

Abstract
H2O2 production by skeletal muscle mitochondria oxidizing palmitoylcarnitine was examined under two conditions: the absence of respiratory chain inhibitors and the presence of myxothiazol to inhibit complex III. Without inhibitors, respiration and H2O2 production were low unless carnitine or malate was added to limit acetyl-CoA accumulation. With palmitoylcarnitine alone, H2O2 production was dominated by complex II (44% from site IIF in the forward reaction); the remainder was mostly from complex I (34%, superoxide from site IF). With added carnitine, H2O2 production was about equally shared between complexes I, II, and III. With added malate, it was 75% from complex III (superoxide from site IIIQo) and 25% from site IF. Thus complex II (site IIF in the forward reaction) is a major source of H2O2 production during oxidation of palmitoylcarnitine ± carnitine. Under the second condition (myxothiazol present to keep ubiquinone reduced), the rates of H2O2 production were highest in the presence of palmitoylcarnitine ± carnitine and were dominated by complex II (site IIF in the reverse reaction). About half the rest was from site IF, but a significant portion, ~40 pmol H2O2 · min−1 · mg protein−1, was not from complex I, II, or III and was attributed to the proteins of β-oxidation (electron-transferring flavoprotein (ETF) and ETF-ubiquinone oxidoreductase). The maximum rate from the ETF system was ~200 pmol H2O2 · min−1 ~ mg protein−1 under conditions of compromised antioxidant defense and reduced ubiqui-none pool. Thus complex II and the ETF system both contribute to H2O2 production during fatty acid oxidation under appropriate conditions.
Keywords: Palmitate, Palmitoylcarnitine, ROS, Complex I, Complex II, Succinate dehydrogenase, Complex III, ETF, Electron-transferring flavoprotein, ETFQOR, Electron transferring flavoprotein–ubiquinone oxidoreductase, Free radicals
Fatty acid β-oxidation by skeletal muscle mitochondria is a major source of ATP under physiological conditions [1,2], and impaired lipid metabolism is associated with several pathological conditions, such as multi-acyl-CoA dehydrogenase deficiency, obesity-related insulin resistance, and type 2 diabetes [3]. Changes in reactive oxygen species (ROS)1 production in these pathological conditions have been reported [4–8].
Palmitoylcarnitine is a major substrate for β-oxidation. It enters mitochondria on the carnitine–acylcarnitine translocase and is converted to palmitoyl-CoA. The fatty acid β-oxidation spiral involves four sequential enzymes: acyl-CoA dehydrogenase, 2-enoyl-CoA hydratase, L-3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase [9]. In contrast to peroxisomal β-oxidation, in which molecular oxygen serves as the electron acceptor of acyl-CoA oxidases, mitochondrial acyl-CoA dehydrogenases transfer single electrons to electron-transferring flavoprotein (ETF) [10]. Singly reduced ETF− is then oxidized by ETF-ubiquinone oxido-reductase (ETFQOR), which donates electrons directly to the ubiquinone (Q) pool in the mitochondrial inner membrane [11–13], to be passed to complex III of the respiratory chain, cytochrome c, complex IV, and finally molecular oxygen. The second dehydrogenation reaction in the β-oxidation spiral is catalyzed by hydroxyacyl-CoA dehydrogenase, which uses NAD+ as its electron acceptor. The reduced NADH is then oxidized by complex I, reducing the Q pool. The end product of β-oxidation, acetyl-CoA, condenses with oxaloacetate to form citrate, which is then oxidized by the Krebs cycle. Thus fatty acid oxidation in mitochondria is linked to oxidative phosphorylation and ATP production.
In contrast to many other substrates, such as malate, glutamate, succinate, or glycerol 3-phosphate, oxidation of fatty acids requires four enzymatic reactions and donates electrons at multiple points in the electron transport chain: complex I, ETFQOR, and complex II (via formation of succinate in the Krebs cycle). This makes fatty acid oxidation a good candidate for high rates of superoxide or H2O2 formation due to possible leaks of electrons to molecular oxygen at several different sites.
There are several sites of superoxide or H2O2 production in the Krebs cycle and electron transport chain [14–16]. In order of maximum capacity in skeletal muscle mitochondria, they are the ubiquinol-oxidizing site of complex III (site IIIQo), the ubiquinone-reducing site of complex I (site IQ) and the flavin site of complex II (site IIF), the flavin site of complex I (site IF), and the Q-binding site of glycerol 3-phosphate dehydrogenase (GPDH) [17,18]. Other sites include the dihydrolipoate moieties of 2-oxoglutarate dehydrogenase and pyruvate dehydrogenase, and the ETF/ETFQOR system, but their maximum rates have not been established. The native rates from various sites in the absence of inhibitors have been measured only during oxidation of glutamate and malate, when sites IF and IIIQo dominate [19]; oxidation of succinate, when site IQ dominates [20,21]; and oxidation of glycerol 3-phosphate, when site IQ dominates and sites IIF and GPDH also contribute [17,18].
The sites of H2O2 production during oxidation of palmitoylcarnitine by skeletal muscle mitochondria have been addressed in earlier studies in isolated mitochondria [20,22–25]. Although mitochondria isolated from the complex cellular environment lose some inputs of metabolic control, this widely used model still provides a detailed understanding of mechanisms of metabolic regulation, including those related to mitochondrial ROS production. These insights then feed forward our understanding of more complex systems such as intact cells, tissues, or animals. Prior studies in isolated mitochondria have identified several different components of the respiratory chain and β-oxidation pathway as sources of H2O2 during palmitoylcarnitine oxidation by isolated mitochondria: site IQ [20,23], site IIIQo [20,22–24], ETF/ETFQOR [20,22–24], and acyl-CoA dehydrogenase [22,25]. One possible source of disagreement is the use of site-specific inhibitors of the respiratory chain. Such inhibitors are great tools for understanding the capacities and mechanisms of H2O2 production at sites of interest. However, inhibition of one center disrupts normal electron flow and may lead to changes in the reduction states of other centers far from the site of inhibition, with consequent changes in their production of superoxide or H2O2. In the presence of inhibitors it becomes impossible to measure changes in native rates of H2O2 production in different metabolic states or disease models, which is crucial for the investigation of ROS-related physiology and pathology. Therefore it is of high importance to resolve the contradicting conclusions and perform measurements in the absence of respiratory chain inhibitors (native rates) that more closely match physiological conditions in vivo.
The disadvantages of site-specific inhibitors can be partially avoided by using endogenous reporters to predict rates of superoxide formation from sites IF and IIIQo[19]. This approach assumes a unique relationship between the reduction state of the superoxide-producing species within the site and its rate of reaction with oxygen to generate superoxide (or H2O2). The reduction state of the superoxide producer can be detected by measuring the reduction state of an endogenous “reporter,” a species that is close to equilibrium with the superoxide-producing moiety. NADH was established as a reporter of rates of superoxide production at site IF (plus any other sites that respond to NADH reduction state) and cytochrome b566 as a reporter of superoxide production at site IIIQo[19].
For the first time we provide a complete analysis and dissection of the specific sites of superoxide/H2O2 production during fatty acid oxidation in the absence of respiratory chain inhibitors. We also investigate the contribution of the ETF/ETFQOR system to the rates of H2O2 production in the absence and presence of complex III inhibitors that lead to reduction of the Q pool and estimate the maximum rate of H2O2 production from this system in skeletal muscle mitochondria. We identify complex II as a new site of superoxide and/or H2O2 production that was not recognized in earlier studies and whose H2O2 production was previously wrongly attributed to other sites.
Experimental procedures
Animals, reagents, and mitochondrial preparation
Female Wistar rats (Harlan Laboratories), age 5–8 weeks, were fed chow ad libitum with free access to water. Skeletal muscle mitochondria were isolated in Chappell–Perry buffer (100 mM KCl, 50 mM Tris, 2 mM EGTA, pH 7.1 at 25 °C) by standard procedures [26]. Protein concentration was determined by the biuret method. The animal protocol was approved by the Buck Institute Animal Care and Use Committee, in accordance with IACUC standards. All reagents were from Sigma (St. Louis, MO, USA) except for Amplex UltraRed (Invitrogen, Carlsbad, CA, USA) and atpenin A5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The L-isomer of palmitoylcarnitine was used in all experiments.
H2O2 production
Rates of superoxide production were measured indirectly as rates of H2O2 production after conversion of superoxide to H2O2 by endogenous superoxide dismutase (SOD) in the matrix. H2O2 was detected using horseradish peroxidase oxidizing Amplex UltraRed to its fluorescent resorufin product [19,27]. Exogenous SOD was added to convert any superoxide released in the medium. Mitochondria (0.3 mg protein · ml−1) were suspended in a medium containing 120 mM KCl, 5 mM Hepes, 5 mM K2HPO4, 1 mM EGTA, and 0.3% (w/v) bovine serum albumin (pH 7.0 at 37 °C), together with 5 U · ml−1 horseradish peroxidase, 25 U · ml−1 SOD, and 50 μM Amplex UltraRed. The fluorescence signal was recorded using a Varian Cary Eclipse spectrofluorimeter (λex560 nm, λem590 nm) with constant stirring. Rates of fluorescence change were calibrated with known amounts of H2O2 and normalized to the amount of protein [27]. The rates of H2O2 production in the calibration curve in Fig. 6B (see below) were obtained by titrating rotenone in the presence of 4 μM FCCP and 5 mM malate [28].
Fig. 6.

H2O2 production during oxidation of palmitoylcarnitine (PC) plus carnitine plus malonate in the presence of the uncoupler FCCP. (A) Typical trace of H2O2 production in the presence of 4 μM FCCP during oxidation of palmitoylcarnitine plus carnitine plus malonate and effect of addition of myxothiazol. The trace shows biphasic behavior: a first phase of high rate (labeled “fast”) and a second phase of slower rate (labeled “slow”). Numbers by the trace represent rates of H2O2 production in pmol · min−1 · mg protein−1 (n=6). (B) Calibration of H2O2 production at site IF (plus any other sites that respond to NADH reduction state) as a function of reduction state of NAD(P)H in the presence of 4 μM FCCP (circles) (see Experimental procedures for details). The corresponding calibration in CDNB-treated mitochondria is also shown for comparison (triangles; data from [19]). (C) Total measured rates of H2O2 production (open bars) and predicted rates from site IF (black bars; predicted using the calibration curve in (B)) during oxidation of palmitoylcarnitine plus carnitine, malonate, and myxothiazol. Values are means ± SEM (n=6). p values were calculated using Welch’s t test.
NAD(P) reduction state
Experiments were performed using 0.3 mg mitochondrial protein · ml−1 at 37 °C in the same medium as for H2O2 measurements. The reduction state of endogenous NAD(P) was determined by autofluorescence [28] using a Shimadzu RF5301-PC spectrofluorimeter at λex 365 nm, λem 450 nm. NAD(P) was assumed to be 0% reduced after 5 min without added substrate and 100% reduced with 5 mM malate and 4 μM rotenone (Fig. 2A). Intermediate values were determined as %NAD(P)H relative to the 0 and 100% values. Although this technique measures contribution from both mitochondrial NADH and mitochondrial NADPH, the content of NAD+ plus NADH in skeletal muscle mitochondria is much greater than the combined NADP+ and NADPH [28,29]. Moreover the enhancement of NADH fluorescence in mitochondria is two- to fourfold greater than it is for mitochondrial NADPH [30]. The higher content and greater fluorescence enhancement of NADH make the autofluorescence signal predominantly a measure of NADH. The NAD(P)+ reduction states in the calibration curve in Fig. 6B (see below) were manipulated by varying the concentration of rotenone in the presence of 5 mM malate and 4 μM FCCP [28].
Fig. 2.
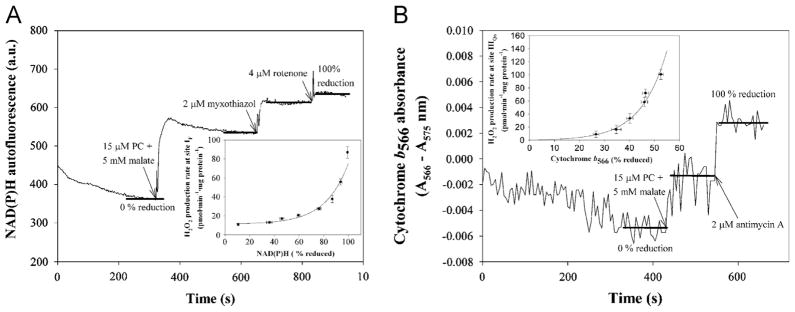
Reduction states of NAD(P) and cytochrome b566 as reporters of superoxide production from sites IF (plus any other sites that respond to NADH reduction state) and IIIQo, respectively. Typical traces of (A) NAD(P)H autofluorescence and (B) cytochrome b566 absorbance in suspensions of skeletal muscle mitochondria (0.3 mg protein · ml−1 in (A) and 1.5 mg protein · ml−1 in (B)). The oxidized signal was established after 5 min incubation in the absence of substrates. Substrates were then added to establish the reduction state to be measured (in the example illustrated, 15 μM palmitoylcarnitine (PC) and 5 mM malate), followed by the addition of inhibitors (2 μM myxothiazol in (A)). 100% reduction was achieved by addition of 4 μM rotenone (A) or 2 μM antimycin A (B) in each run to calibrate the scale. 100% reduction in (B) was sometimes measured instead by adding 5 mM succinate plus 4 μM rotenone and 2 μM antimycin A and this value was used to calculate intermediate % reduction with other substrates during the experimental day. Insets are from [19]. Inset (A), calibration of the rate of H2O2 production at site IF (pmol · min−1 · mg protein−1) as a function of % reduction of NAD(P). Inset (B), calibration of the rate of H2O2 production at site IIIQo(pmol · min−1 · mg protein−1) as a function of % reduction of cytochrome b566.
Cytochrome b566 reduction state
Experiments were performed at 1.5 mg mitochondrial protein · ml−1 in the same medium as for H2O2 measurements. The reduction state of endogenous cytochrome b566 was measured with constant stirring at 37 °C in an Olis DW-2 dual-wavelength spectrophotometer as A566 nm–A575 nm [27]. The signal at this wavelength pair reports ~75% cytochrome b566 and ~25% cytochrome b562 [27,31]. Cytochrome b566 was assumed to be 0% reduced after 5 min without added substrate and 100% reduced with 5 mM succinate and 4 μM rotenone or with 15 μM palmitoylcarnitine plus 5 mM malate, both in the presence of 2 μM antimycin A (Fig. 2B, see below). Intermediate values were determined as %b566 reduced relative to the 0 and 100% values. At least 15 data points were used to calculate the average percentage reduction under each condition.
Respiration
Experiments were performed in the same medium as H2O2 measurements at 0.3 mg mitochondrial protein · ml−1. Oxygen consumption was recorded at 37 °C using a Clark electrode. State 2 respiration was in the presence of substrates only. Phosphorylating (State 3) respiration was initiated by addition of 0.5 mM ADP. Nonphosphorylating (State 4) respiration was recorded after addition of 1 μg · ml−1 oligomycin and uncoupled respiration was measured after subsequent addition of 1 μM FCCP.
Complex II activity
Experiments were performed strictly as in [17] but with the buffer used in the present study. Activity was measured as the rate of reduction of 50 μM dichlorophenolindophenol (DCPIP) in the presence of 1 mM phenazine methosulfate instigated by addition of 10 mM succinate at 15 °C to freeze the activation state.
Endogenous reporters of superoxide production
The method is described in [19]. Briefly, the reduction state of NAD(P)+ was used to predict superoxide production from site IF from the calibration curve in Fig. 2A (inset, see below) for all data except Figs. 6C and 7 (ETF/ETFQOR system), which used the calibration in Fig. 6B (see below). The reduction state of cytochrome b566 was used to predict superoxide production from site IIIQo using the calibration curve in Fig. 2B (inset, see below).
Fig. 7.

Maximum rates of H2O2 production from characterized sites of the mitochondrial respiratory chain. IF, flavin site of complex I (plus any other sites that respond to NADH reduction state); IQ, ubiquinone binding site of complex I; IIF, flavin site of complex II; IIIQo, Qo binding site of complex III; mGPDH, mitochondrial glycerol 3-phosphate dehydrogenase; ETF/ETFQOR, electron-transferring flavoprotein/electron-transferring flavoprotein–ubiquinone oxidoreductase system. Data are from [19] except that for ETF/ETFQOR, which is from Fig. 6C.
Statistics and curve fitting
Data are presented as means ± SEM. Differences between groups were assessed by Student’s t test using SigmaPlot version 11. One-way ANOVA was used to compare differences in H2O2 production after antimycin A treatment (Fig. 4A and B). Calibration curves in Fig. 2A and B were single exponential fits [19]. The calibration curve in Fig. 6B was fit poorly by a single exponential, so it was fitted empirically using Eq. (1), which assumes that the reporter (NADH) and the superoxide producer (FADH2) have different potentials but are close to equilibrium:
| (1) |
where y is the rate of H2O2 production (pmol · min−1 · mg protein−1); x is the reduction state of the NAD+ pool (% NAD(P)H); k=0.0033 · pmol · %−1 · min−1 · mg protein−1, a scaling factor expressing the rate constant of H2O2 formation; and a = 2.05, a parameter proportional to the difference in midpoint potentials. The errors caused by the calibrations were calculated for each x value as the root mean square of the linearly interpolated SEM of the x value propagated through the calibration equation and linearly interpolated SEM of the y value.
Fig. 4.
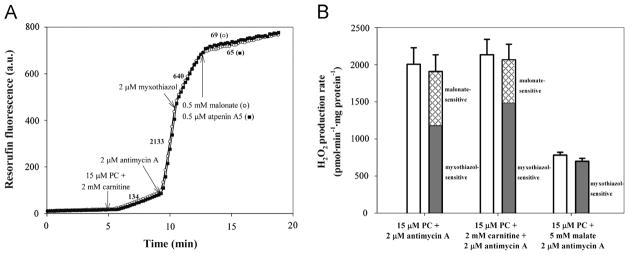
H2O2 production during oxidation of palmitoylcarnitine (PC) in the presence of the complex IIIQi inhibitor antimycin A. (A) Typical traces of H2O2 production during oxidation of palmitoylcarnitine plus carnitine and effects of inhibitors. Substrates and inhibitors were added as shown. Numbers by the traces represent rates of H2O2 production in pmol · min−1 · mg protein−1 (n=6). (B) Total (open bars), myxothiazol-sensitive (gray bars), and malonate-sensitive (cross-hatched bars) rates of H2O2 production during oxidation of palmitoylcarnitine alone and in combination with carnitine or malate, all in the presence of antimycin A. Values are means ± SEM (n=6).
Errors for predicted rates of H2O2 production (Figs. 3, 5B, 5C, and 6C) were calculated as the root mean square of the calibration errors above and the propagated SEMs of the reporter level through the calibration equation. Exact equations are in [19]. Welch’s t test was used to compare measured and predicted values in Figs. 3, 5B, 5C, and 6C.
Fig. 3.

Comparison of the measured rate of H2O2 production and the sum of the predicted rates from sites IF, IIF, and IIIQo under native conditions. Measured rates are from Fig. 1B and predicted rates are from Table 2. p values were determined by Welch’s t test. The contribution of site IIF was corrected for changes at the other sites using the changes in the reduction states of NAD(P)+ and cytochrome b566 upon addition of malonate. PC, palmitoylcarnitine.
Fig. 5.

Measured and predicted rates of H2O2 production during oxidation of palmitoylcarnitine (PC) in the presence of the complex IIIQo inhibitor myxothiazol. (A) Typical traces of H2O2 production during oxidation of palmitoylcarnitine plus carnitine and effects of inhibitors. Substrates and inhibitors were added as shown. Numbers by the traces represent rates of H2O2 production in pmol · min−1 · mg protein−1 (n=6). (B) Total measured rates of H2O2 production (open bars) and predicted rates from sites IF (plus any other sites that respond to NADH reduction state) (black bars; predicted from the NAD(P) reduction state using the calibration in Fig. 2A inset) and IIF (striped bars, predicted from the decrease in rate caused by addition of malonate after correction for changes in the predicted rate from site IF after addition of malonate) during oxidation of palmitoylcarnitine alone and in combination with carnitine or malate, all in the presence of myxothiazol. Values are means ± SEM (n=6). p values were calculated by Welch’s t test. (C) Total measured rates of H2O2 production (open bar) and predicted rates from site IF (black bar; predicted as in (B)) during oxidation of palmitoylcarnitine alone and in combination with carnitine or malate, all in the presence of myxothiazol and malonate. Values are means ± SEM (n=6). p values were calculated by Welch’s t test.
Results
Oxygen consumption by skeletal muscle mitochondria oxidizing palmitoylcarnitine
Table 1 summarizes the respiration rates of mitochondria on succinate plus rotenone and on three substrate combinations commonly used to assess β-oxidation. Respiration on succinate had appropriate high rates and respiratory control. Palmitoylcarnitine alone supported very slow respiration that was not stimulated by ADP or uncoupler, presumably because 3-ketoacyl thiolase, the last reaction of β-oxidation, was limited by the high acetyl-CoA/CoA ratio [32].
Table 1.
Oxygen consumption by rat skeletal muscle mitochondria.
| Substrate | Oxygen consumption (nmol O · min−1 · mg protein−1)
|
|||
|---|---|---|---|---|
| State 2 | State 3 | State 4O | Uncoupled | |
| 5 mM succinate + 4 μM rotenone | 133±14 | 654±39 | 212±18 | 478±24 |
| 15 μM palmitoylcarnitine | 11±1 | Unchanged | Unchanged | Unchanged |
| 15 μM palmitoylcarnitine + 2 mM carnitine | 57±6 | 96±15 | 72±8 | 100±15 |
| 15 μM palmitoylcarnitine + 5 mM malate | 71±10 | 376±28 | 102±6 | 374±34 |
Mitochondria were incubated at 0.3 mg protein · ml−1 as described under Experimental procedures. State 2 respiration was measured after substrate addition. Subsequent additions were 0.5 mM ADP (to initiate State 3), 1 μg · ml−1 oligomycin (State 4O), and 1 μM FCCP (uncoupled respiration). Values are means ±SEM for n = 3 independent experiments.
Addition of carnitine rescues this condition by accepting acetyl groups from acetyl-CoA to generate acetylcarnitine and CoA in a reaction catalyzed by carnitine acetyltransferase. The acetylcarnitine is then exported in exchange for incoming carnitine. Carnitine acetyltransferase activity is much higher in mitochondria from skeletal muscle (410 μmol · min−1 · mg protein−1) than from liver (5 μmol · min−1 · mg protein−1) [33], and there is correspondingly increased respiration on palmitoylcarnitine after addition of carnitine in mitochondria from skeletal muscle but not liver [24]. Table 1 shows that inclusion of carnitine stimulated oxidation of palmitoylcarnitine sufficiently to allow respiratory control. The combination of palmitoylcarnitine plus carnitine was attractive for this study because its metabolism may generate H2O2 without overt involvement of Krebs cycle enzymes.
Addition of malate also rescues oxidation of palmitoylcarnitine, by generating oxaloacetate through malate dehydrogenase. Citrate synthase then condenses oxaloacetate and acetyl-CoA to form citrate, releasing CoA. Table 1 shows that palmitoylcarnitine plus malate supported substantial phosphorylating and uncoupled respiration and good respiratory control.
Thus, oxidation of palmitoylcarnitine, supplemented by either carnitine or malate, provides sufficient electron flux to establish these physiologically relevant substrate combinations as suitable for further investigation.
H2O2 production during oxidation of palmitoylcarnitine in the absence of respiratory chain inhibitors
Fig. 1A shows that addition of palmitoylcarnitine at 5 min caused a measureable increase in the rate of Amplex UltraRed oxidation to resorufin; this was subsequently calibrated by additions of H2O2 to give the rate of H2O2 production. The open bars in Fig. 1B show the overall rates of H2O2 production measured in this way during oxidation of each of the fatty acid substrate combinations in Table 1. Oxidation of palmitoylcarnitine alone produced H2O2 at a low rate. Addition of malate or carnitine increased the rate several fold. The maximum rate was found with palmitoylcarnitine plus carnitine.
Fig. 1.
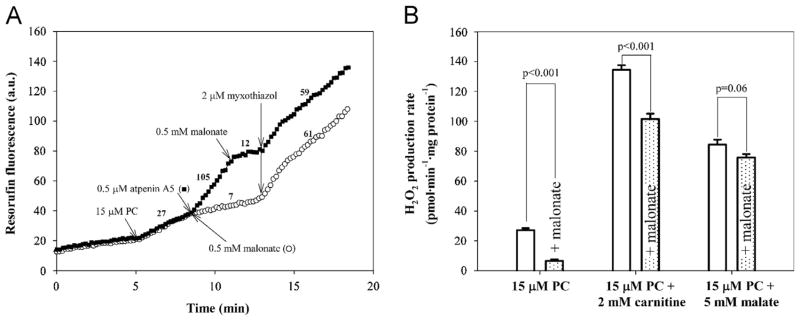
Effects of complex II inhibitors, malonate and atpenin A5, on rates of H2O2 production by rat skeletal muscle mitochondria oxidizing palmitoylcarnitine alone and in combination with carnitine or malate. (A) Typical traces of H2O2 production during palmitoylcarnitine oxidation and effects of complex II inhibitors. Palmitoylcarnitine (PC) was added after 5 min incubation of mitochondria (0.3 mg protein · ml−1) with the Amplex UltraRed detection system plus exogenous SOD. The background rates in the absence of substrate were subtracted from all rates after substrate addition. Numbers by the traces represent rates of H2O2 production in pmol · min−1 · mg protein−1 after calibration by addition of known amounts of H2O2. The addition of atpenin A5 increased the rate of H2O2 production during oxidation of palmitoylcarnitine from 27 ±1 (n=6) to 105±5 (n=4) pmol · min−1 · mg protein−1. After addition of malonate it decreased to 7±1 pmol · min−1 · mg protein−1 (n=4). Subsequent numbers are rates for the presented traces (n=1). (B) Effects of malonate addition on the rates of H2O2 production during oxidation of substrates from Table 1. Open bars, no malonate added; shaded bars, 0.5 mM malonate added after substrate. Values are means ± SEM. n=4 (PC ± malonate), n=6 (PC + carnitine ± malonate), n=5 (PC + malate ± malonate). p values were calculated using an unpaired Student t test.
Next, we determined the individual sites of H2O2 production with these substrate combinations. Even though respiration on palmitoylcarnitine alone was slow, we included it because, surprisingly, site IIF of complex II was the major H2O2 producer under this condition (Figs. 3 and 5B below).
Complex II generates superoxide and/or H2O2 in the forward reaction during palmitoylcarnitine oxidation in the absence of respiratory inhibitors
Addition of malonate, a specific inhibitor of succinate oxidation, significantly decreased the rates of H2O2 production with palmitoylcarnitine (Fig. 1A and B) and with palmitoylcarnitine plus carnitine, but had little effect on the rate with palmitoylcarnitine plus malate (Fig. 1B, shaded bars). These observations can be explained by generation of superoxide or H2O2 at complex II, which is sensitive to malonate during oxidation of succinate or glycerol3-phosphate in the presence of rotenone (to inhibit complex I) and myxothiazol (to inhibit complex III) in skeletal muscle mitochondria [17,18]. Malonate sensitivity of H2O2production during oxidation of palmitoylcarnitine has not been described before and is explored in more detail below.
We propose that under these conditions addition of palmitoylcarnitine (±carnitine) triggers superoxide or H2O2 production from site IIF using electrons from endogenous succinate, but addition of palmitoylcarnitine plus malate does not. This hypothesis is supported by several observations.
First, measurements of complex II activity showed that palmitoylcarnitine± carnitine indirectly activated the enzyme, explaining the trigger mechanism. To calibrate, incubation of mitochondria with 5 mM succinate plus rotenone and myxothiazol at 37 °C gave high complex II activity (80±6 nmol DCPIP · min−1 · mg · protein−1), and incubation without succinate gave low activity (40±4 units). Incubation with palmitoylcarnitine alone without rotenone or myxothiazol gave an activity of 64±8 units and incubation with palmitoylcarnitine plus carnitine gavean activity of 61±8 units (all values mean±SEM, n=3). This substantial activation of complex II by palmitoylcarnitine can be explained by removal of inhibitory oxaloacetate by condensation with acetyl-CoA formed by β-oxidation. In contrast, malate oxidation produces excess inhibitory oxaloacetate and prevented superoxide and/or H2O2 production from site IIF when palmitoylcarnitine plus malate was used as substrate.
Second, atpenin A5, an inhibitor of the ubiquinone binding site of complex II [34], did not prevent H2O2 production with palmitoylcarnitine as substrate, but stimulated it. It is known that H2O2 production from site IIF in the forward reaction from succinate is not prevented byatpenin A5 (unlike the reverse reaction from Q), but instead is stimulated as the flavin of complex II becomes more reduced [17]. Fig. 1A shows that addition of atpenin A5 increased the rate of H2O2 production in the presence of palmitoylcarnitine alone, whereas malonate decreased the rate. The stimulated rate in the presence of atpenin A5 was abolished by subsequent addition of malonate (Fig. 1A), showing that it emanated from site IIF operating in the forward reaction from succinate (because the reverse reaction from QH2 was inhibited by atpenin A5). The stimulation of H2O2 production by subsequent addition of myxothiazol is addressed below (Fig. 5). Therefore, malonate but not atpenin A5 was used under native conditions below to inhibit H2O2 production from site IIF.
We conclude from Fig. 1 that site IIF in the forward reaction produced ~25% of the observed H2O2 during oxidation of palmitoylcarnitine plus carnitine and ~75% during oxidation of palmitoylcarnitine alone. When malate was present and the substrate-binding site of complex II was inhibited by oxaloacetate, oxidation of palmitoylcarnitine did not result in significant H2O2 production from site IIF. After correction for small changes in H2O2 production from sites IF and IIIQo after addition of malonate (see Fig. 3 below), the contribution of site IIF become 36% for palmitoylcarnitine plus carnitine and 44% for palmitoylcarnitine alone.
Quantitative contributions of sites IF, IIIQo, and IIF to superoxide and/or H2O2 production during oxidation of palmitoylcarnitine
Endogenous reporters can predict the rates of superoxide production from sites IF and IIIQo in the absence of site-specific respiratory inhibitors (e.g., rotenone, antimycin A, myxothiazol, or stigmatellin) [19]. We used this technique in this work. We measured the reduction state of endogenous NAD(P) (Fig. 2A), to report superoxide production from site IF (plus any other sites that respond to NADH reduction state), and cytochrome b566 (Fig. 2B), to report superoxide production from site IIIQo. The corresponding calibration curves are shown as insets in Fig. 2A and B. Table 2 presents the reduction states of NAD(P) and cytochrome b566under the conditions of Fig. 1B and the rates of H2O2 production from sites IF and IIIQo predicted from the calibration curves.
Table 2.
Rates of H2O2 production from sites IF (plus any other sites that respond to NADH reduction state), IIIQo, and IIF predicted from the reduction states of NAD(P)H and cytochrome b566 and the inhibition by malonate in rat skeletal muscle mitochondria with various substrate combinations.
| Substrate | Site IF
|
Site IIIQo
|
Site IIF corrected H2O2 (pmol · min−1 · mg protein−1) | ||
|---|---|---|---|---|---|
| NAD(P)H (% reduced) | H2O2 predicted (pmol · min−1 · mg protein−1) | Cytochrome b566 (% reduced) | H2O2 predicted (pmol · min−1 · mg protein−1) | ||
| 15 μM palmitoylcarnitine | 15.1±0.7 (6) | 11.9±1.4 | 26.1±2.4 (6) | 7.8±5.4 | 15.5±7.5 |
| 15 μM palmitoylcarnitine + 2 mM carnitine | 79.9±0.7 (6) | 32.9±2.8 | 42.2±3.0 (6) | 39.5±14.5 | 39.6±21.0 |
| 15 μM palmitoylcarnitine + 5 mM malate | 65.7±1.4 (6) | 21.3±1.8 | 47.5±2.3 (6) | 66.2±18.0 | |
| 15 μM palmitoylcarnitine + 0.5 mM malonate | 13.8±0.8 (5) | 11.8±1.4 | 16.6±1.0 (4) | 2.7±4.9 | |
| 15 μM palmitoylcarnitine + 2 mM carnitine + 0.5 mM malonate | 79.3±1.1 (6) | 32.2±2.8 | 43.8±2.4 (6) | 46.2±14.4 | |
The reduction states of NAD(P) and cytochrome b566 were measured under the conditions of Figs. 1 and 2. The rates of H2O2 production from sites IF and IIIQo were predicted from the calibration curves in Fig. 2A and B (insets). To calculate the corrected rates from site IIF, the measured decreases in H2O2 production rate caused by addition of malonate in Fig. 1B were corrected for the changes in rate from sites IF and IIIQo predicted from the changes in reduction state of the endogenous reporters ± malonate. Values are means ± SEM (n independent experiments). The SEM of the predicted rates of H2O2 production from sites IF and IIIQo were calculated by propagation of calibration and measurement errors using the equations in [19]. The SEM of the corrected rate from site IIF was calculated as SEM = √(SEMa2 + SEMb2 + SEMc2), where SEMa, SEMb, and SEMc represent the errors of the predicted rates from sites IF and IIIQo before and after malonate addition and the SEM of the rate from site IIF assessed as the malonate-sensitive rate in Fig. 1B.
The effect of malonate addition on the reduction of the reporters and the predicted rates of H2O2 production is also presented in Table 2. Malonate had no discernible effect on the predicted rate from site IF, but it lowered the predicted rate from site IIIQo with palmitoylcarnitine alone and raised it with palmitoylcarnitine plus carnitine (although these effects were not statistically significant). These data allowed the raw decrease in H2O2 production rate at site IIF caused by addition of malonate (Fig. 1B) to be corrected for consequent changes in H2O2 production rate at sites IF and IIIQo. The corrected values for site IIF are shown in Table 2.
For each substrate combination, Fig. 3 shows the predicted rates of H2O2 production from sites IF, IIIQo, and IIF from Table 2 (black, gray, and striped bars) and compares them to the empirical rates (open bars) from Fig. 1B. During oxidation of palmitoylcarnitine alone, site IIF was the dominant site, responsible for 44% of the total predicted rate of H2O2 production. Site IF produced 34% and site IIIQo produced 22% of the total predicted rate. When palmitoylcarnitine was oxidized with carnitine supplementation, the contributions from sites IF (29%), IIIQo (35%), and IIF (36%) were approximately equal. When palmitoylcarnitine plus malate was used as substrate, site IIF did not contribute (because it was inhibited by the added malate). Site IIIQo was the major superoxide producer (75%), with the remaining 25% contributed by site IF.
In each case, the predicted rates fully accounted within experimental error for the empirical rates, demonstrating that essentially all the H2O2 produced during oxidation of palmitoylcarnitine in the absence of respiratory chain inhibitors came from these three sites, and any contribution from other sites was below the detection limit of this methodology.
Thus, we have identified and quantified the native sites of superoxide or H2O2 production during fatty acid oxidation by rat skeletal muscle mitochondria in the absence of respiratory chain inhibitors and absence of ATP synthesis. Site IIF of complex II was identified as one of the major contributors. The ETF/ETFQOR system, other enzymes of the β-oxidation spiral, and site IQ were not shown to be major sources of H2O2 under any condition examined, because there were no significant differences between the observed rates and the sum of the rates predicted by the endogenous reporters or prevented by inhibition of site IIF.
Rates of H2O2 production with palmitoylcarnitine plus antimycin A
We now switch from conditions with the Q pool relatively oxidized to studies with complex III inhibited to keep the QH2/Q ratio high. This condition was used in earlier studies and revealed very high rates of H2O2 production from unknown sites. It was proposed to originate from complexes upstream of the Q pool such as ETF, ETFQOR, and acyl-CoA dehydrogenase [20,22,24]. We did not find that these enzymes produced measureable superoxide or H2O2 in the absence of respiratory inhibitors (above). Here we explore if reduction of the Q pool can cause high rates of H2O2 production from these or other upstream centers to reveal the mechanism of superoxide and/or H2O2 production from fatty acid oxidation-specific enzymes.
Fig. 4A shows that addition of antimycin A, an inhibitor of the Qi site of complex III, caused very rapid H2O2 production with palmitoylcarnitine plus carnitine as substrate. Rates with palmitoylcarnitine alone were similar, but rates with palmitoylcarnitine plus malate were lower (Fig. 4B, open bars).
We estimated the contributions of sites IIIQo and IIF in each case. Antimycin A inhibition of the Qi site enhances superoxide production from site IIIQo, which is prevented by Qo site inhibitors, including myxothiazol [27,35–37]. Fig. 4 shows that addition of myxothiazol strongly inhibited H2O2 production with all three substrate combinations, showing that site IIIQo was a major contributor under these conditions, as expected. However, with palmitoylcarnitine ± carnitine, myxothiazol inhibition was incomplete, suggesting that sites other than IIIQo were also generating H2O2. The contribution of site IIF to the residual rates was estimated by adding malonate. This largely abolished H2O2 production (Fig. 4), suggesting that most of the myxothiazol-insensitive rate originated not from the ETF system but from complex II.
Thus, the majority of H2O2 production by muscle mitochondria incubated with palmitoylcarnitine ± carnitine in the presence of antimycin A arose from sites IIIQo and IIF. The important contribution from site IIF explains why there was a lower overall rate and a greater proportional contribution from site IIIQo in the presence of malate (Fig. 4B): malate completely inhibited this contribution of site IIF.
To quantify fully the contributions of each site in the presence of antimycin A, any effects of myxothiazol and malonate on the reduction state of site IF should be measured using the changes in NAD(P)H and corrected for, as in Table 2 and Fig. 3 (complex III stays fully reduced under these conditions, so its contribution to the signal does not change when malonate is added). Unfortunately, the strong fluorescence of antimycin A at 450 nm overlaps NAD(P)H autofluorescence, making it hard to correct for changes at site IF accurately with antimycin A present. Therefore, we used myxothiazol alone in all following experiments.
Rates of H2O2 production with palmitoylcarnitine plus myxothiazol
Fig. 5 shows the rates of H2O2 production with myxothiazol alone (to prevent superoxide formation at site IIIQo yet still allow correction for any changes in superoxide production at site IF after addition of inhibitors). These rates were slower than those with antimycin A plus myxothiazol (Fig. 4), which can be explained by more complete inhibition of complex III and higher QH2/Q ratio when both inhibitors were present. Addition of malonate very strongly inhibited the rates with palmitoylcarnitine ± carnitine, but not with palmitoylcarnitine plus malate. Measurement of NAD(P)+ reduction state allowed prediction of the rates from site IF under each condition using the calibration curve in Fig. 2A (inset) and correction of the rates from site IIF for the small changes in the rate from site IF on addition of malonate.
Fig. 5B reports the contributions of sites IF and IIF to H2O2 production in the presence of myxothiazol. With palmitoylcarnitine± carnitine, H2O2 production was dominated by site IIF, which accounted for ~70% of the total measured rates. Site IF produced only a small amount,~4% of the totals. When palmitoylcarnitine plus malate was used as substrate, site IIF did not contribute (because it was inhibited by the added malate), and site IF was the major superoxide producer (68% of the total measured rate).
In the presence of myxothiazol, H2O2 production from site IIF with palmitoylcarnitine was depressed by addition of carnitine (Fig. 5B). This can be explained by a measured decrease in the activity of complex II from 58.2±6.1 to 45.6±6.5 nmol DCPIP min−1 mg protein−1(n=3) under these conditions. This was presumably because decreased flux through the ETF pathway in the presence of myxothiazol stalled β-oxidation and lowered acetyl-CoA production, allowing removal of acetyl-CoA by carnitine and accumulation of inhibitory oxaloacetate.
From Fig. 1A we concluded that site IIF produced H2O2 in the forward reaction from succinate during palmitoylcarnitine oxidation under native conditions, because the rate was inhibited by malonate (which inhibits at the flavin site) but stimulated by atpenin A5 (which inhibits at the Q-binding site of complex II). However, the situation was different in the presence of antimycin A (Fig. 4A) or myxothiazol (Fig. 5A), when the Qpool was much more reduced. In each case, atpenin A5 inhibited H2O2 production as effectively as malonate, showing that under these conditions the route of electrons into site IIF was by the reverse reaction, from the Q pool.
Importantly, unlike the native condition (Fig. 3), in the presence of myxothiazol there was a clear and significant difference between the empirical rate and the sum of the predicted rates of H2O2 production, with palmitoylcarnitine plus either carnitine or malate as substrate (Fig. 5B). This H2O2 production was not from site IF, IQ, IIF, or IIIQo, because these sites were either accounted for or inhibited directly (or, for site IQ, inhibited indirectly by abolishing proton-motive force).
To investigate this unassigned rate more closely, in Fig. 5C we analyzed the residual rate after myxothiazol addition when complex II was inhibited from the start of the experiment (i.e., malonate added together with substrate). This avoided errors introduced in correcting for contributions from other sites and led to a more significant difference between the empirical and the predicted rates, even though NAD(P) was more reduced after myxothiazol addition (because the escape of electrons to complex II was prevented), and superoxide production from site IF was increased (Fig. 5C).
Thus, complex II produces superoxide and/or H2O2 at very high rates by the reverse reaction at site IIF using electrons from the Q pool when mitochondria oxidize palmitoylcarnitine ± carnitine in the presence of antimycin A or myxothiazol. This novel finding has to be taken into account in interpreting other studies using fatty acids as substrate. In the presence of palmitoylcarnitine plus carnitine, malonate, and myxothiazol there was a highly significant rate of H2O2 production that was not accounted for by site IF, IQ, IIF, or IIIQo (Fig. 5C); we argue below that it originated in the ETF/ETFQOR system.
Maximum rate of H2O2 production by the putative ETF/ETFQOR system
We searched for conditions that maximized H2O2 production dependent on the presence of palmitoylcarnitine that was unattributed to characterized sites. High rates were obtained in the presence of the uncoupler FCCP. This approach had two benefits. (i) Uncoupling oxidized the electron transport chain and therefore enzymes involved in electron supply were not inhibited by their products. Subsequent addition of myxothiazol caused a strong reduction of the Q pool and upstream redox centers (NAD(P)+, ETFQOR, ETF). (ii) Uncoupling compromised the antioxidant defense systems by decreasing NAD(P)H formation by energy-dependent transhydrogenase and reduction of the glutathione pool [38].
In uncoupled mitochondria respiring on palmitoylcarnitine plus carnitine in the presence of malonate to inhibit site IIF, myxothiazol addition caused a high initial rate of H2O2 production. This decreased to a lower rate after a few minutes (Fig. 6A). The possible mechanism of this biphasic kinetics is discussed below. Calculation of the contribution of site IF required a calibration curve obtained under the same conditions as a compromised antioxidant defense system. Fig. 6B shows the dependence of H2O2 production from site IF on NAD(P)+ reduction state in the presence of FCCP. The calibration curve superimposed that obtained after treatment of mitochondria with1-chloro-2,4-dintrobenzene (CDNB) to deplete glutathione [19], consistent with the interpretation that FCCP also compromised the glutathione pool.
Fig. 6C shows the empirical rates in the fast and slow phases and the predicted contributions of site IF, the only characterized site that was active under these conditions. The final nonpredicted rate of H2O2 production in the fast phase was 210±11 pmol · min−1 · mg protein−1, which probably originated from the ETF/ETFQOR system.
Fig. 7 compares the rate that we have assigned to ETF/ETFQOR to the maximum rates of superoxide/H2O2 production from the other characterized sites. The maximum rate from ETF/ETFQOR was much less than the maximum capacities of sites IIIQo, IQ, and IIF, but comparable to the maximum capacities of site IF and glycerol 3-phosphate dehydrogenase.
Discussion
We have characterized the rates and sites of origin of H2O2 production in isolated rat skeletal muscle mitochondria oxidizing substrates of β-oxidation under two conditions: the absence of respiratory inhibitors (Fig. 3) and with complex III inhibited to reduce the Q pool (Figs. 4 and 5B).
In contrast to previous studies of fatty acid oxidation (see the introduction) we used endogenous reporters in the absence of respiratory inhibitors to pinpoint sites of superoxide and/or H2O2 production under native conditions. We found that the flavin site of complex I (site IF) (plus any other sites that respond to NADH reduction state) and the Qo site of complex III (site IIIQo) produced much of the superoxide during oxidation of palmitoylcarnitine. For the first time, the flavin site of complex II (site IIF) was found to produce superoxide and/or H2O2 in the forward reaction at substantial rates during oxidation of palmitoylcarnitine ± carnitine (Fig. 3). There was no significant contribution by any other site.
With complex III inhibited using antimycin A, site IIIQo was the major site, and site IIF produced superoxide and/or H2O2 at a high rate in the reverse reaction from the reduced Q pool (unless it was inhibited by added malate; Fig. 4B). When complex III was inhibited by myxothiazol to eliminate site IIIQo, site IIF was dominant, with only a small contribution from site IF (Fig. 5B). Another site, probably the ETF/ETFQOR system, contributed significantly to H2O2 production under these conditions, with a maximum rate of 210 pmol · min−1· mg protein−1under optimal conditions (Fig. 6C).
Now we will consider each site in turn:
Site IF
Superoxide production by complex I (plus any other sites that respond to NADH reduction state) increases at higher NADH/NAD+ ratio [19,28,39–41]. During β-oxidation, hydroxyacyl-CoA dehydrogenase converts one NAD+ to NADH per turn of the cycle, making site IF an appealing superoxide-producing candidate during fatty acid oxidation. However, oxidation of palmitoylcarnitine alone reduces NAD+ to a much lower extent than that of NAD-linked substrates such as glutamate plus malate, so the contribution of site IF has been thought to be negligible [24]. We found similar results (15% NAD(P)+ reduction with palmitoylcarnitine (Table 2) and 85% NAD(P)+ reduction with glutamate plus malate [19]), presumably because oxidation of palmitoylcarnitine was restricted by a high acetyl-CoA/CoA ratio, which inhibits the last enzyme of β-oxidation, 3-ketoacyl thiolase [32]. Nonetheless, Fig. 3 shows that site IF was a major contributor to the low rate of superoxide production with palmitoylcarnitine alone as substrate.
Addition of carnitine or malate to mitochondria respiring on palmitoylcarnitine removes acetyl-CoA. This greatly increased NAD(P) reduction (Table 2) and thus superoxide production from site IF. However, the relative contribution of site IF was not much changed because of increased production at other sites as well (Fig. 3).
When complex III was inhibited, strong reduction of the Q pool caused other sites to increase superoxide or H2O2 production greatly, and the relative contribution of site IF was correspondingly decreased (Figs. 4B, 5B, and 6C). However, when these other sites were inhibited by addition of myxothiazol and malate or malonate, site IF was a major contributor to the remaining low rates (Figs. 5B and 5C).
Site IQ
Superoxide is produced at site IQ a thigh rates when the proton-motive force and pH gradient are large, both during reverse electron flow from a reduced Q pool during oxidation of succinate [21,42] or glycerol3-phosphate [17,18,43]and during forward electron flow from NADH in the presence of certain IQ inhibitors [42,44]. Because oxidation of fatty acids can reduce NAD+ through hydroxyacyl-CoA dehydrogenase and the Q pool through acyl-CoA dehydrogenase and maintain a high proton-motive force and pH gradient, it has been speculated that oxidation of fatty acids smaysupport superoxide production at site I Q [20,23,42]. We found no evidence for production of superoxide at site IQ during oxidation of fatty acids, because there was no significant rate unattributable to sites IF, IIF, and IIIQo under native conditions (Fig. 3). In the presence of complex III inhibitors the proton-motive force will have collapsed, so site IQ is not a candidate for the unattributed H2O2 production rates in Figs. 5B, C, and 6C. Our results do not support the hypothesis that fatty acid oxidation generates superoxide at site IQ, in agreement with the conclusions of others [24,45].
Site IIIQo
Superoxide is produced during oxidation of QH2 at the Qo site of complex III [46,47] at a rate that depends on the QH2/Q ratio [27]. During oxidation of fatty acids the Q pool can be reduced by complex I, complex II, and ETFQOR. Cytochrome b566, which reports the reduction state of the Q pool [19,27], was more reduced with palmitoylcarnitine plus malate (48%; Table 2) than with glutamate plus malate (37%) [19], leading to high rates of superoxide production from site IIIQo. In this study superoxide production from IIIQo made the greatest individual contribution to total H2O2 production under native conditions with palmitoylcarnitine plus carnitine or malate (Fig. 3) and when complex III was inhibited by antimycin A (Fig. 4B). Its high contribution had to be eliminated by addition of myxothiazol to allow analysis of the smaller rates from the ETF/ETFQOR system (Figs. 5 and 6). Site IIIQo was also identified in previous studies using antimycin A [20,22–24].
Site IIF
This is the first report of H2O2 production from complex II during fatty acid oxidation. Under native conditions, inhibition by malonate was diagnostic of the contribution of complex II to the observed rate of H2O2 production (Fig. 1). Site IIF was a major contributor unless it was inhibited by addition of malate (Fig. 3). In contrast, the Q-binding-site inhibitor atpenin A5 increased H2O2 production with palmitoylcarnitine. This H2O2 production was abolished by subsequent addition of malonate (Fig. 1A), implicating forward electron flow into the flavin site of complex II. This effect can be explained by activation of complex II by the added palmitoylcarnitine generating acetyl-CoA and consuming endogenous inhibitory oxaloacetate, allowing endogenous succinate to cause H2O2 production at site IIF in the forward reaction under our experimental conditions.
When complex III was inhibited, site IIF was a major contributor to the high observed rates of H2O2 production (Fig. 4B) and the dominant contributor when site IIIQo was inhibited by myxothiazol (Fig. 5B). Both malonate and atpenin A5 strongly inhibited myxothiazol-induced H2O2 production (Figs. 4 and 5A), strongly supporting the idea that complex II can produce superoxide and/or H2O2 at high rates in the reverse reaction from a reduced Q pool during fatty acid oxidation.
Other sites
When electron flow through complex III was abolished and sites IF, IQ, IIF, and IIIQo were inhibited or fully accounted for, there remained a significant rate of H2O2 production in the presence of palmitoylcarnitine plus carnitine or malate (Figs. 5B, 5C, and 6C). The possible candidates are other enzymes directly connected to the Q pool, including ETFQOR, glycerol 3-phosphate dehydrogenase, dihydroorotate dehydrogenase, sulfide ubiquinone oxidoreductase, and proline dehydrogenase, as well as enzymes of the Krebs cycle or β-oxidation, including pyruvate and 2-oxoglutarate dehydrogenases, ETF, and acyl-CoA dehydrogenase.
The best candidate for the unassigned H2O2 production is the ETF/ETFQOR system, for the following reasons. (i) There is a requirement for substrates of β-oxidation, and these will reduce ETF/ETFQOR but not the other candidates by forward electron flow. (ii) Most candidates that might use electrons from the Q pool are present at rather low concentrations in skeletal muscle mitochondria. This is not true for glycerol 3-phosphate dehydrogenase, but it is not known to use electrons from the Q pool to generate H2O2 in the absence of glycerol 3-phosphate [18], making its participation unlikely. Skeletal muscle energy metabolism relies on β-oxidation, and therefore muscle mitochondria have high expression of the appropriate enzymes [48–50]. There is relatively high expression of ETFQOR (about 7% of total FAD-containing enzymes in the mitochondrial inner membrane [48]) and a larger ETF pool (63 pmol · mg protein−1 in skeletal muscle compared to brain mitochondria, which contain only 9 pmol · mg protein−1 of ETF [49]). Altogether, flavoproteins of β-oxidation account for 80% of the total content of mitochondrial flavoenzymes [50]. (iii) Compared to other complexes in the respiratory chain (complex I [51,52], complex II [53], glycerol 3-phosphate dehydrogenase [54,55], and dihydroorotate dehydrogenase [56,57]), ETFQOR has the most positive redox potentials (+27 mV for FAD1e-/FADox and +47 mV for [FeS]2+/[FeS]1+) [58,59]. Therefore it is thermodynamically favorable for a reduced Q pool to reduce ETFQOR and possibly ETF. (iv) Isolated ETFQOR is kinetically reversible [13] and submitochondrial particles catalyze the reduction of ETF by NADH and succinate under anaerobic conditions [60]. Thus ETFQOR can reduce ETF using electrons from the Qpool.
We now discuss the possible sites within the ETF/ETFQOR system.
Acyl-CoA dehydrogenase
Acyl-CoA dehydrogenases, in contrast to acyl-CoA oxidases, have low reactivity toward molecular oxygen [10], because of desolvation of the active site [61]. Moreover, the reduced medium-chain acyl-CoA dehydrogenase complexed with enoyl-CoA product is essentially devoid of oxygen reactivity [61,62]. Thus, acyl-CoA dehydrogenase is not a likely candidate.
ETF and ETFQOR
Identification of the species within the ETF/ETFQOR system that generates superoxide or H2O2 is challenging. We were unable to obtain the most potent known inhibitor of ETFQOR, 2-(3-methylpentyl)-4,6-dinitrophenol [63], to test this question directly.
We hypothesize that ETF is the site of superoxide and/or H2O2 production. Several observations support this hypothesis. (i) The one-electron reduction of ETF by acyl-CoA dehydrogenase is fast [12,64]. (ii) The half-reduced species of isolated ETF produces superoxide and H2O2 at high rates [4]. (iii) The redox couple FAD1e-/FADox in ETF has a more negative redox potential (+4 mV [65]) than the FAD in ETFQOR (+27 mV, see above), making ETF the better reductant of oxygen to superoxide or H2O2. (iv) The crystal structure of ETFQOR predicts low reactivity towards oxygen at all redox centers. The hydrophobic Q-binding pocket of the enzyme allows Q to penetrate deeply. If electron transfer to Q involves semiquinone as a transient intermediate, the semiquinone is protected from molecular oxygen. The FAD and FeS clusters are deeply buried and therefore unlikely to be reactive [66].
The biphasic kinetics of H2O2 production in Fig. 6A are probably explained by semiflavin ETF− being the source of superoxide and H2O2. As ETF gradually becomes reduced in the presence of a high QH2/Q ratio, ETF− builds up, causing high initial rates of H2O2 production. This is followed by an abrupt switch to a second phase of slower H2O2 production, probably caused by fully reduced flavin ETF2− as the whole ETF pool becomes fully reduced by acyl-CoA dehydrogenase. Two-electron reduction of ETF may occur either by acyl-CoA dehydrogenase at a much lower rate [12,64] or by disproportionation catalyzed by ETFQOR [12,59]. However, further experiments are required to establish the superoxide/H2O2-producing species within the ETF/ETFQOR system.
Conclusions
We have identified multiple sites of H2O2 and/or superoxide production during fatty acid oxidation in isolated skeletal muscle mitochondria. We conclude that when the Q pool is fairly oxidized, fatty acid oxidation in nonphosphorylating mitochondria produces superoxide and/or H2O2 mainly at site IF, site IIF in the forward reaction, and site IIIQo, and the contribution of ETF/ETFQOR is negligible. However, reduction of the Q pool by inhibition of complex III causes high rates of superoxide and/or H2O2 production from site IIIQo, site IIF in the reverse reaction, ETF/ETFQOR, and site IF. The contribution of each of these sites to superoxide and H2O2 production in cells during fatty acid oxidation under physiological or pathological conditions remains to be determined.
The reduction state of the Q pool was found to be crucial for the identification of the sites and mechanisms of superoxide and/or H2O2 production in isolated skeletal muscle mitochondria. However, it is difficult to correlate our in vitro conditions with in vivo studies because of the limited number of in vivo measurements of the reduction state of the Q pool in relevant systems. Direct measurements of flash-frozen samples of spontaneously contracting atria indicate that the Q pool is 40% oxidized under this condition [67]. The reduction states of cytochromes bl/h, c, and aa3 were measured in isolated pulmonary artery smooth muscle cells under normoxia and hypoxia [68]. Under hypoxia, cytochrome c and cytochrome aa3 were reduced, whereas cytochrome bl/h was oxidized. This is in a good agreement with a mechanism of ubiquinol oxidation by complex III [27] and suggests that the Q pool stays nearly fully reduced as it cannot be oxidized by complex III because of the lack of oxidized cytochrome c. Interestingly, under the hypoxia condition, higher rates of mitochondrial superoxide production were observed using the superoxide-sensitive dye MitoSOX [68]. Therefore, our novel in vitro findings can provide a firm foundation for future investigations of site-specific ROS production in muscle under physiological conditions (e.g., rest vs exercise) and in diseases featuring altered lipid metabolism.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 AG033542 and TL1 AG032116 and by The Ellison Medical Foundation, Grant AG-SS-2288-09.
Abbreviations
- PC
palmitoylcarnitine
- ETF
electron-transferring flavoprotein
- ETFQOR
electron-transferring flavoprotein ubiquinone oxidoreductase
- Q
ubiquinone
- QH2
ubiquinol
- ROS
reactive oxygen species
- Site IF
flavin site of complex I
- Site IQ
ubiquinone-binding site of complex I
- Site IIF
flavin site of complex II
- Site IIIQo
outer ubiquinone-binding site of complex III
- GPDH
glycerol 3-phosphate dehydrogenase
- SOD
superoxide dismutase
- DCPIP
dichlorophenolindophenol
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- CDNB
1-chloro-2,4-dinitrobenzene
References
- 1.Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E, et al. Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol. 1993;265:E380–E391. doi: 10.1152/ajpendo.1993.265.3.E380. [DOI] [PubMed] [Google Scholar]
- 2.Hoppeler H. Skeletal muscle substrate metabolism. Int J Obes Relat Metab Disord. 1999;23(Suppl 3):S7–10. doi: 10.1038/sj.ijo.0800878. [DOI] [PubMed] [Google Scholar]
- 3.Laforet P, Vianey-Saban C. Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscul Disord. 2010;20:693–700. doi: 10.1016/j.nmd.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigues JV, Gomes CM. Mechanism of superoxide and hydrogen peroxide generation by human electron-transfer flavoprotein and pathological variants. Free Radic Biol Med. 2012;53:12–19. doi: 10.1016/j.freeradbiomed.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 6.Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. 2010;31:364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefort N, Glancy B, Bowen B, Willis WT, Bailowitz Z, De Filippis EA, et al. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes. 2010;59:2444–2452. doi: 10.2337/db10-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Kim JJ, Battaile KP. Burning fat: the structural basis of fatty acid beta-oxidation. Curr Opin Struct Biol. 2002;12:721–728. doi: 10.1016/s0959-440x(02)00390-1. [DOI] [PubMed] [Google Scholar]
- 10.Mackenzie J, Pedersen L, Arent S, Henriksen A. Controlling electron transfer in acyl-CoA oxidases and dehydrogenases: a structural view. J Biol Chem. 2006;281:31012–31020. doi: 10.1074/jbc.M603405200. [DOI] [PubMed] [Google Scholar]
- 11.Ruzicka FJ, Beinert H. A new iron–sulfur flavoprotein of the respiratory chain:a component of the fatty acid beta oxidation pathway. J Biol Chem. 1977;252:8440–8445. [PubMed] [Google Scholar]
- 12.Ramsay RR, Steenkamp DJ, Husain M. Reactions of electron-transfer flavoprotein and electron-transfer flavoprotein: ubiquinone oxidoreductase. Biochem J. 1987;241:883–892. doi: 10.1042/bj2410883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beckmann JD, Frerman FE. Electron-transfer flavoprotein–ubiquinone oxidoreductase from pig liver: purification and molecular, redox, and catalytic properties. Biochemistry. 1985;24:3913–3921. doi: 10.1021/bi00336a016. [DOI] [PubMed] [Google Scholar]
- 14.Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287:27255–-27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orr AL, Quinlan CL, Perevoshchikova IV, Brand MD. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J Biol Chem. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinlan CL, Treberg JR, Perevoshchikova IV, Orr AL, Brand MD. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radic Biol Med. 2012;53:1807–1817. doi: 10.1016/j.freeradbiomed.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 21.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman DL, Brookes PS. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J Biol Chem. 2009;284:16236–16245. doi: 10.1074/jbc.M809512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem. 2010;285:5748–5758. doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schonfeld P, Wojtczak L. Brown adipose tissue mitochondria oxidizing fatty acids generate high levels of reactive oxygen species irrespective of the uncoupling protein-1 activity state. Biochim Biophys Acta. 2012;1817:410–418. doi: 10.1016/j.bbabio.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Affourtit C, Quinlan CL, Brand MD. Measurement of proton leak and electron leak in isolated mitochondria. Methods Mol Biol. 2012;810:165–182. doi: 10.1007/978-1-61779-382-0_11. [DOI] [PubMed] [Google Scholar]
- 27.Quinlan CL, Gerencser AA, Treberg JR, Brand MD. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem. 2011;286:31361–31372. doi: 10.1074/jbc.M111.267898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Treberg JR, Quinlan CL, Brand MD. Hydrogen peroxide efflux from muscle mitochondria underestimates matrix superoxide production—a correction using glutathione depletion. FEBS J. 2010;277:2766–2778. doi: 10.1111/j.1742-4658.2010.07693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klingenberg M, Pette D. Proportions of mitochondrial enzymes and pyridine nucleotides. Biochem Biophys Res Commun. 1962;7:430–432. doi: 10.1016/0006-291x(62)90329-7. [DOI] [PubMed] [Google Scholar]
- 30.Estabrook RW. Fluorometric measurement of reduced pyridine nucleotide in cellular and subcellular particles. Anal Biochem. 1962;4:231–245. doi: 10.1016/0003-2697(62)90006-4. [DOI] [PubMed] [Google Scholar]
- 31.Crofts AR, Meinhardt SW, Jones KR, Snozzi M. The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides: a modified Q-cycle mechanism. Biochim Biophys Acta. 1983;723:202–218. doi: 10.1016/0005-2728(83)90120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz H. Regulation of fatty acid oxidation in heart. J Nutr. 1994;124:165–171. doi: 10.1093/jn/124.2.165. [DOI] [PubMed] [Google Scholar]
- 33.Marquis NR, Fritz IB. The distribution of carnitine, acetylcarnitine, and carnitine acetyltransferase in rat tissues. J Biol Chem. 1965;240:2193–2196. [PubMed] [Google Scholar]
- 34.Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, et al. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase) Proc Natl Acad Sci USA. 2003;100:473–477. doi: 10.1073/pnas.0237315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 36.Ksenzenko M, Konstantinov AA, Khomutov GB, Tikhonov AN, Ruuge EK. Effect of electron transfer inhibitors on superoxide generation in the cytochrome bc1 site of the mitochondrial respiratory chain. FEBS Lett. 1983;155:19–24. doi: 10.1016/0014-5793(83)80200-2. [DOI] [PubMed] [Google Scholar]
- 37.Starkov AA, Fiskum G. Myxothiazol induces H2O2 production from mitochondrial respiratory chain. Biochem Biophys Res Commun. 2001;281:645–-650. doi: 10.1006/bbrc.2001.4409. [DOI] [PubMed] [Google Scholar]
- 38.Rydstrom J. Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta. 2006;1757:721–726. doi: 10.1016/j.bbabio.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 39.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 40.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation–reduction state. Biochem J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 42.Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) J Biol Chem. 2011;286:27103–27110. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic Biol Med. 2003;35:938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 44.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 45.Schonfeld P, Wieckowski MR, Lebiedzinska M, Wojtczak L. Mitochondrial fatty acid oxidation and oxidative stress: lack of reverse electron transfer-associated production of reactive oxygen species. Biochim Biophys Acta. 2010;1797:929–938. doi: 10.1016/j.bbabio.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 46.Boveris A, Cadenas E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 1975;54:311–314. doi: 10.1016/0014-5793(75)80928-8. [DOI] [PubMed] [Google Scholar]
- 47.Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry. 2002;41:7866–7874. doi: 10.1021/bi025581e. [DOI] [PubMed] [Google Scholar]
- 48.Voltti H, Hassinen IE. Oxidation–reduction midpoint potentials of mitochondrial flavoproteins and their intramitochondrial localization. J Bioenerg Biomembr. 1978;10:45–58. doi: 10.1007/BF00743226. [DOI] [PubMed] [Google Scholar]
- 49.Kunz WS, Gellerich FN. Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain. Biochem Med Metab Biol. 1993;50:103–110. doi: 10.1006/bmmb.1993.1051. [DOI] [PubMed] [Google Scholar]
- 50.Kunz WS, Kunz W. Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim Biophys Acta. 1985;841:237–246. doi: 10.1016/0304-4165(85)90064-9. [DOI] [PubMed] [Google Scholar]
- 51.Treberg JR, Brand MD. A model of the proton translocation mechanism of complex I. J Biol Chem. 2011;286:17579–17584. doi: 10.1074/jbc.M111.227751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verkhovskaya ML, Belevich N, Euro L, Wikstrom M, Verkhovsky MI. Real-time electron transfer in respiratory complex I. Proc Natl Acad Sci USA. 2008;105:3763–3767. doi: 10.1073/pnas.0711249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonomi F, Pagani S, Cerletti P, Giori C. Modification of the thermodynamic properties of the electron-transferring groups in mitochondrial succinate dehydrogenase upon binding of succinate. Eur J Biochem. 1983;134:439–445. doi: 10.1111/j.1432-1033.1983.tb07586.x. [DOI] [PubMed] [Google Scholar]
- 54.Unden G, Bongaerts J. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim Biophys Acta. 1997;1320:217–234. doi: 10.1016/s0005-2728(97)00034-0. [DOI] [PubMed] [Google Scholar]
- 55.Barron JT, Gu L, Parrillo JE. NADH/NAD redox state of cytoplasmic glycolytic compartments in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2000;279:H2872–H2878. doi: 10.1152/ajpheart.2000.279.6.H2872. [DOI] [PubMed] [Google Scholar]
- 56.Mohsen AW, Rigby SE, Jensen KF, Munro AW, Scrutton NS. Thermodynamic basis of electron transfer in dihydroorotate dehydrogenase B from Lactococcus lactis: analysis by potentiometry, EPR spectroscopy, and ENDOR spectroscopy. Biochemistry. 2004;43:6498–6510. doi: 10.1021/bi036179i. [DOI] [PubMed] [Google Scholar]
- 57.Zameitat E, Pierik AJ, Zocher K, Loffler M. Dihydroorotate dehydrogenase from Saccharomyces cerevisiae: spectroscopic investigations with the recombinant enzyme throw light on catalytic properties and metabolism of fumarate analogues. FEMS Yeast Res. 2007;7:897–904. doi: 10.1111/j.1567-1364.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 58.Paulsen KE, Orville AM, Frerman FE, Lipscomb JD, Stankovich MT. Redox properties of electron-transfer flavoprotein ubiquinone oxidoreductase as determined by EPR–spectroelectrochemistry. Biochemistry. 1992;31:11755–11761. doi: 10.1021/bi00162a012. [DOI] [PubMed] [Google Scholar]
- 59.Usselman RJ, Fielding AJ, Frerman FE, Watmough NJ, Eaton GR, Eaton SS. Impact of mutations on the midpoint potential of the [4Fe–4S]+1,+2 cluster and on catalytic activity in electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO) Biochemistry. 2008;47:92–100. doi: 10.1021/bi701859s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frerman FE. Reaction of electron-transfer flavoprotein ubiquinone oxidoreductase with the mitochondrial respiratory chain. Biochim Biophys Acta. 1987;893:161–169. doi: 10.1016/0005-2728(87)90035-1. [DOI] [PubMed] [Google Scholar]
- 61.Wang R, Thorpe C. Reactivity of medium-chain acyl-CoA dehydrogenase toward molecular oxygen. Biochemistry. 1991;30:7895–7901. doi: 10.1021/bi00246a004. [DOI] [PubMed] [Google Scholar]
- 62.Ghisla S, Thorpe C. Acyl-CoA dehydrogenases:a mechanistic overview. Eur J Biochem. 2004;271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 63.Simkovic M, Frerman FE. Alternative quinone substrates and inhibitors of human electron-transfer flavoprotein-ubiquinone oxidoreductase. Biochem J. 2004;378:633–640. doi: 10.1042/BJ20031272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hall CL, Lambeth JD. Studies on electron transfer from general acyl-CoA dehydrogenase to electron transfer flavoprotein. J Biol Chem. 1980;255:3591–3595. [PubMed] [Google Scholar]
- 65.Husain M, Stankovich MT, Fox BG. Measurement of the oxidation–reduction potentials for one-electron and two-electron reduction of electron-transfer flavoprotein from pig liver. Biochem J. 1984;219:1043–1047. doi: 10.1042/bj2191043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, Frerman FE, Kim JJ. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc Natl Acad Sci USA. 2006;103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu MS, Siess M, Hoffmann PC. The effect of changes in functional activity on ubiquinone redox status in isolated atria. Eur J Biochem. 1973;37:259–269. doi: 10.1111/j.1432-1033.1973.tb02984.x. [DOI] [PubMed] [Google Scholar]
- 68.Sommer N, Pak O, Schorner S, Derfuss T, Krug A, Gnaiger E, et al. Mitochondrial cytochrome redox states and respiration in acute pulmonary oxygen sensing. Eur Respir J. 2010;36:1056–1066. doi: 10.1183/09031936.00013809. [DOI] [PubMed] [Google Scholar]