

Abstract
During infection, Yersinia, a facultative intracellular bacterial species, exhibits the ability to first invade host cells and then counteract phagocytosis by the host cells. During these two distinct stages, invasion or antiphagocytic factors assist bacteria in manipulating host cells to accomplish each of these functions; however, the mechanism through which Yersinia regulates these functions during each step remains unclear. Here, we discuss those factors that seem to function reversely and give some hypothesis about how bacteria switch between the two distinct status.
Keywords: invasion, anti-phagocytosis, intracellular survival, T3SS
Introduction
Three Yersinia species are known to be pathogenic to humans: Yersinia enterocolitis, Y. pseudotuberculosis, and Y. pestis. Y. enterocolitis and Y. pseudotuberculosis are enteropathogenic bacteria causing enteritis, ileitis, and mesenteric lymphadenitis, whereas Y. pestis is the causative agent of bubonic plague, among the most deadly human infectious disease in history. All three species harbor a virulence plasmid, which encodes a type III secretion system (T3SS) for secreting Yop protein substrates, to establish a successful infection. Six Yops, including YopE, YopH, YopM, YopO/YpkA, YopP/J, and YopT, are delivered by T3SS into host cells and these then inhibit phagocytosis and block pro-inflammatory signals (Shao, 2008). In cases involving infection with enteropathogenic Yersinia species other than Y. pestis, two adhesins, invasin and YadA, have been shown to be important for mediating contact with host cells (Isberg et al., 1987; Paerregaard et al., 1991). However, although both adhesins are inactive in Y. pestis due to an IS1541 element insertion within inv and a frameshift mutation in yadA (Parkhill et al., 2001; Song et al., 2004; Chain et al., 2006), Y. pestis maintains the ability to attach to and enter into host cells (Davis et al., 1996; Perry and Fetherston, 1997; Cowan et al., 2000), indicating that these adhesins are not necessary for the virulence of Y. pestis and that other adhesins and invasins are required for mediating the association with host cells. Y. pestis is also known for its ability to survive in macrophages during its early invasion process. After arming itself in the macrophage, Y. pestis becomes resistant to phagocytosis and is then capable of surviving outside the cell, which is critical for its pathogenesis. In this review, we will summarize what is known regarding the mechanisms through which Y. pestis survives in a host, inside or outside the cell.
Life cycle of Y. pestis during infection
Y. pestis is a facultative intracellular gram-negative bacterium. During the early stages of infection, Y. pestis can enter both macrophages and neutrophils through mechanisms of active or passive entry (Lukaszewski et al., 2005). However, Y. pestis is typically killed in neutrophils, whereas in macrophages, it can survive and acquire antiphagocytic capabilities, which enables its extracellular survival in vivo (Lukaszewski et al., 2005). Interestingly, Y. pestis can also enter into non-professional phagocytes, such as epithelial cells (Cowan et al., 2000; Leigh et al., 2005), which indicates that Y. pestis can not only be passively phagocytized by the professional phagocytes, but can also evolve a mechanism allowing it to invade host cells that generally do not possess phagocytic ability. The invasion of Y. pestis into host cells, including phagocytes and non-professional phagocytes, may be mediated by binding of adhesive factors present on their surface, including Ail, Pla, and Psa, to receptors on the membranes of host cells (Lahteenmaki et al., 2001; Miller et al., 2001; Benedek et al., 2004; Liu et al., 2006; Galvan et al., 2007; Felek et al., 2010).
After arming itself inside the macrophages, Y. pestis escapes from the cell and develops resistance to phagocytosis by both macrophages and neutrophils. Therefore, during the late stage of infection, phagocytes cannot ingest Y. pestis, and the bacteria exists mainly in an extracellular environment, which has been confirmed through autopsies of human pneumonic plague victims, wherein the samples examined exhibited abundant extracellular bacteria but little evidence of phagocytosis. The mechanism of release from macrophages is largely unknown but may be associated with apoptosis or necrosis observed through in vitro cell models. Although the mechanism through which Y. pestis relocates from an intracellular compartment to an extracellular environment is unclear, the antiphagocytic ability of Yersinia has been attributed to Caf1, F1 antigen, and Yops (e.g., YopH, YpkA, YopE, and YopT) (Figure 1). Many in vitro studies have demonstrated that these virulence factors act synergistically to promote evasion or inhibit ingestion by host cells, including professional phagocytes and some non-professional phagocytes.
Figure 1.
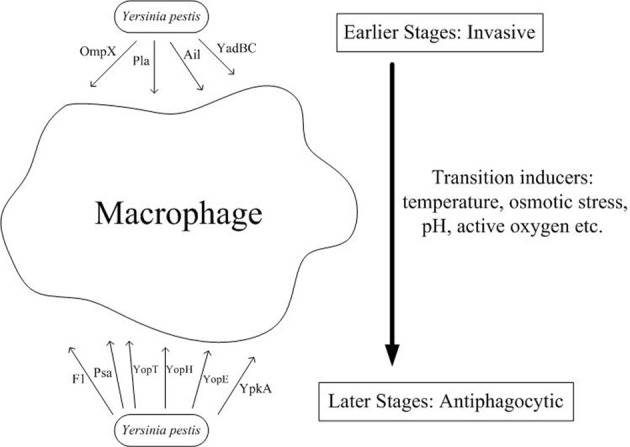
Both invasive and antiphagocytic factors are involved in Y. pestis pathogenesis. During the initial encounter with macrophages during infection, Y. pestis enters the macrophage through binding of its surface proteins, such as Pla, Ail, YadBC, and OmpX, to undetermined receptors present on the macrophage surface. However, following release from the macrophage through another undefined mechanism, Y. pestis expresses several virulence factors, including F1 antigen, Psa, and four Yops (YopT, YopH, YopE, and YpkA) which are employed to resist phagocytosis by the surrounding professional phagocytes. An additional mechanism subsequently arms Y. pestis against host phagocytes of the innate immune system, thereby allowing it to invade more tissues and organs and cause more severe impairment.
Y. pestis carries both invasive factors, which promote contact with and entry into host cells, and antiphagocytic factors that inhibit uptake by host cells. The mechanism through which this bacterium balances these contradictory factors and utilizes them in the different stages of infection to manipulate host cells remains an interesting topic. Here, we have discussed all of the known factors used by Y. pestis to enter into or escape from host cells.
Phagocytic receptors of host cells and invasive ligands of bacteria
The process of bacterial entry into host cells can be divided into two types. The first type, bacterial invasion, involves active bacterial entry, during which bacteria are not welcomed and grudgingly accepted by the host cells. Many bacteria initially depend more on their own invasive factors to enter into the host cells in order to escape the harsh extracellular environment conditions, including low pH and shear stress, and host defensive mechanisms, such as recognition and killing by macrophages and cytotoxic T cells, thereby allowing them to survive in the relatively mild intracellular milieu. The second type, phagocytosis of bacteria, involves passive entry during which bacteria are resistant to internalization and are initially ingested by the host cells through a surface mechanism. Phagocytosis plays important roles in reducing the threat from bacterial pathogens; therefore, it is used by host cells to degrade and clear hazardous exogenous material, avoid disruption of normal functions by bacteria, and present signals for activation of the innate and adaptive immune systems. In some cases, these two types of entry are indistinguishable, as both modes of entry involve initial binding of bacterial ligands to phagocytic receptors (Greenberg, 1999). Although these two types of entry mechanisms are very popular in most bacterial pathogens and are easy to differentiate, some bacterial pathogens, such as Mycobacteria and Coxiella, utilize phagocytic signaling pathways to enter into the host cells, which may be hard to define their types of entry (Méresse et al., 1999; Friedrich et al., 2012).
Phagocytosis, classically defined as the cellular engulfment of particles larger than 0.5 μm in diameter (Jaumouille and Grinstein, 2011), is a highly conserved process that has evolved to counteract the threat of altered “self” molecular or “non-self” molecular. Phagocytosis encompasses an orchestrated cascade of events that involves particle recognition, signal transduction, cytoskeleton rearrangement, membrane remodeling, and phagosome maturation (Groves et al., 2008). In higher organisms, phagocytosis is mainly performed by professional phagocytes, such as macrophages, neutrophils, and dendritic cells (DCs), but several other cell types, including epithelial cells, endothelial cells, and fibroblasts, collectively identified as non-professional phagocytes, can also perform phagocytosis under certain conditions (Cannon and Swanson, 1992; Lowry et al., 1998; Sousa et al., 2005; Kim et al., 2010). Although non-professional phagocytes do not express FcRs and CRs, which are unique feature of professional phagocytes, the downstream molecular pathways of phagocytic receptors are generally expressed and once surface receptors are acquired, they can initiate the same phagocytic signaling pathway as professional phagocytes (Ezekowitz et al., 1991; Odin et al., 1991; van Zon et al., 2009).
The phagocytosis process begins with binding of a phagocytic receptor, directly or indirectly through opsonins, to their ligands on the surface of pathogens, which mediates the adherence of bacteria to the host cells (Pizarro-Cerda and Cossart, 2006; Huang et al., 2011). The best characterized phagocytic receptors are immunoglobulin receptors (FcRs) and complement receptors (CRs), which bind to bacteria, opsonized by antibody and complement respectively, and which activate downstream signaling pathways in a similar manner (May and Machesky, 2001; Groves et al., 2008; Nimmerjahn and Ravetch, 2008). A growing number of other cell surface receptors mediating phagocytic uptake of particles have been identified, such as scavenger receptors (e.g., SR-A, MARCO, CD36, and SR-B) and C-type lectins (e.g., mannose receptor, DC-SIGN, and dectin-1) (East and Isacke, 2002; Peiser et al., 2002; Cambi and Figdor, 2003; McGreal et al., 2005; Pluddemann et al., 2006; Vachon et al., 2006; Areschoug and Gordon, 2009). Additionally, members of the integrin family, excluding CR3, which belongs to the β2 subfamily, can also mediate entry of bacteria into host cells (Cambi and Figdor, 2003; Schraven and Reth, 2007). In particular, some receptors such as CD14 participate in the uptake of bacteria, but act only to tether the particles and cannot initiate phagocytic signaling alone, as this requires accessory receptors to deliver the phagocytic signal and co-start the phagocytic process (Wright et al., 1990).
A number of bacterial components, specifically ligands presented on their surface, are engaged during entry into host cells. Various components found on the plasma membrane of most gram-positive and gram-negative bacteria are likely involved in entry into host cells, including lipopolysaccharides, peptidoglycans, lipoteichoic acid, capsules, pili, and even CpG DNA (Pluddemann et al., 2006). In the past few years, a number of bacterial surface proteins have been shown to bind to the extracellular or surface receptors of the host cells or to be delivered into the cytosol of host cells, where they then assist in the invasive progress (Pluddemann et al., 2006; Areschoug and Gordon, 2008). The active entry of bacteria can be classified into two different mechanisms: “zippering” and “triggering” (Swanson and Baer, 1995; Cossart and Sansonetti, 2004). Zippering means that bacteria present ligands on their surface allowing them to bind to host cells and initiate the entry process (Cossart and Sansonetti, 2004). This zippering mechanism is exemplified by FcR- and CR3-mediated phagocytosis, which is characterized by the formation of inclusion shaped by the bacteria they ingest. Triggering is a mechanism through which bacteria inject effectors into host cells via T3SS to regulate phagocytosis (Swanson and Baer, 1995; Cossart and Sansonetti, 2004). The triggering mechanism enables host cells to internalize bacteria and fluids together (i.e., macropinocytosis) (Cossart and Sansonetti, 2004). For Yersinia, the zippering mechanism is the primary mode of entry since surface molecules of Yersinia mediate contact with, and adhesion of, host cells (Cossart and Sansonetti, 2004).
Invasion factors
Bacterial pathogens can utilize many kinds of receptors present on the host cell surface, including the following, to facilitate invasion into host cells: FcγR; CR3; β1, β3, and β5 integrins; Toll-like receptors; mannose receptor; galactose receptor; and scavenger receptors (Taylor et al., 2005; Pluddemann et al., 2006). In addition to common strategies used by many other bacterial pathogens, Yersinia also employs several kinds of specific molecules on its outer membrane through which it can bind to and invade various host cells, including epithelial cells, endothelial cells, and fibroblasts, in addition to professional phagocytes. Indeed, Y. pestis has three identified adhesins: Ail (attachment-invasion locus), Pla, and Psa, in which Ail and Pla acts as invasive factors, but results of mediating adherence by Psa to host cells is not invasion, but prevention of invasion(as discussed below).
Ail
Ail in Y. pestis is a 17.5 kDa outer membrane protein, encoded chromosomally, which is predicted to have eight transmembrane domains and four short extracellular loops extending from the surface of the bacterial outer membrane (Miller et al., 1990; Beer and Miller, 1992). Ail from all three species demonstrates cell adhesion and serum-resistance activities (Miller and Falkow, 1988; Miller et al., 2001; Kolodziejek et al., 2007; Bartra et al., 2008; Felek and Krukonis, 2009; Kolodziejek et al., 2010). In a mouse model of infection, Ail mutant Y. pestis exhibited a >3000-fold increase in the 50% lethal dose and a lower rate of colonization to the host tissue than the wild type (Felek and Krukonis, 2009).
In in vitro cell cultures, Ail-deficient strains have demonstrated reduced epithelial cell association and internalization, that is, at approximately 90 and 98%, respectively (Kolodziejek et al., 2007). Escherichia coli carrying Ail is highly invasive to CHO cells (Peiser et al., 2002) and moderately invasive to Hep-2 cells (Groves et al., 2008). Similar to YadA, Ail was also recently found to bind to fibronectin (Tsang et al., 2010), an extracellular matrix component. Additionally, treatment of cultivated cells with antifibronectin antibody decreased Ail-mediated adherence and inhibited KIM5-mediated cytotoxicity of host cells in an Ail-dependent manner (Tsang et al., 2010). Therefore, fibronectin, which has many integrin-binding sites, may act as a bridging molecular to engage host cells with this pathogen (Tsang et al., 2010). Moreover, because biochemical data and genetic analysis suggest that the C-terminal half of extracellular loop 2 of Ail mediates interaction with host cell surface components (Kirjavainen et al., 2008), it is possible that this domain interacts with fibronectin and is subsequently involved in attachment and entry into host cells.
Interestingly, despite sharing 100% homology with Y. pestis, Ail from Y. pseudotuberculosis fails to confer the same adhesion and invasion functions, which may be due to two amino acid substitutions in extracellular loop 3 (Yang et al., 1996; Miller et al., 2001; Tsang et al., 2010). However, Ail from Y. enterocolitis exhibits the same activity as that from Y. pestis, although they share only 70% sequence identity (Miller et al., 2001). These data provide strong evidence that some other factors contribute to Ail-mediated phagocytosis.
Plasminogen activator
Plasminogen activator (Pla), encoded by the Y. pestis-specific plasmid pPCP1, is a membrane protein of the omptin family of bacterial outer membrane proteases (Kukkonen and Korhonen, 2004). Pla exhibits different phenotypes in inducing plague. In comparison with the wild type, Y. pestis lacking Pla has been reported to have greatly reduced virulence and was found to be one of the highly expressed genes when inoculated subcutaneously, but produced equivalent or nearly equivalent virulence when introduced by aerosols or directly into the blood stream (Sodeinde et al., 1992; Welkos et al., 1997). In models of pneumonic plague, dissemination of Pla-deficient Y. pestis to the circulation system was found to be unaffected, but restricted outgrowth was observed in lungs (Sebbane et al., 2006; Lathem et al., 2007). However, Y. pestis lacking Pla in mouse models of bubonic plague was observed to grow normally at the subcutaneous sites of inoculation but was not disseminated to the lymphatic system and deeper tissues (Sebbane et al., 2006; Lathem et al., 2007).
Pla is a 10-strand antiparallel β-barrel with four short periplasmic turns and has five surface-exposed loops where catalytic residues are located (Kukkonen and Korhonen, 2004). A striking feature of Pla is its ability to activate plasminogen to plasmin, which then dissolves fibrin clots and digests laminin that further impairs tissue barriers (Degen et al., 2007). In addition to proteolytic functions, Pla also plays a role in adhesion and invasion to epithelial cells, probably by binding to extracellular matrix components such as laminin and reconstituted basement membrane (Lobo, 2006). Escherichia coli expressing Pla is capable of invading HUVECs (Kukkonen et al., 2004), ECV304 (Lahteenmaki et al., 2001; Kukkonen et al., 2004), HeLa cells (Benedek et al., 2004), and macrophages (Zhang et al., 2008), and this process appears to be independent of residues S99 and D206, which are required for proteolytic activity of Plas (Lahteenmaki et al., 2001), thus providing evidence that the adhesion activity of Pla is independent of its protease activity. Pla-mediating internalization of bacteria by HeLa cells can be inhibited by phagocytic signaling inhibitors, such as wortmannin, staurosprin, genistein, C3 exoenzymes, and NDGA, and actin polymerization inhibitors, such as cytochalasin D, although bacterial association was not affected (Benedek et al., 2004). Additionally, the adhesion of Pla in Y. pestis is more efficient than that of its counterparts in other bacteria, such as PgtE of Samonella, OmpT of Escherichia coli, and Epo of Erwinia (Kukkonen et al., 2004).
Recently, Zhang et al. found that Pla is a ligand for a macrophage and DC surface receptor DEC-205, which is usually thought to be related to antigen presentation (Zhang et al., 2008). Using alveolar macrophage and CHO cells stably expressing DEC-205, the authors determined that Pla interacted with DEC-205 and that this interaction mediated adherence of Y. pestis to host cells and promoted early Yersinia dissemination from lungs to spleens during pneumonic plague (Zhang et al., 2008). These results suggested a new target of Pla activity, which may help elucidate the rapid progression of primary pneumonic plague.
Other
YadBC, representing two surface proteins, is thought to be able to help bacteria to invade into WI-26 type1 pneumocytes and HeLa epithelioid cells (Forman et al., 2008). Although the YadBC double mutant of Y. pestis exhibited similar attachment to host cells, the invasion defect was small but significant (Forman et al., 2008).
Antiphagocytic factor
Psa
Psa, a homopolymer macromolecular complex known as an adhesion pilus, is assembled into a molecular mass of 15 kDa subunits, forming a capsule-like structure on the surface of Y. pestis (Lindler et al., 1990; Lindler and Tall, 1993). It is ideally expressed in an environment where the temperature remains at 37°C and pH ranges from 5.8 to 6.0 (Bichowsky-Slomnicki and Ben-Efraim, 1963). The expression of Psa is positively regulated by a global transcription regulator, RovA (Cathelyn et al., 2006) and is negatively regulated by Fur (Zhou et al., 2006). Mutated Psa is slightly attenuated by the intravenous mode of infection but exhibits a significant dissemination defect (Lathem et al., 2007). Recent studies have reported that the virulence of Y. pestis KIM5 lacking Psa was either unaffected or only slightly affected during subcutaneous challenge of Swiss Webster mice (Bearden et al., 1997) and was unaltered in native or immunized BALB/C mice (Anisimov et al., 2009).
Psa is known to bind to β1-linked galactosyl residues in glycosphingolipids (Payne et al., 1998) and apoB-containing LDL in human plasma (Makoveichuk et al., 2003), and these interactions could prevent binding of purified Psa to macrophages or fibroblasts (Payne et al., 1998; Makoveichuk et al., 2003). Indeed, Psa acts as a antiphagocytic factor independent of Yops (Huang and Lindler, 2004). Although Psa mediates the association of Y. pestis with human epithelial cells or mouse macrophages, it cannot resist uptake by host cells (Huang and Lindler, 2004; Liu et al., 2006; Grabenstein et al., 2006). In contrast to wild-type strains, Psa-mutated Y. pestis binding to human respiratory tract epithelial cells has been found to be significant, although the number of internalized bacteria was equivalent for both (Grabenstein et al., 2006). Additionally, Psa-modified Escherichia coli or polystyrene beads were observed to bind to host cells to a greater extent than either the control bacteria or beads, respectively, and their internalization by host cells was similarly poor (Huang and Lindler, 2004; Liu et al., 2006). These results indicated that Psa confers adhesive but not invasive activity to bacteria. Psa-mediating inhibition of phagocytosis may be caused by binding to lipoprotein, which could prevent recognition of bacterial pathogens by host cells.
F1
Another fimbrial structure expressed by Y. pestis is fraction 1 (F1) antigen, which is composed of linear fibers of the Caf1 subunit (Zavialov et al., 2003). F1 is produced at high yields at 35–37°C to cover the bacterial surface. After the initial intracellular stage during infection, Y. pestis is released from macrophages and expresses large amounts of F1 (Du et al., 2002). Together with other anti-phagocytic factors, F1 efficiently limits phagocytosis of Y. pestis by host cells and contributes to the extracellular survival of Y. pestis in vivo, although F1-negative strains exhibit similar virulence as the wild type (Du et al., 2002).
A recent study provided direct evidence of the inhibiting effect of F1 on phagocytosis by epithelial cells (Liu et al., 2006). Using human respiratory tract epithelial cells as models, binding of F1-coated latex beads to A549 cells was reduced when compared with that to BSA-coated latex beads (Benedek et al., 2004). Y. pestis deficient in Caf1 was better internalized by host cells, and complementing this strain with Caf-containing plasmid significantly reduced internalization by host cells, confirming the F1-mediated inhibition of uptake of Y. pestis by respiratory tract epithelial cells (Benedek et al., 2004). Additionally, the authors of this study reported that the Caf-Psa double mutant was internalized more efficiently than the single mutant, thereby indicating that both F1 and Psa contribute to inhibition of phagocytosis (Benedek et al., 2004).
YopH
YopH, a 468-amino-acid protein secreted by T3SS, exhibits the most potent activity of all PTPase(Protein Tyrosine Phosphatase) enzymes isolated to date (Zhang et al., 1992). The crystal structure of YopH indicates that its N- and C-terminal domains are linked by a proline-rich sequence. The N-terminal domain contains a type III secretion signal, a chaperon-binding region and a substrate-binding domain (Khandelwal et al., 2002), while the C-terminal domain includes a PTPase catalytic domain and an additional substrate-binding domain (Phan et al., 2003; Bahta and Burke, 2012). These two substrate recognition domains cooperate to reinforce binding of YopH to its substrate and enhance its activity and virulence.
The main function of YopH is anti-phagocytosis, which is executed by dephosphorylating its substrates, including p130Cas, FAk, and paxillin in epithelial cells and p130Cas, SKAP-HOM and Fyb in macrophages (Hamid et al., 1999; Deleuil et al., 2003; Aepfelbacher, 2004). These form the so-called focal adhesion complexes, which play pivotal roles in β1 integrin-mediating phagocytosis. By targeting and dephosphorylating these proteins, YopH may antagonize β1 integrin-mediated uptake of bacteria by host cells at an early stage. Antiphagocytosis activity is essential for the virulence of Yersinia since strains lacking YopH are efficiently ingested by host cell phagocytosis (Kerschen et al., 2004).
YopE
YopE is a 219-amino-acid protein that exhibits eukaryotic GAP activity (Rosqvist et al., 1990; Black and Bliska, 2002; Pawel-Rammingen et al., 2002). YopE is comprised of three domains: an N terminal type III secretion signal followed by an intracellular membrane targeting domain and a C terminal Rho GAP domain (Black and Bliska, 2002; Pawel-Rammingen et al., 2002). In infected cells, translocated YopE has been found to cause cell rounding up and detachment through its ability to disrupt actin microfilaments (Black and Bliska, 2002; Pawel-Rammingen et al., 2002).
YopE inactivates small RhoA-like G proteins by inducing their GDPase activity, which results in conversion of the active GTP-bound state to an inactive GDP-bound state, and blocking of downstream signaling cascades (Aili et al., 2006). Given the importance of Rho GTPase in regulating actin polymerization, which is an important event in phagocytosis, YopE can antagonize phagocytosis of Yersinia by host cells (Viboud et al., 2006). Indeed, research has shown that YopE-deficient mutants can be efficiently internalized by macrophages and neutrophils (Songsungthong et al., 2010).
A recent study also demonstrated that YopE from Y. enterocolitis could bind to and inactivate RhoG, an upstream regulator of Rac1 and other Rho GTPases, both in vivo and in vitro (Roppenser et al., 2009). YopE colocalized with RhoG in the ER and Golgi, and this localization determined its substrate specificity and activity (Roppenser et al., 2009). As RhoG can be activated by the β1 integrin-mediating pathway of host cells when challenged by bacteria, inhibition of RhoG by YopE may contribute to the overall antiphagocytic activity in a new manner (Mohammadi and Isberg, 2009; Roppenser et al., 2009).
YopT
YopT (322 amino acids) was identified as the second Yop, in addition to YopE, that can disrupt the actin cytoskeleton (Iriarte and Cornelis, 1998). YopT is similar to YopE in that its N-terminal region includes a type III secretion signal and a chaperon-binding domain (Sorg et al., 2003), although its C-terminus comprises a novel cysteine protease belonging to clan CE (Shao et al., 2003). YopT is expressed in Y. pestis, but in more than 50% cases, Y. pseudotuberculosis does not carry a functional YopT due to deletions in the region of the yopT gene (Schmidt, 2011).
YopT specifically recognizes and cleaves the phenyl groups of lipid-modified RhoA, Rac, and Cdc42, which results in detachment of Rho GTPase from the plasma membrane (Zumbihl et al., 1999; Shao et al., 2003). Mislocalization of Rho GTPase impairs its function and paralyzes various downstream signaling pathways (Iriarte and Cornelis, 1998). In cultured cells, ectopic expression of YopT leads to disruption of the actin cytoskeleton, cell rounding up, and inhibition of phagocytosis (Iriarte and Cornelis, 1998). Phagocytosis by macrophages and neutrophils of Yersinia mutants lacking YopT has been reported to be significantly greater than that by the wild type (Viboud et al., 2006).
However, the virulence of YopT-mutated Yersinia is not altered in mouse oral infection, and bacterial dissemination to the liver is not affected, suggesting that YopT is not required for virulence (Trülzsch et al., 2004; Mohammadi and Isberg, 2009). Additional studies have reported that YopT enhances virulence only in the absence of YopE (Viboud et al., 2006). Considering their similar activity, YopT may be a redundancy due to the presence of YopE, and this may partially explain why YopT is inactivated in certain strains.
YpkA/YopO
YpkA (732 amino acids) is a multifunctional protein containing an N-terminal Ser/Thr kinase catalytic domain and C-terminal GDI domain (Navarro et al., 2007). The first 150 amino acids in the sequence comprise a type III secretion signal and membrane localization domain, while the last 20 amino acids comprise an actin-binding domain (Juris et al., 2000), which localizes to the inner face of the plasma membrane. YpkA is produced in bacteria as an inactive kinase and, when translated into host cells, is activated by binding to actin (Juris et al., 2000; Trasak et al., 2007). Upon activation, YpkA undergoes autophosphorylation, and then subsequently phosphorylates both native and artificial substrates, such as MBP and histone (Juris et al., 2000; Trasak et al., 2007).
Surprisingly, a recent study showed that the C-terminal domain of YpkA mimics the host GDI function to inhibit GDP-GTP exchange of small G proteins, like RhoA and Rac1, and to keep them in their inactive states (Prehna et al., 2006). This activity enables YpkA to repress Rho activity and to limit functions of its downstream signaling pathway, such as actin stress fiber formation (Barz et al., 2000; Dukuzumuremyi et al., 2000; Prehna et al., 2006). On the other hand, the kinase domain of YpkA can bind to and phosphorylate a G protein subunit Gαq, thereby efficiently impairing guanine nucleotide binding and preventing activation of Gαq (Navarro et al., 2007). Because both Rho proteins and Gαq control actin stress fiber formation, simultaneous inactivation of these two targets enables YpkA to more efficiently usurp downstream signaling pathways of the host cells (Navarro et al., 2007).
Considering its obvious effect on the actin cytoskeleton, YpkA is believed to act as another important virulent factor, in addition to YopH, YopE, and YopT, in inhibiting phagocytosis of Y. pestis by host cells during infection (Wiley et al., 2006), as YpkA can sequester Rac and block Rac-dependent Fcγ receptor-mediated phagocytosis at the plasma membrane; however, this is not observed with RhoA and RhoA-dependent complement-mediated phagocytosis (Groves et al., 2010) as RhoA is trapped in the cytosol by endogenous RhoGDIα (Groves et al., 2010).
Switch from invasive to antiphagocytic status
How does Y. pestis switch from the earlier invasive status to the later antiphagocytic status? After migrating to the inside of macrophages, Y. pestis bacteria would encounter harsher microenvironments, including higher temperature, low pH values, osmotic pressure, and reactive oxygen species (as shown in Figure 1). Consequently, Y. pestis may have evolved the ability to adapt to these challenges. Triggering by various kinds of environmental signals may have up- or down-regulated their survival- or virulence-associated genes, thereby changing surface components and presenting heterogeneous phenotypes. This transition may enhance their survival within host cells and their virulence once escaped.
First, and most importantly, the temperature shift from 28°C in vitro to 37°C in vivo could induce the expression of F1, Psa, and Yops, all of which work in coordination to block phagocytosis by host cells (Lawson et al., 2006; Fukuto et al., 2010). Second, the low pH value of the phagosome inside the macrophage allows for upregulation of the expression of PsA (Lawson et al., 2006; Fukuto et al., 2010). Although Psa mediates adhesion to host cells, this binding does not initiate the progress of uptake, and in contrast, inhibits the internalization of bacteria through an unknown mechanism(as discussed above). In addition, the Y.pestis-Containing Vacuole (YCV) may provide a niche for bacteria to make this transition. Around 8 h postinfection, the YCV acquired markers of late endosomes or lysosomes and had a spacious morphology, but it could prevent vacuole acidification and avoid bactericidal autophagy (Grabenstein et al., 2006; Pujol et al., 2009). Residence in the YCV and responses to environmental stress may promote bacteria to initiate the transcription regulation of invasive or anti-phagocytic factors, resulting into the final anti-phagocytic status.
Although the adhesive factors Ail and Pla are still present on the surface of bacteria, owing to their relatively short outer membrane regions, they are unable to escape the masking effects of the F1 capsule, which would block contact between Ail and Pla and their respective receptors, leading to further activation of the uptake signaling pathway. Conversely, the Psa is long enough to overcome the masking effects of the F1 capsule, but binding to its receptor does not prime the internalization progress and produces the opposite effect, which inhibits phagocytosis by host cells. Furthermore, adhesion of Psa to the surface of host cells facilitates the delivery of Yops into host cells, which further blocks the phagocytic ability of the host cells.
Conclusions
Deficiency in invasin and YadA does not hamper the invasion of Y. pestis into host cells, which is necessary for infection during the earlier stage, which suggests that Y. pestis must utilize other invasive factors to aid its entry into host cells. Loss of virulence of these invasive-factor mutants indicates that Y. pestis must acquire the ability to tip the balance of entry into host cells and to subsequently have antiphagocytic activities. During the earlier stages of infection, Y. pestis requires invasive factors to enter into host cells, and during the later stages, it must produce antiphagocytic factors to escape uptake by host cells. Although the mechanism through which Y. pestis fine-tunes the temporal expression of these seemingly contradictory factors remains unknown, the simultaneous presence of these opposing functions in this bacterium represents a unique capability to survive both inside and outside host cells. Further analysis of the regulation of invasion and antiphagocytic associated virulence factors will improve our understanding of the pathogenesis of Y. pestis and will provide greater insights into the in vivo survival mechanism of facultative intracellular or extracellular bacteria.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support was provided by the National Natural Science Foundation of China (30930001, 81301405) and by the National Basic Research Program of China (2009CB522600, 2012CB518704).
References
- Aepfelbacher M. (2004). Modulation of Rho GTPases by type III secretion system translocated effectors of yersinia. Ergebnisse Physiol. Biol. Chem. Exp. Pharmakol. 152, 65–77 10.1007/s10254-004-0035-3 [DOI] [PubMed] [Google Scholar]
- Aili M., Isaksson E. L., Hallberg B., Wolf-Watz H., Rosqvist R. (2006). Functional analysis of the YopE GTPase-activating protein (GAP) activity of Yersinia pseudotuberculosis. Cell Microbiol. 8, 1020–1033 10.1111/j.1462-5822.2005.00684.x [DOI] [PubMed] [Google Scholar]
- Anisimov A. P., Bakhteeva I. V., Panfertsev E. A., Tat'yana E. S., Tat'yana B. K., Platonov M. E., et al. (2009). The subcutaneous inoculation of pH 6 antigen mutants of Yersinia pestis does not affect virulence and immune response in mice. J. Med. Microbiol. 58, 26–36 10.1099/jmm.0.005678-0 [DOI] [PubMed] [Google Scholar]
- Areschoug T., Gordon S. (2008). Pattern recognition receptors and their role in innate immunity: focus on microbial protein ligands. Contrib. Microbiol. 15, 45–60 10.1159/000135685 [DOI] [PubMed] [Google Scholar]
- Areschoug T., Gordon S. (2009). Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell Microbiol. 11, 1160–1169 10.1111/j.1462-5822.2009.01326.x [DOI] [PubMed] [Google Scholar]
- Bahta M., Burke T. R. (2012). Yersinia pestis and approaches to targeting its outer protein H protein-tyrosine phosphatase (YopH). Curr. Med. Chem. 19, 5726–5734 10.2174/092986712803988866 [DOI] [PubMed] [Google Scholar]
- Bartra S. S., Styer K. L., O'Bryant D. M., Nilles M. L., Hinnebusch B. J., Aballay A., et al. (2008). Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect. Immun. 76, 612–622 10.1128/IAI.01125-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barz C., Abahji T. N., Trülzsch K., Heesemann J. (2000). The Yersinia Ser/Thr protein kinase YpkA/YopO directly interacts with the small GTPases RhoA and Rac-1. FEBS Lett. 482, 139–143 10.1016/S0014-5793(00)02045-7 [DOI] [PubMed] [Google Scholar]
- Bearden S. W., Fetherston J. D., Perry R. D. (1997). Genetic organization of the yersiniabactin biosynthetic region and construction of avirulent mutants in Yersinia pestis. Infect. Immun. 65, 1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer K., Miller V. (1992). Amino acid substitutions in naturally occurring variants of ail result in altered invasion activity. J. Bacteriol. 174, 1360–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedek O., Nagy G., Emody L. (2004). Intracellular signalling and cytoskeletal rearrangement involved in Yersinia pestis plasminogen activator (Pla) mediated HeLa cell invasion. Microb. Pathog. 37, 47–54 10.1016/j.micpath.2004.04.001 [DOI] [PubMed] [Google Scholar]
- Bichowsky-Slomnicki L., Ben-Efraim S. (1963). Biological activities in extracts of Pasteurella pestis and their relation to the “pH 6 antigen”. J. Bacteriol. 86, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D. S., Bliska J. B. (2002). The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37, 515–527 10.1046/j.1365-2958.2000.02021.x [DOI] [PubMed] [Google Scholar]
- Cambi A., Figdor C. G. (2003). Dual function of C-type lectin-like receptors in the immune system. Curr. Opin. Cell Biol. 15, 539–546 10.1016/j.ceb.2003.08.004 [DOI] [PubMed] [Google Scholar]
- Cannon G. J., Swanson J. A. (1992). The macrophage capacity for phagocytosis. J. Cell Sci. 101, 907–913 [DOI] [PubMed] [Google Scholar]
- Cathelyn J. S., Crosby S. D., Lathem W. W., Goldman W. E., Miller V. L. (2006). RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc. Natl. Acad. Sci. U.S.A. 103, 13514–13519 10.1073/pnas.0603456103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain P. S., Hu P., Malfatti S. A., Radnedge L., Larimer F., Vergez L. M., et al. (2006). Complete genome sequence of Yersinia pestis strains antiqua and Nepal516: evidence of gene reduction in an emerging pathogen. J. Bacteriol. 188, 4453–4463 10.1128/JB.00124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P., Sansonetti P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248 10.1126/science.1090124 [DOI] [PubMed] [Google Scholar]
- Cowan C., Jones H. A., Kaya Y. H., Perry R. D., Straley S. C. (2000). Invasion of epithelial cells by Yersinia pestis: evidence for a Y. pestis-specific invasin. Infect. Immun. 68, 4523–4530 10.1128/IAI.68.8.4523-4530.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K. J., Fritz D. L., Pitt M. L., Welkos S. L., Worsham P. L., Friedlander A. M. (1996). Pathology of experimental pneumonic plague produced by fraction 1-positive and fraction 1-negative Yersinia pestis in African green monkeys (Cercopithecus aethiops). Arch. Pathol. Lab. Med. 120, 156–163 [PubMed] [Google Scholar]
- Degen J., Bugge T., Goguen J. (2007). Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 5, 24–31 10.1111/j.1538-7836.2007.02519.x [DOI] [PubMed] [Google Scholar]
- Deleuil F., Mogemark L., Francis M. S., Wolf-Watz H., Fällman M. (2003). Interaction between the Yersinia protein tyrosine phosphatase YopH and eukaryotic Cas/Fyb is an important virulence mechanism. Cell. Microbiol. 5, 53–64 10.1046/j.1462-5822.2003.00236.x [DOI] [PubMed] [Google Scholar]
- Du Y., Rosqvist R., Forsberg Å. (2002). Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect. Immun. 70, 1453–1460 10.1128/IAI.70.3.1453-1460.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukuzumuremyi J.-M., Rosqvist R., Hallberg B., Åkerström B., Wolf-Watz H., Schesser K. (2000). The Yersinia protein kinase A is a host factor inducible RhoA/Rac-binding virulence factor. J. Biol. Chem. 275, 35281–35290 10.1074/jbc.M003009200 [DOI] [PubMed] [Google Scholar]
- East L., Isacke C. M. (2002). The mannose receptor family. Biochim. Biophys. Acta 1572, 364–386 10.1016/S0304-4165(02)00319-7 [DOI] [PubMed] [Google Scholar]
- Ezekowitz R. A., Williams D. J., Koziel H., Armstrong M. Y., Warner A., Richards F. F., et al. (1991). Uptake of pneumocystis carinii mediated by the macrophage mannose receptor. Nature 351, 155–158 10.1038/351155a0 [DOI] [PubMed] [Google Scholar]
- Felek S., Krukonis E. S. (2009). The Yersinia pestis ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect. Immun. 77, 825–836 10.1128/IAI.00913-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felek S., Tsang T. M., Krukonis E. S. (2010). Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect. Immun. 78, 4134–4150 10.1128/IAI.00167-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman S., Wulff C. R., Myers-Morales T., Cowan C., Perry R. D., Straley S. C. (2008). yadBC of Yersinia pestis, a new virulence determinant for bubonic plague. Infect. Immun. 76, 578–587 10.1128/IAI.00219-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich N., Hagedorn M., Soldati-Favre D., Soldati T. (2012). Prison break: pathogens' strategies to egress from host cells. Microbiol. Mol. Biol. Rev. 76, 707–720 10.1128/MMBR.00024-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuto H. S., Svetlanov A., Palmer L. E., Karzai A. W., Bliska J. B. (2010). Global gene expression profiling of Yersinia pestis replicating inside macrophages reveals the roles of a putative stress-induced operon in regulating type III secretion and intracellular cell division. Infect. Immun. 78, 3700–3715 10.1128/IAI.00062-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan E. M., Chen H., Schifferli D. M. (2007). The Psa fimbriae of Yersinia pestis interact with phosphatidylcholine on alveolar epithelial cells and pulmonary surfactant. Infect. Immun. 75, 1272–1279 10.1128/IAI.01153-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabenstein J. P., Fukuto H. S., Palmer L. E., Bliska J. B. (2006). Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect. Immun. 74, 3727–3741 10.1128/IAI.00255-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S. (1999). Modular components of phagocytosis. J. Leukoc. Biol. 66, 712–717 [DOI] [PubMed] [Google Scholar]
- Groves E., Dart A. E., Covarelli V., Caron E. (2008). Molecular mechanisms of phagocytic uptake in mammalian cells. Cell. Mol. Life Sci. 65, 1957–1976 10.1007/s00018-008-7578-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves E., Rittinger K., Amstutz M., Berry S., Holden D. W., Cornelis G. R., et al. (2010). Sequestering of rac by the Yersinia effector YopO blocks Fcγ receptor-mediated phagocytosis. J. Biol. Chem. 285, 4087–4098 10.1074/jbc.M109.071035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid N., Gustavsson A., Andersson K., McGee K., Persson C., Rudd C. E., et al. (1999). YopH dephosphorylates cas and Fyn-binding protein in macrophages. Microb. Pathog. 27, 231 10.1006/mpat.1999.0301 [DOI] [PubMed] [Google Scholar]
- Huang X. Z., Lindler L. E. (2004). The pH 6 antigen is an antiphagocytic factor produced by Yersinia pestis independent of Yersinia outer proteins and capsule antigen. Infect. Immun. 72, 7212–7219 10.1128/IAI.72.12.7212-7219.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z. Y., Hunter S., Chien P., Kim M. K., Han-Kim T. H., Indik Z. K., et al. (2011). Interaction of two phagocytic host defense systems: fcgamma receptors and complement receptor 3. J. Biol. Chem. 286, 160–168 10.1074/jbc.M110.163030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriarte M., Cornelis G. R. (1998). YopT, a new Yersinia Yop effector protein, affects the cytoskeleton of host cells. Mol. Microbiol. 29, 915–929 10.1046/j.1365-2958.1998.00992.x [DOI] [PubMed] [Google Scholar]
- Isberg R. R., Voorhis D. L., Falkow S. (1987). Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell 50, 769–778 10.1016/0092-8674(87)90335-7 [DOI] [PubMed] [Google Scholar]
- Jaumouille V., Grinstein S. (2011). Receptor mobility, the cytoskeleton, and particle binding during phagocytosis. Curr. Opin. Cell Biol. 23, 22–29 10.1016/j.ceb.2010.10.006 [DOI] [PubMed] [Google Scholar]
- Juris S. J., Rudolph A. E., Huddler D., Orth K., Dixon J. E. (2000). A distinctive role for the Yersinia protein kinase: actin binding, kinase activation, and cytoskeleton disruption. Proc. Natl. Acad. Sci.U.S.A. 97, 9431–9436 10.1073/pnas.170281997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschen E. J., Cohen D. A., Kaplan A. M., Straley S. C. (2004). The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect. Immun. 72, 4589–4602 10.1128/IAI.72.8.4589-4602.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal P., Keliikuli K., Smith C. L., Saper M. A., Zuiderweg E. R. (2002). Solution structure and phosphopeptide binding to the N-terminal domain of Yersinia YopH: comparison with a crystal structure. Biochemistry 41, 11425–11437 10.1021/bi026333l [DOI] [PubMed] [Google Scholar]
- Kim M., Ashida H., Ogawa M., Yoshikawa Y., Mimuro H., Sasakawa C. (2010). Bacterial interactions with the host epithelium. Cell Host Microbe 8, 20–35 10.1016/j.chom.2010.06.006 [DOI] [PubMed] [Google Scholar]
- Kirjavainen V., Jarva H., Biedzka-Sarek M., Blom A. M., Skurnik M., Meri S. (2008). Yersinia enterocolitica serum resistance proteins YadA and Ail bind the complement regulator C4b-binding protein. PLoS Pathog. 4:e1000140 10.1371/journal.ppat.1000140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejek A. M., Schnider D. R., Rohde H. N., Wojtowicz A. J., Bohach G. A., Minnich S. A., et al. (2010). Outer membrane protein X (Ail) contributes to Yersinia pestis virulence in pneumonic plague and its activity is dependent on the lipopolysaccharide core length. Infect. Immun. 78, 5233–5243 10.1128/IAI.00783-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejek A. M., Sinclair D. J., Seo K. S., Schnider D. R., Deobald C. F., Rohde H. N., et al. (2007). Phenotypic characterization of OmpX, an Ail homologue of Yersinia pestis KIM. Microbiology 153, 2941–2951 10.1099/mic.0.2006/005694-0 [DOI] [PubMed] [Google Scholar]
- Kukkonen M., Korhonen T. K. (2004). The omptin family of enterobacterial surface proteases/adhesins: from housekeeping in escherichia coli to systemic spread of Yersinia pestis. Int. J. Med. Microbiol. 294, 7–14 10.1016/j.ijmm.2004.01.003 [DOI] [PubMed] [Google Scholar]
- Kukkonen M., Suomalainen M., Kyllönen P., Lähteenmäki K., Lång H., Virkola R., et al. (2004). Lack of O−antigen is essential for plasminogen activation by Yersinia pestis and salmonella enterica. Mol. Microbiol. 51, 215–225 10.1046/j.1365-2958.2003.03817.x [DOI] [PubMed] [Google Scholar]
- Lahteenmaki K., Kukkonen M., Korhonen T. K. (2001). The pla surface protease/adhesin of Yersinia pestis mediates bacterial invasion into human endothelial cells. FEBS Lett. 504, 69–72 10.1016/S0014-5793(01)02775-2 [DOI] [PubMed] [Google Scholar]
- Lathem W. W., Price P. A., Miller V. L., Goldman W. E. (2007). A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315, 509–513 10.1126/science.1137195 [DOI] [PubMed] [Google Scholar]
- Lawson J. N., Lyons C. R., Johnston S. A. (2006). Expression profiling of Yersinia pestis during mouse pulmonary infection. DNA Cell Biol. 25, 608–616 10.1089/dna.2006.25.608 [DOI] [PubMed] [Google Scholar]
- Leigh S. A., Forman S., Perry R. D., Straley S. C. (2005). Unexpected results from the application of signature-tagged mutagenesis to identify Yersinia pestis genes required for adherence and invasion. Microb. Pathog. 38, 259–266 10.1016/j.micpath.2005.02.004 [DOI] [PubMed] [Google Scholar]
- Lindler L. E., Klempner M. S., Straley S. C. (1990). Yersinia pestis pH 6 antigen: genetic, biochemical, and virulence characterization of a protein involved in the pathogenesis of bubonic plague. Infect. Immun. 58, 2569–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindler L. E., Tall B. D. (1993). Yersinia pestis pH 6 antigen forms fimbriae and is induced by intracellular association with macrophages. Mol. Microbiol. 8, 311–324 10.1111/j.1365-2958.1993.tb01575.x [DOI] [PubMed] [Google Scholar]
- Liu F., Chen H., Galvan E. M., Lasaro M. A., Schifferli D. M. (2006). Effects of psa and F1 on the adhesive and invasive interactions of Yersinia pestis with human respiratory tract epithelial cells. Infect. Immun. 74, 5636–5644 10.1128/IAI.00612-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo L. A. (2006). Adhesive properties of the purified plasminogen activator pla of Yersinia pestis. FEMS Microbiol. Lett. 262, 158–162 10.1111/j.1574-6968.2006.00382.x [DOI] [PubMed] [Google Scholar]
- Lowry M. B., Duchemin A. M., Robinson J. M., Anderson C. L. (1998). Functional separation of pseudopod extension and particle internalization during Fc gamma receptor-mediated phagocytosis. J. Exp. Med. 187, 161–176 10.1084/jem.187.2.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukaszewski R. A., Kenny D. J., Taylor R., Rees D. G., Hartley M. G., Oyston P. C. (2005). Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect. Immun. 73, 7142–7150 10.1128/IAI.73.11.7142-7150.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makoveichuk E., Cherepanov P., Lundberg S., Forsberg Å., Olivecrona G. (2003). pH6 antigen of Yersinia pestis interacts with plasma lipoproteins and cell membranes. J. Lipid Res. 44, 320–330 10.1194/jlr.M200182-JLR200 [DOI] [PubMed] [Google Scholar]
- May R. C., Machesky L. M. (2001). Phagocytosis and the actin cytoskeleton. J. Cell Sci. 114, 1061–1077 [DOI] [PubMed] [Google Scholar]
- McGreal E. P., Miller J. L., Gordon S. (2005). Ligand recognition by antigen-presenting cell C-type lectin receptors. Curr. Opin. Immunol. 17, 18–24 10.1016/j.coi.2004.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méresse S., Steele-Mortimer O., Moreno E., Desjardins M., Finlay B., Gorvel J.-P. (1999). Controlling the maturation of pathogen-containing vacuoles: a matter of life and death. Nat. Cell Biol. 1, E183–E188 10.1038/15620 [DOI] [PubMed] [Google Scholar]
- Miller V., Bliska J., Falkow S. (1990). Nucleotide sequence of the Yersinia enterocolitica ail gene and characterization of the Ail protein product. J. Bacteriol. 172, 1062–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller V., Falkow S. (1988). Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect. Immun. 56, 1242–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller V. L., Beer K. B., Heusipp G., Young B. M., Wachtel M. R. (2001). Identification of regions of Ail required for the invasion and serum resistance phenotypes. Mol. Microbiol. 41, 1053–1062 10.1046/j.1365-2958.2001.02575.x [DOI] [PubMed] [Google Scholar]
- Mohammadi S., Isberg R. R. (2009). Yersinia pseudotuberculosis virulence determinants invasin, YopE, and YopT modulate RhoG activity and localization. Infect. Immun. 77, 4771–4782 10.1128/IAI.00850-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro L., Koller A., Nordfelth R., Wolf-Watz H., Taylor S., Dixon J. E. (2007). Identification of a molecular target for the Yersinia protein kinase a. Mol. Cell 26, 465–477 10.1016/j.molcel.2007.04.025 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F., Ravetch J. V. (2008). Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- Odin J. A., Edberg J. C., Painter C. J., Kimberly R. P., Unkeless J. C. (1991). Regulation of phagocytosis and [Ca2+]i flux by distinct regions of an Fc receptor. Science 254, 1785–1788 10.1126/science.1837175 [DOI] [PubMed] [Google Scholar]
- Paerregaard A., Espersen F., Skurnik M. (1991). Role of the Yersinia outer membrane protein YadA in adhesion to rabbit intestinal tissue and rabbit intestinal brush border membrane vesicles. APMIS 99, 226–232 10.1111/j.1699-0463.1991.tb05143.x [DOI] [PubMed] [Google Scholar]
- Parkhill J., Wren B. W., Thomson N. R., Titball R. W., Holden M. T., Prentice M. B., et al. (2001). Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413, 523–527 10.1038/35097083 [DOI] [PubMed] [Google Scholar]
- Pawel-Rammingen V., Telepnev M. V., Schmidt G., Aktories K., Wolf-Watz H., Rosqvist R. (2002). GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol. Microbiol. 36, 737–748 10.1046/j.1365-2958.2000.01898.x [DOI] [PubMed] [Google Scholar]
- Payne D., Tatham D., Williamson E. D., Titball R. W. (1998). The pH 6 antigen of Yersinia pestis binds to β1-linked galactosyl residues in glycosphingolipids. Infect. Immun. 66, 4545–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiser L., Mukhopadhyay S., Gordon S. (2002). Scavenger receptors in innate immunity. Curr. Opin. Immunol. 14, 123–128 10.1016/S0952-7915(01)00307-7 [DOI] [PubMed] [Google Scholar]
- Perry R. D., Fetherston J. D. (1997). Yersinia pestis–etiologic agent of plague. Clin. Microbiol. Rev. 10, 35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan J., Lee K., Cherry S., Tropea J. E., Burke T. R., Jr., Waugh D. S. (2003). High-resolution structure of the Yersinia pestis protein tyrosine phosphatase YopH in complex with a phosphotyrosyl mimetic-containing hexapeptide. Biochemistry 42, 13113–13121 10.1021/bi030156m [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda J., Cossart P. (2006). Bacterial adhesion and entry into host cells. Cell 124, 715–727 10.1016/j.cell.2006.02.012 [DOI] [PubMed] [Google Scholar]
- Pluddemann A., Mukhopadhyay S., Gordon S. (2006). The interaction of macrophage receptors with bacterial ligands. Expert Rev. Mol. Med. 8, 1–25 10.1017/S1462399406000159 [DOI] [PubMed] [Google Scholar]
- Prehna G., Ivanov M. I., Bliska J. B., Stebbins C. (2006). Yersinia virulence depends on mimicry of host rho-family nucleotide dissociation inhibitors. Cell 126, 869–880 10.1016/j.cell.2006.06.056 [DOI] [PubMed] [Google Scholar]
- Pujol C., Klein K. A., Romanov G. A., Palmer L. E., Cirota C., Zhao Z., et al. (2009). Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect. Immun. 77, 2251–2261 10.1128/IAI.00068-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roppenser B., Röder A., Hentschke M., Ruckdeschel K., Aepfelbacher M. (2009). Yersinia enterocolitica differentially modulates RhoG activity in host cells. J. Cell Sci. 122, 696–705 10.1242/jcs.040345 [DOI] [PubMed] [Google Scholar]
- Rosqvist R., Forsberg Å., Rimpiläinen M., Bergman T., Wolf-Watz H. (1990). The cytotoxic protein YopE of Yersinia obstructs the primary host defence. Mol. Microbiol. 4, 657–667 10.1111/j.1365-2958.1990.tb00635.x [DOI] [PubMed] [Google Scholar]
- Schmidt G. (2011). Yersinia enterocolitica outer protein T (YopT). Eur. J. Cell Biol. 90, 955–958 10.1016/j.ejcb.2010.12.005 [DOI] [PubMed] [Google Scholar]
- Schraven B., Reth M. (2007). Immunoreceptors and integrins. Immunol. Rev. 218, 5–8 10.1111/j.1600-065X.2007.00549.x [DOI] [PubMed] [Google Scholar]
- Sebbane F., Jarrett C. O., Gardner D., Long D., Hinnebusch B. J. (2006). Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci.U.S.A. 103, 5526–5530 10.1073/pnas.0509544103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao F. (2008). Biochemical functions of Yersinia type III effectors. Curr. Opin. Microbiol. 11, 21–29 10.1016/j.mib.2008.01.005 [DOI] [PubMed] [Google Scholar]
- Shao F., Vacratsis P. O., Bao Z., Bowers K. E., Fierke C. A., Dixon J. E. (2003). Biochemical characterization of the Yersinia YopT protease: cleavage site and recognition elements in Rho GTPases. Proc. Natl. Acad. Sci.U.S.A. 100, 904–909 10.1073/pnas.252770599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeinde O. A., Subrahmanyam Y., Stark K., Quan T., Bao Y., Goguen J. D. (1992). A surface protease and the invasive character of plague. Science 258, 1004–1007 10.1126/science.1439793 [DOI] [PubMed] [Google Scholar]
- Song Y., Tong Z., Wang J., Wang L., Guo Z., Han Y., et al. (2004). Complete genome sequence of Yersinia pestis strain 91001, an isolate avirulent to humans. DNA Res. 11, 179–197 10.1093/dnares/11.3.179 [DOI] [PubMed] [Google Scholar]
- Songsungthong W., Higgins M. C., Rolán H. G., Murphy J. L., Mecsas J. (2010). ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell. Microbiol. 12, 988–1001 10.1111/j.1462-5822.2010.01448.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg I., Hoffmann C., Dumbach J., Aktories K., Schmidt G. (2003). The C terminus of YopT is crucial for activity and the N terminus is crucial for substrate binding. Infect. Immun. 71, 4623–4632 10.1128/IAI.71.8.4623-4632.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa S., Lecuit M., Cossart P. (2005). Microbial strategies to target, cross or disrupt epithelia. Curr. Opin. Cell Biol. 17, 489–498 10.1016/j.ceb.2005.08.013 [DOI] [PubMed] [Google Scholar]
- Swanson J. A., Baer S. C. (1995). Phagocytosis by zippers and triggers. Trends Cell Biol. 5, 89–93 10.1016/S0962-8924(00)88956-4 [DOI] [PubMed] [Google Scholar]
- Taylor P. R., Martinez-Pomares L., Stacey M., Lin H. H., Brown G. D., Gordon S. (2005). Macrophage receptors and immune recognition. Annu. Rev. Immunol. 23, 901–944 10.1146/annurev.immunol.23.021704.115816 [DOI] [PubMed] [Google Scholar]
- Trasak C., Zenner G., Vogel A., Yüksekdag G., Rost R., Haase I., et al. (2007). Yersinia protein kinase YopO is activated by a novel G-actin binding process. J. Biol. Chem. 282, 2268–2277 10.1074/jbc.M610071200 [DOI] [PubMed] [Google Scholar]
- Trülzsch K., Sporleder T., Igwe E. I., Rüssmann H., Heesemann J. (2004). Contribution of the major secreted yops of Yersinia enterocolitica, O: 8 to pathogenicity in the mouse infection model. Infect. Immun. 72, 5227–5234 10.1128/IAI.72.9.5227-5234.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang T. M., Felek S., Krukonis E. S. (2010). Ail binding to fibronectin facilitates Yersinia pestis binding to host cells and Yop delivery. Infect. Immun. 78, 3358–3368 10.1128/IAI.00238-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon E., Martin R., Plumb J., Kwok V., Vandivier R. W., Glogauer M., et al. (2006). CD44 is a phagocytic receptor. Blood 107, 4149–4158 10.1182/blood-2005-09-3808 [DOI] [PubMed] [Google Scholar]
- van Zon J. S., Tzircotis G., Caron E., Howard M. (2009). A mechanical bottleneck explains the variation in cup growth during FcgammaR phagocytosis. Mol. Syst. Biol. 5, 298 10.1038/msb.2009.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viboud G. I., Mejía E., Bliska J. B. (2006). Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell. Microbiol. 8, 1504–1515 10.1111/j.1462-5822.2006.00729.x [DOI] [PubMed] [Google Scholar]
- Welkos S., Friedlander A., Davis K. (1997). Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain C092. Microb. Pathog. 23, 211–223 10.1006/mpat.1997.0154 [DOI] [PubMed] [Google Scholar]
- Wiley D. J., Nordfeldth R., Rosenzweig J., DaFonseca C. J., Gustin R., Wolf-Watz H., et al. (2006). The Ser/Thr kinase activity of the Yersinia protein kinase A (YpkA) is necessary for full virulence in the mouse, mollifying phagocytes, and disrupting the eukaryotic cytoskeleton. Microb. Pathog. 40, 234–243 10.1016/j.micpath.2006.02.001 [DOI] [PubMed] [Google Scholar]
- Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. (1990). CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431–1433 10.1126/science.1698311 [DOI] [PubMed] [Google Scholar]
- Yang Y., Merriam J. J., Mueller J. P., Isberg R. R. (1996). The psa locus is responsible for thermoinducible binding of Yersinia pseudotuberculosis to cultured cells. Infect. Immun. 64, 2483–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavialov A. V., Berglund J., Pudney A. F., Fooks L. J., Ibrahim T. M., MacIntyre S., et al. (2003). Structure and biogenesis of the capsular f1 antigen from Yersinia pestis: preserved folding energy drives fiber formation. Cell 113, 587–596 10.1016/S0092-8674(03)00351-9 [DOI] [PubMed] [Google Scholar]
- Zhang S., Park C. G., Zhang P., Bartra S. S., Plano G. V., Klena J. D., et al. (2008). Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J. Biol. Chem. 283, 31511 10.1074/jbc.M804646200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Y., Clemens J., Schubert H., Stuckey J., Fischer M., Hume D., et al. (1992). Expression, purification, and physicochemical characterization of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem. 267, 23759–23766 [PubMed] [Google Scholar]
- Zhou D., Qin L., Han Y., Qiu J., Chen Z., Li B., et al. (2006). Global analysis of iron assimilation and fur regulation in Yersinia pestis. FEMS Microbiol. Lett. 258, 9–17 10.1111/j.1574-6968.2006.00208.x [DOI] [PubMed] [Google Scholar]
- Zumbihl R., Aepfelbacher M., Andor A., Jacobi C. A., Ruckdeschel K., Rouot B., et al. (1999). The cytotoxin YopT of Yersinia enterocolitica induces modification and cellular redistribution of the small GTP-binding protein RhoA. J. Biol. Chem. 274, 29289–29293 10.1074/jbc.274.41.29289 [DOI] [PubMed] [Google Scholar]