pyramidal neurons following prenatal exposure to VPA [17]. These authors stated that the increased ratio of synaptic excitation/inhibition in the amygdala might be associated with the characteristic behaviour in this ASD model [17], which was reiterated by Kim et al. based on their own work showing increased expression of glutamatergic proteins in prefrontal networks of postnatal brains of rat offspring exposed to VPA in utero [84]. Finally, as impairments in the GABAergic system may critically contribute to an increased synaptic excitation/inhibition ratio, an additional mechanism may also involve reduced GABAergic inhibition as recently shown in the temporal cortex of rat pups following prenatal VPA exposure [85].
Although functional imaging studies of the autistic brain have long suggested decreased activity, these recent findings from the VPA model propose an alternate explanation wherein electrophysiological abnormalities may cause network hyperactivity within hyperconnected microcircuits [22, 42, 57]. Indeed, a recent imaging study has shown hyperconnectivity in several large-scale brain networks of children with ASD [86]. While previous functional magnetic resonance imaging (fMRI) studies have been somewhat contradictory, these recent findings suggest that further research into brain network connectivity may lead to a better understanding of the pathology of ASD in the brain [86].
Furthermore, prenatal VPA exposure has been shown to increase NMDA receptor NR2A and NR2B subunits [22], with NR2A being important in epileptogenesis [87]. Consequently, this enhanced activity may subsequently lead to greater activity-dependent processes involved in controlling structural aspects neuronal and synaptic network maturation and growth (Figure 1). For example, it has been shown that activity-induced NR2A activation can lead to increased brain-derived neurotrophic factor (BDNF) expression [87]. Thus, given that BDNF is important in neuronal development [88–90] and epileptogenesis [91], this may be one of several signaling pathways involved in the clinicopathology of ASD.
While activity modulates not only structural aspects of neuronal connectivity such as dendritic branching and spine formation, it can also influence the neurochemical landscape of the neural network. For example, an emerging body of the literature is now providing mechanistic insight into neurotransmitter specification and the role of electrical activity in the developing nervous system [92–94]. Consequently, alterations in the proportions of excitatory and inhibitory neural transmitter phenotypes may play a very important role in the pathophysiology associated with ASD [95]. A number of neurodevelopmental processes ranging from early events of cell proliferation and differentiation, to late events involving maturation of the dendritic arbors and synapses, can all be influenced by activity [92, 94]. Thus, further study on the electrophysiological changes associated with rodent brains exposed to VPA is likely to uncover convergent molecular pathway(s) that drive these changes.
6. Conclusion
Based on accumulating clinical data, it is becoming increasingly clear that VPA is an important risk factor associated with ASD. In addition, a growing body of animal literature is also showing that VPA can lead to ASD-like features in rodents. Moreover, published work suggests that the VPA model of ASD appears to exhibit all elements of a relevant model, namely, construct, face, and predictive validity, and therefore is likely to prove useful in elucidating mechanisms of ASD clinicopathology. It is also important to note that the significance of the two recent epidemiological studies discussed in this paper may also lie in the potential of revealing VPA-like environmental or nutritional substances that could increase the risk of ASD. For example, it was recently shown that in certain cell types global histone acetylation and HDAC activity could be regulated by metabolites of intermediate metabolism [96]. Based on studies primarily in nonneuronal cells, it is also now evident that other agents, including those in the human diet, can be converted by metabolism to intermediates that can influence HDAC activity [96, 97]. Thus, improving our understanding of how VPA and VPA-like compounds can alter early nervous system development will ultimately improve our understanding of ASD.
Acknowledgments
The authors would like to thank Johanna Hung for helpful comments on an earlier version of the paper and Dr. Jeffrey Buchhalter for insightful discussion. This study was supported by Canadian Institutes of Health Research and CIHR Regenerative Medicine Initiative. Nathanael Turner is supported by a PURE studentship. Taylor Chomiak and Nathanael Turner are cofirst authors.
References
- 1.Chomiak T, Hu B. Alterations of neocortical development and maturation in autism: insight from valproic acid exposure and animal models of autism. Neurotoxicology and Teratology. 2013;36:57–66. doi: 10.1016/j.ntt.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Courchesne E, Pierce K, Schumann CM, et al. Mapping early brain development in autism. Neuron. 2007;56(2):399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 3.Schumann CM, Bloss CS, Barnes CC, et al. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. Journal of Neuroscience. 2010;30(12):4419–4427. doi: 10.1523/JNEUROSCI.5714-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends in Neurosciences. 2008;31(3):137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Adolphs R. The neurobiology of social cognition. Current Opinion in Neurobiology. 2001;11(2):231–239. doi: 10.1016/s0959-4388(00)00202-6. [DOI] [PubMed] [Google Scholar]
- 6.Hazlett HC, Poe MD, Gerig G, et al. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Archives of General Psychiatry. 2011;68(5):467–476. doi: 10.1001/archgenpsychiatry.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raznahan A, Wallace GL, Antezana L, et al. Compared to what? Early brain overgrowth in autism and the perils of population norms. Biol Psychiatry. 2013;74(8):563–575. doi: 10.1016/j.biopsych.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morhardt DR, Barrow W, Jaworski M, Accardo PJ. Head circumference in young children with autism: the impact of different head circumference charts. Journal of Child Neurology. 2013 doi: 10.1177/0883073812469827. [DOI] [PubMed] [Google Scholar]
- 9.Ecker C, Suckling J, Deoni SC, et al. Brain anatomy and its relationship to behavior in adults with autism spectrum disorder: a multicenter magnetic resonance imaging study. Archives of General Psychiatry. 2012;69(2):195–209. doi: 10.1001/archgenpsychiatry.2011.1251. [DOI] [PubMed] [Google Scholar]
- 10.Balfour JA, Bryson HM. Valproic acid. CNS Drugs. 1994;2(2):144–173. [Google Scholar]
- 11.Christianson AL, Chesler N, Kromberg JGR. Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Developmental Medicine and Child Neurology. 1994;36(4):361–369. doi: 10.1111/j.1469-8749.1994.tb11858.x. [DOI] [PubMed] [Google Scholar]
- 12.Williams G, King J, Cunningham M, Stephan M, Kerr B, Hersh JH. Fetal valproate syndrome and autism: additional evidence of an association. Developmental Medicine and Child Neurology. 2001;43(3):202–206. [PubMed] [Google Scholar]
- 13.Williams PG, Hersh JH. A male with fetal valproate syndrome and autism. Developmental Medicine and Child Neurology. 1997;39(9):632–634. doi: 10.1111/j.1469-8749.1997.tb07500.x. [DOI] [PubMed] [Google Scholar]
- 14.Moore SJ, Turnpenny P, Quinn A, et al. A clinical study of 57 children with fetal anticonvulsant syndromes. Journal of Medical Genetics. 2000;37(7):489–497. doi: 10.1136/jmg.37.7.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasalam AD, Hailey H, Williams JHG, et al. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Developmental Medicine and Child Neurology. 2005;47(8):551–555. doi: 10.1017/s0012162205001076. [DOI] [PubMed] [Google Scholar]
- 16.Laegreid L, Kyllerman M, Hedner T, Hagberg B, Viggedahl G. Benzodiazepine amplification of valproate teratogenic effects in children of mothers with absence epilepsy. Neuropediatrics. 1993;24(2):88–92. doi: 10.1055/s-2008-1071520. [DOI] [PubMed] [Google Scholar]
- 17.Lin HC, Gean PW, Wang C-C, Chan YH, Chen P. The amygdala excitatory/inhibitory balance in a valproate-induced rat autism model. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0055248.e55248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bambini-Junior V, Rodrigues L, Behr GA, Moreira JCF, Riesgo R, Gottfried C. Animal model of autism induced by prenatal exposure to valproate: behavioral changes and liver parameters. Brain Research. 2011;1408:8–16. doi: 10.1016/j.brainres.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 19.Binkerd PE, Rowland JM, Nau H, Hendrickx AG. Evaluation of valproic acid (VPA) developmental toxicity and pharmacokinetics in Sprague-Dawley rats. Fundamental and Applied Toxicology. 1988;11(3):485–493. doi: 10.1016/0272-0590(88)90112-1. [DOI] [PubMed] [Google Scholar]
- 20.Dufour-Rainfray D, Vourc’h P, Le Guisquet A, et al. Behavior and serotonergic disorders in rats exposed prenatally to valproate: a model for autism. Neuroscience Letters. 2010;470(1):55–59. doi: 10.1016/j.neulet.2009.12.054. [DOI] [PubMed] [Google Scholar]
- 21.Kim KC, Kim P, Go HS, et al. The critical period of valproate exposure to induce autistic symptoms in Sprague-Dawley rats. Toxicology Letters. 2011;201(2):137–142. doi: 10.1016/j.toxlet.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(33):13501–13506. doi: 10.1073/pnas.0704391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markram K, Rinaldi T, Mendola DL, Sandi C, Markram H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacology. 2008;33(4):901–912. doi: 10.1038/sj.npp.1301453. [DOI] [PubMed] [Google Scholar]
- 24.Roullet FI, Wollaston L, deCatanzaro D, Foster JA. Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience. 2010;170(2):514–522. doi: 10.1016/j.neuroscience.2010.06.069. [DOI] [PubMed] [Google Scholar]
- 25.Schneider T, Przewłocki R. Behavioral alterations in rats prenatally to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30(1):80–89. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- 26.Schneider T, Turczak J, Przewłocki R. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology. 2006;31(1):36–46. doi: 10.1038/sj.npp.1300767. [DOI] [PubMed] [Google Scholar]
- 27.Schneider T, Roman A, Basta-Kaim A, et al. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology. 2008;33(6):728–740. doi: 10.1016/j.psyneuen.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 28.Vorhees CV. Behavioral teratogenicity of valproic acid: selective effects on behavior after prenatal exposure to rats. Psychopharmacology. 1987;92(2):173–179. doi: 10.1007/BF00177911. [DOI] [PubMed] [Google Scholar]
- 29.Vorhees CV. Teratogenicity and developmental toxicity of valproic acid in rats. Teratology. 1987;35(2):195–202. doi: 10.1002/tera.1420350205. [DOI] [PubMed] [Google Scholar]
- 30.Wagner GC, Reuhl KR, Cheh M, McRae P, Halladay AK. A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. Journal of Autism and Developmental Disorders. 2006;36(6):779–793. doi: 10.1007/s10803-006-0117-y. [DOI] [PubMed] [Google Scholar]
- 31.Felix-Ortiz AC, Febo M. Gestational valproate alters BOLD activation in response to complex social and primary sensory stimuli. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037313.e37313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mychasiuk R, Richards S, Nakahashi A, Kolb B, Gibb R. Effects of rat prenatal exposure to valproic acid on behaviour and neuro-anatomy. Developmental Neuroscience. 2012;34:268–276. doi: 10.1159/000341786. [DOI] [PubMed] [Google Scholar]
- 33.Bromley RL, Mawer GE, Briggs M, et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. Journal of Neurology, Neurosurgery & Psychiatry. 2013;84(6):637–643. doi: 10.1136/jnnp-2012-304270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen J, Grønborg TK, Sørensen MJ, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. Journal of the American Medical Association. 2013;309(16):1696–1703. doi: 10.1001/jama.2013.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dean JC, Hailey H, Moore SJ, Lloyd DJ, Turnpenny PD, Little J. Long term health and neurodevelopment in children exposed to antiepileptic drugs before birth. Journal of Medical Genetics. 2002;39(4):251–259. doi: 10.1136/jmg.39.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodier PM, Ingram JL, Tisdale B, Nelson S, Romano J. Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. Journal of Comparative Neurology. 1996;370(2):247–261. doi: 10.1002/(SICI)1096-9861(19960624)370:2<247::AID-CNE8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Rodier PM, Ingram JL, Tisdale B, Croog VJ. Linking etiologies in humans and animal models: studies of autism. Reproductive Toxicology. 1997;11(2-3):417–422. doi: 10.1016/s0890-6238(97)80001-u. [DOI] [PubMed] [Google Scholar]
- 38.Roullet FI, Lai JK, Foster JA. In Utero exposure to valproic acid and autism—a current review of clinical and animal studies. Neurotoxicology and Teratology. 2013;36:47–56. doi: 10.1016/j.ntt.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Dufour-Rainfray D, Vourc’h P, Tourlet S, Guilloteau D, Chalon S, Andres CR. Fetal exposure to teratogens: evidence of genes involved in autism. Neuroscience and Biobehavioral Reviews. 2011;35(5):1254–1265. doi: 10.1016/j.neubiorev.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 40.Bringas ME, Carvajal-Floresa FN, López-Ramírez TA, Atzori M, Flores G. Rearrangement of the dendritic morphology in limbic regions and altered exploratory behavior in a rat model of autism spectrum disorder. Neuroscience. 2013;241:170–187. doi: 10.1016/j.neuroscience.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 41.Markram K, Markram H. The intense world theory-a unifying theory of the neurobiology of autism. Frontiers in Human Neuroscience. 2010;4:p. 224. doi: 10.3389/fnhum.2010.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markram H, Rinaldi T, Markram K. The intense world syndrome-an alternative hypothesis for autism. Frontiers in Neuroscience. 2007;1(1):77–96. doi: 10.3389/neuro.01.1.1.006.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belzung C, Leman S, Vourc’h P, Andres C. Rodent models for autism: a critical review. Drug Discovery Today. 2005;2(2):93–101. [Google Scholar]
- 44.Woo CC, Leon M. Environmental enrichment as an effective treatment for autism: a randomized controlled trial. Behavioral Neuroscience. 2013;127(4):487–497. doi: 10.1037/a0033010. [DOI] [PubMed] [Google Scholar]
- 45.Lazic SE, Essioux L. Improving basic and translational science by accounting for litter-to-litter variation in animal models. BMC Neuroscience. 2013;14:p. 37. doi: 10.1186/1471-2202-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oguchi-Katayama A, Monma A, Sekino Y, Moriguchi T, Sato K. Comparative gene expression analysis of the amygdala in autistic rat models produced by pre- and post-natal exposures to valproic acid. Journal of Toxicological Sciences. 2013;38(3):391–402. doi: 10.2131/jts.38.391. [DOI] [PubMed] [Google Scholar]
- 47.Reynolds S, Millette A, Devine DP. Sensory and motor characterization in the postnatal valproate rat model of autism. Developmental Neuroscience. 2012;34(2-3):258–267. doi: 10.1159/000336646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yochum CL, Dowling P, Reuhl KR, Wagner GC, Ming X. VPA-induced apoptosis and behavioral deficits in neonatal mice. Brain Research. 2008;1203:126–132. doi: 10.1016/j.brainres.2008.01.055. [DOI] [PubMed] [Google Scholar]
- 49.Chomiak T, Karnik V, Block E, Hu B. Altering the trajectory of early postnatal cortical development can lead to structural and behavioural features of autism. BMC Neuroscience. 2010;11:p. 102. doi: 10.1186/1471-2202-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jager-Roman E, Deichl A, Jakob S. Fetal growth, major malformations, and minor anomalies in infants born to women receiving valproic acid. Journal of Pediatrics. 1986;108(6):997–1004. doi: 10.1016/s0022-3476(86)80949-0. [DOI] [PubMed] [Google Scholar]
- 51.Go HS, Kim KC, Choi CS, et al. Prenatal exposure to valproic acid increases the neural progenitor cell pool and induces macrocephaly in rat brain via a mechanism involving the GSK-3beta/beta-catenin pathway. Neuropharmacology. 2012;63(6):1028–1041. doi: 10.1016/j.neuropharm.2012.07.028. [DOI] [PubMed] [Google Scholar]
- 52.Akhtar MW, Raingo J, Nelson ED, et al. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. Journal of Neuroscience. 2009;29(25):8288–8297. doi: 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balasubramaniyan V, Boddeke E, Bakels R, et al. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 2006;143(4):939–951. doi: 10.1016/j.neuroscience.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 54.Hsieh J, Gage FH. Epigenetic control of neural stem cell fate. Current Opinion in Genetics and Development. 2004;14(5):461–469. doi: 10.1016/j.gde.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 55.Hsieh J, Gage FH. Chromatin remodeling in neural development and plasticity. Current Opinion in Cell Biology. 2005;17(6):664–671. doi: 10.1016/j.ceb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Sui L, Chen M. Prenatal exposure to valproic acid enhances synaptic plasticity in the medial prefrontal cortex and fear memories. Brain Research Bulletin. 2012;87(6):556–563. doi: 10.1016/j.brainresbull.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 57.Rinaldi T, Perrodin C, Markram H. Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic Acid animal model of autism. Frontiers in Neural Circuits. 2008;2:p. 4. doi: 10.3389/neuro.04.004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukuchi M, Nii T, Ishimaru N, et al. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neuroscience Research. 2009;65(1):35–43. doi: 10.1016/j.neures.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 59.Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Current Molecular Pharmacology. 2009;2(1):95–109. doi: 10.2174/1874467210902010095. [DOI] [PubMed] [Google Scholar]
- 60.Jessberger S, Nakashima K, Clemenson GD, Jr., et al. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. Journal of Neuroscience. 2007;27(22):5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yasuda S, Liang M-H, Marinova Z, Yahyavi A, Chuang D-M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Molecular Psychiatry. 2009;14(1):51–59. doi: 10.1038/sj.mp.4002099. [DOI] [PubMed] [Google Scholar]
- 62.MacDonald JL, Roskams AJ. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Developmental Dynamics. 2008;237(8):2256–2267. doi: 10.1002/dvdy.21626. [DOI] [PubMed] [Google Scholar]
- 63.Foley AG, Gannon S, Rombach-Mullan N, et al. Class I histone deacetylase inhibition ameliorates social cognition and cell adhesion molecule plasticity deficits in a rodent model of autism spectrum disorder. Neuropharmacology. 2012;63(4):750–760. doi: 10.1016/j.neuropharm.2012.05.042. [DOI] [PubMed] [Google Scholar]
- 64.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. Journal of Biological Chemistry. 2001;276(39):36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 65.Krämer OH, Zhu P, Ostendorff HP, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO Journal. 2003;22(13):3411–3420. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hao Y, Creson T, Zhang L, et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. Journal of Neuroscience. 2004;24(29):6590–6599. doi: 10.1523/JNEUROSCI.5747-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geier CF, Garver K, Terwilliger R, Luna B. Development of working memory maintenance. Journal of Neurophysiology. 2009;101(1):84–99. doi: 10.1152/jn.90562.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Golarai G, Ghahremani DG, Whitfield-Gabrieli S, et al. Differential development of high-level visual cortex correlates with category-specific recognition memory. Nature Neuroscience. 2007;10(4):512–522. doi: 10.1038/nn1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luna B, Garver KE, Urban TA, Lazar NA, Sweeney JA. Maturation of cognitive processes from late childhood to adulthood. Child Development. 2004;75(5):1357–1372. doi: 10.1111/j.1467-8624.2004.00745.x. [DOI] [PubMed] [Google Scholar]
- 70.Scherf KS, Behrmann M, Humphreys K, Luna B. Visual category-selectivity for faces, places and objects emerges along different developmental trajectories. Developmental Science. 2007;10(4):F15–F30. doi: 10.1111/j.1467-7687.2007.00595.x. [DOI] [PubMed] [Google Scholar]
- 71.Shaw P, Eckstrand K, Sharp W, et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19649–19654. doi: 10.1073/pnas.0707741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gogtay N, Giedd JN, Lusk L, et al. Dynamic mapping of human cortical development during childhood through early adulthood. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(21):8174–8179. doi: 10.1073/pnas.0402680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Knudsen EI, Heckman JJ, Cameron JL, Shonkoff JP. Economic, neurobiological, and behavioral perspectives on building America’s future workforce. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(27):10155–10162. doi: 10.1073/pnas.0600888103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elman JL. Learning and development in neural networks: the importance of starting small. Cognition. 1993;48(1):71–99. doi: 10.1016/0010-0277(93)90058-4. [DOI] [PubMed] [Google Scholar]
- 75.Quartz SR. The constructivist brain. Trends in Cognitive Sciences. 1999;3(2):48–57. doi: 10.1016/s1364-6613(98)01270-4. [DOI] [PubMed] [Google Scholar]
- 76.Penzes P, Cahill ME, Jones KA, Vanleeuwen J, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nature Neuroscience. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annual Review of Biochemistry. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 78.Tung EW, Winn LM. Epigenetic modifications in valproic acid-induced teratogenesis. Toxicology and Applied Pharmacology. 2010;248(3):201–209. doi: 10.1016/j.taap.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 79.Handel AE, Ebers GC, Ramagopalan SV. Epigenetics: molecular mechanisms and implications for disease. Trends in Molecular Medicine. 2010;16(1):7–16. doi: 10.1016/j.molmed.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 80.Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurology. 2002;1(6):352–358. doi: 10.1016/s1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 81.Chez MG, Chang M, Krasne V, Coughlan C, Kominsky M, Schwartz A. Frequency of epileptiform EEG abnormalities in a sequential screening of autistic patients with no known clinical epilepsy from 1996 to 2005. Epilepsy and Behavior. 2006;8(1):267–271. doi: 10.1016/j.yebeh.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 82.Dalton KM, Nacewicz BM, Johnstone T, et al. Gaze fixation and the neural circuitry of face processing in autism. Nature Neuroscience. 2005;8(4):519–526. doi: 10.1038/nn1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nordahl CW, Scholz R, Yang X, et al. Increased rate of amygdala growth in children aged 2 to 4 years with autism spectrum disorders: a longitudinal study. Archives of General Psychiatry. 2012;69(1):53–61. doi: 10.1001/archgenpsychiatry.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim KC, Lee DK, Go HS, et al. Pax6-dependent cortical glutamatergic neuronal differentiation regulates autism-like behavior in prenatally valproic acid-exposed rat offspring. Molecular Neurobiology. 2013 doi: 10.1007/s12035-013-8535-2. [DOI] [PubMed] [Google Scholar]
- 85.Banerjee A, García-Oscos F, Roychowdhury S, et al. Impairment of cortical GABAergic synaptic transmission in an environmental rat model of autism. International Journal of Neuropsychopharmacology. 2013;16(6):1309–1318. doi: 10.1017/S1461145712001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Uddin LQ, Supekar K, Lynch CJ, et al. Salience network-based classification and prediction of symptom severity in children with autism. JAMA Psychiatry. 2013;70(8):869–879. doi: 10.1001/jamapsychiatry.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Q, He S, Hu X, et al. Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. Journal of Neuroscience. 2007;27(3):542–552. doi: 10.1523/JNEUROSCI.3607-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nature Neuroscience. 2002;5(11):1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- 89.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15(4):791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 90.McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17(6):1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 91.Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing? Trends in Neurosciences. 2001;24(1):47–53. doi: 10.1016/s0166-2236(00)01682-9. [DOI] [PubMed] [Google Scholar]
- 92.Stamou M, Streifel KM, Goines PE, Lein PJ. Neuronal connectivity as a convergent target of gene x environment interactions that confer risk for autism spectrum disorders. Neurotoxicology and Teratology. 2013;36:3–16. doi: 10.1016/j.ntt.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spitzer NC. Activity-dependent neurotransmitter respecification. Nature Reviews Neuroscience. 2012;13(2):94–106. doi: 10.1038/nrn3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spitzer NC. Electrical activity in early neuronal development. Nature. 2006;444(7120):707–712. doi: 10.1038/nature05300. [DOI] [PubMed] [Google Scholar]
- 95.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain and Behavior. 2003;2(5):255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vogelauer M, Krall AS, McBrian MA, Li J-Y, Kurdistani SA. Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. Journal of Biological Chemistry. 2012;287(38):32006–32016. doi: 10.1074/jbc.M112.362467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rajendran P, Williams DE, Ho E, Dashwood RH. Metabolism as a key to histone deacetylase inhibition. Critical Reviews in Biochemistry and Molecular Biology. 2011;46(3):181–199. doi: 10.3109/10409238.2011.557713. [DOI] [PMC free article] [PubMed] [Google Scholar]