

Abstract
Purpose
The prognosis of small cell lung cancer (SCLC) is poor, and there has been very little progress in the medical treatment of SCLC in the past two decades. We investigated the potential of janus kinases (JAKs) inhibitor AZD1480 for treatment of SCLC in vitro and in vivo.
Experimental Design
JAK1 or JAK2 were inhibited by AZD1480 or siRNAs, and the effect of inhibition of JAK gene family on SCLC cell viability was evaluated. The effect of AZD1480 on cell cycle distribution and apoptosis induction were studied. Antitumor effects of AZD1480 in tumor xenografts were assessed.
Results
AZD1480 significantly inhibited growth of 6 out of 13 SCLC cells with IC50s ranging from 0.73 to 3.08 μM. Knocking-down of JAK2 and JAK1 inhibited proliferation of Jak2-positive/Jak1-negative H82 cells and Jak1-positive/Jak2-negative GLC4 cells, respectively. Treatment of SCLC cells with AZD1480 for 24 hours resulted in increase of 4N DNA content and Histone 3 serine 10 phosphorylation, indicative of G2/M phase arrest. Moreover, SCLCs underwent apoptosis after AZD1480 treatment as exemplified by the downregulation of MCL1, the accumulation of cleaved-Caspase 3, cleaved PARP and increase of annexin-V positive cells. Finally, xenograft experiments showed that AZD1480 attenuated the growth of H82 and GLC4 tumors in mice, and we observed stronger apoptosis as well as decreased CD31 positive endothelial cells in H82 and GLC4 xenografts upon AZD1480 treatment.
Conclusions
Janus kinases inhibitor AZD1480 attenuated growth of SCLC cells in vitro and in vivo. Clinical development of anti-JAKs therapies in SCLC warrants further investigation.
Keywords: Small cell lung carcinoma, Janus kinases, Therapy
Introduction
Small-cell lung cancer (SCLC), a smoking related cancer, accounts for 10–15% of lung cancers (1). SCLC has the propensity to develop early and widespread metastatic disease. Although approximately 20% of patients with SCLC limited to the thorax can be cured by concurrent chemo-radiotherapy, patients with metastatic disease are incurable. There has been very little progress in the medical treatment for SCLC for the past two decades (2, 3), and no major drug discovery after establishing the role of cisplatin and etoposide as first-line treatment and topotecan as second-line treatment. Several cytotoxic therapies, such as irinotecan (4) and amrubicin (5), have failed to demonstrate their superiority over current standards. A relatively limited effort has been made to develop targeted agents in SCLC. The clinical experimentation of tyrosine kinase inhibitors (6) and other small molecules (7, 8) has been disappointing. More recently, the investigation of inhibitors of the bcl-2 family has yielded disappointing results in clinical trials of navitoclax (ABT-263) and obatoclax (9, 10), despite promising preclinical efficacy (11, 12).
The genomic alterations present in SCLC have been recently described in detail (13–15). We previously reported copy number alterations of the SCLC genome from 33 SCLC tumors using array-based comparative genomic hybridization (aCGH) analysis (16). A high copy gain of the JAK2 gene, defined as the copy number of the gene in the tumor was more than 8 folds the copy of the gene in the reference genome, was observed in one SCLC tumor, and copy number gain of the JAK2 gene, defined as the copy number of gene in the tumor was more than the copy number in the reference genome, was observed in around 30% of SCLC tumors and cell lines. In an independent aCGH analysis across several tumor types (17) copy number gain of the JAK2 gene was observed in 42.5% of SCLC specimens, and high-level amplification of the JAK2 gene were observed in 5% of SCLC (17). In addition, copy number gain of the JAK2 gene, as determined by exome sequencing, was observed in 21.4% of SCLC tumors (15). Additionally, Pfeiffer et al. showed that phosphorylated STAT3, an indicator of JAK activity, was strongly expressed in 10 out of 10 SCLC tumors, but in 0 out of 13 non-SCLC tumors (18). Yang et al. further demonstrated that knocking-down STAT3, by STAT3 siRNA, resulted in cell death of NCI-H446 and NCI-H1688 SCLC cell lines, both of which express high endogenous STAT3 (19). The JAK-STAT signal transduction pathway may therefore be important for the survival of SCLC.
Janus kinases (JAKs) and signal transducer and activator of transcription (STAT) are major mediators of cytokine signaling (20, 21). The JAK gene family is composed of JAK1, JAK2, JAK3, and TYK2 (tyrosine kinase 2). Mutations in JAK2 have been reported in malignancies (22). JAK2V617F mutation is an oncogenic driver in many myeloproliferative disorders and in nearly all polycythemia vera cases (22–24). Ruxolitinib, an ATP binding competitive inhibitor of both JAK1 and JAK2 (25), inhibits proliferation of JAK2V617F transfected Ba/F3 cells, suppresses colony formation of the erythroid progenitor from polycythemia vera patients, and reduces splenomegaly in a JAK2V617F mouse model (25). Ruxolitinib provided significant clinical benefit in patients with myelofibrosis, a myeloproliferative disorder with JAK2 mutations in 35–50% of cases (26), and has been recently approved by FDA for the treatment of intermediate and high risk myelofibrosis, including primary myelofibrosis, post-polycythemia vera myelofibrosis, and post-essential thrombocythemia myelofibrosis (27, 28). AZD1480 is a multi-kinase inhibitor with potent activity inhibiting Trk-A, JAK1, JAK2, Aurora-A, Flt4, and FGFR1 (29). AZD1480 reduces xenograft formation in various solid tumor cell lines (29), although no cytotoxic effect was observed in solid cancer cell lines lacking mutation in the JAK2 gene. The effect of JAK inhibitors on SCLC has not been addressed before.
Here we investigated the activity of the JAK inhibitor AZD1480 and JAK1/2 siRNA inhibition in SCLC models.
Materials and Methods
Cancer cell lines
The following SCLC cells have been studied: GLC4, NCI-H69, NCI-H82, NCI-H128, NCI-H146, NCI-H187, NCI-H526, NCI-N592, NCI-H620, NCI-H678, NCI-H792, NCI-H1173, DMS-114, and AC-3. DMS-114 was obtained from American Type Culture Collection (ATCC, Manassas, VA), and the other cell lines were obtained as previously described (16). Cells were maintained in RPMI containing 10% fetal bovine serum (FBS) with the exception of DMS-114, which was maintained in Waymouth's media containing 10% FBS. The cells were not tested and authenticated by the authors and were passed less than 10 times since obtaining the cells.
Real-time polymerase chain reaction (PCR)
The mRNA expression of the JAK2 gene in SCLC cells was evaluated using Taqman gene expression assay (Applied Biosystems, Foster City, CA) following manufacturer's instruction. The GAPDH gene was used as endogenous control. Relative expression of the JAK2 gene of each cell line was analyzed using the 2−ΔΔCt value method and was calibrated to the expression level of the NCI-H69 cell line.
The copy number of the JAK2 gene was determined using Taqman copy number assay (Applied Biosystems), following manufacturer's instruction. The ribonuclease P RNA component H1 (RPPH1) gene was used as endogenous control, which was labeled by VIC probe. The copy number was analyzed using CopyCaller version 1 software (Applied Biosystems). The copy number of the JAK2 gene in the reference genomic DNA was defined as 2.
Western blot
Western blot was performed as described previously (30). Antibodies were obtained from Cell Signaling Technology (Danvers, MA) (β-tubulin, JAK1, JAK2, STAT3, phospho-STAT3, PARP, cleaved caspase 3, phospho-histone 3 serine 10, and MCL1), and Sigma Aldrich (St. Louis MI) (Actin).
Growth inhibition assays
AZD1480 was provided by Astrazeneca (Manchester, UK) and INCB16562 by Incyte Corporation (Wilmington, DE). Cisplatin and etoposide were obtained from Sigma Aldrich (St. Louis, MI). SCLC cells were treated with various concentrations up to 10 μM of AZD1480, cisplatin, or etoposide for 72 hours. Cell viability was determined by the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI). The IC50 was the drug concentration at which 50% of cells were viable comparing to the untreated cells.
To determine the synergistic effect of AZD1480 and cisplatin or AZD1480 and etoposide, GLC4 cells were treated with AZD1480 and cisplatin or AZD1480 and etoposide at 1:1 ratio. The combination index (CI) value was determined using CompuSyn software 1.0 (ComboSyn Inc. Paramus, NJ). Synergy was defined as a CI value less than 1.0 (31).
Sequencing JAK2 V617F in SCLC cells
JAK2 V617F were sequenced in the AC3, GLC4, NCI-H128, NCI-H187, NCI-N592, NCI-H620, NCI-H678, NCI-H792 and NCI-H1173 SCLC cells using real-time PCR assay as described previously (32). The forward primer was 5'-TGTAAAACGACGGCCAGTGACTACGGTCAACTGCATGAAA-3' and the reverse primer was 5'-CAGGAAACAGCTATGACCCATGCCAACTGTTTAGCAAC-3'. The JAK2 mutational status of the NCI-H69, NCI-H82, NCI-H146, NCI-H526, and DMS-114 SCLC was available from the Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) database (33).
siRNA transfection
siRNAs were obtained from Ambion (Carlsbad, CA) (JAK1 and JAK2) and Dharmacon (Lafayette, CO) (JAK2 and control siRNA). siRNA transfection was performed using Pepmute reagent (SignaGen Laboratoires, Rockville, MD), following manufacturer's instructions with a final siRNA concentration of 5nM. Protein was extracted 48 hours after transfection, and cell cycle analyses were performed 72 hours after transfection.
Flow-cytometry
Flow cytometry was used to study cell cycle changes and apoptosis in SCLC cells upon AZD1480 treatment. Cell cycle analysis was performed as previously described (30). In brief, cells were treated with AZD1480 at various concentrations for 24 hours, collected and stained with propidium iodide before analysis on FACSCalibur (Becton, Dickinson and Company, Franklin Lakes, NJ).
The occurrence of apoptosis was determined by the FITC Annexin V Apoptosis Detection Kit (Becton, Dickinson and Company, Franklin Lakes, NJ) after cell treatment with AZD1480 at various concentrations for 48 hours.
Immunofluorescence study
Cells were incubated with various concentrations of AZD1480 for 24 hours. 200,000 SCLC cells were collected, washed with PBS, then spun onto a slide at 800rpm for 5 min using cytospin. Spun cells were fixed with 2.5% paraformaldehyde for 10 min, permeated with cold 100% methanol for 10 min, and washed with 0.5% Triton X-100 for 2 min. Anti-phospho-histone 3 serine 10 rabbit antibody (1:1000, Cell Signaling Technology, Danvers, MA) was applied at room temperature for 3 hours, followed by anti-rabbit secondary antibody labeled by FITC for 90 min. Cellular DNA was then stained with 0.01% DAPI, and then cover slips were mounted.
Xenograft studies
Four million NCI-H82 or GLC-4 cells were injected subcutaneously in the flank of athymic nude mice. Once the xenograft reached 5mm in diameter, mice were treated with either vehicle containing 0.5% Hypermellose and 0.1% Tween-80 or AZD1480 60mg/kg per day by oral gavage. Tumor size was measured every three days and calculated using the V = 1/2(L × W2) formula. Use of animals for study was approved by NIH under protocol number MOB005.
Hematoxylin and eosin (H&E) stain and immunohistochemistry study of Ki-67 and CD31 in xenografts were performed as described previously (34). Anti-Ki-67 and anti-CD31 antibodies were from Santa Cruz (Dallas, TX) and Novus Biologicals (Littleton, CO). Quantitations of necrosis (% necrotic cells in tumors: vehicle [n = 7] and AZD1480 [n = 6]) and Ki-67 positive cells (% Ki67 positive cells in tumors; vehicle [n = 5] and AZD1480 [n = 6]) were performed by a pathologist (B.K.)
To evaluate the apoptosis, xenografts were lysed and extent of apoptosis was determined using Caspase-Glo® 3/7 Assay Systems (Promega, Madison, WI).
Statistical analysis
We used Spearman's method to analyze correlations between variables. Comparisons of variables between 2 groups were performed using student t test and among 3 or more groups were performed using one-way ANOVA, followed by LSD test for post-hoc analysis. p-values less than 0.05 were regarded as significant.
Results
Expression of JAK2 is related to copy number of the gene in SCLC cells
We evaluated the expression of JAK1, JAK2 and their downstream molecule STAT3 as well as phospho-STAT3 in SCLC cells (Figure 1A). Expression of JAK1 and JAK2 proteins was variable: little JAK1 expression was detected in H82 and H620 cells, and little JAK2 expression was detected in H187, GLC4, and AC3 cells. The expression of JAK2 mRNA and JAK2 protein were significantly associated with the copy number of the JAK2 gene (Figure 1B and 1C, table S1). Although JAK2 V617F mutations have been frequently observed in myeloproliferative disorders (23), mutations in JAK genes (JAK1, JAK2, JAK3, and TYK2) were reported in less than 5 % of lung cancer specimens (Figure 1D), and we did not observe JAK2 V617F mutation in the 14 SCLC cells studied.
Figure 1. JAK gene family in SCLC cells.
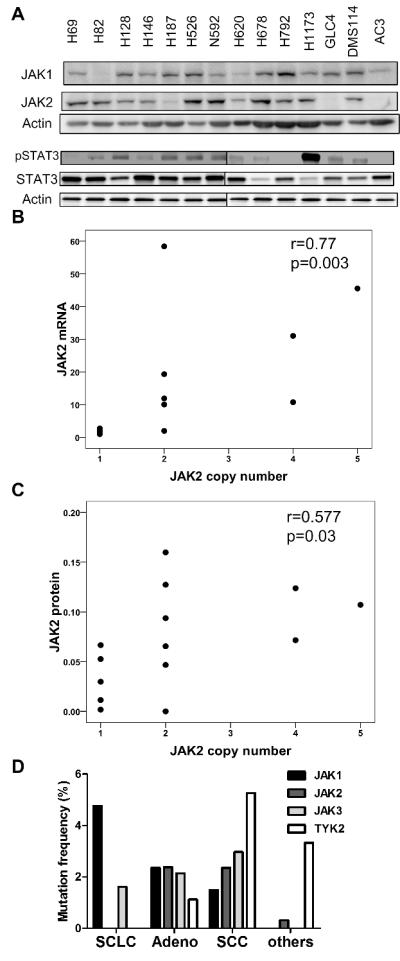
(A) Protein expression of JAK1, JAK2, p-STAT3, and STAT3 in 14 SCLC cell lines. (B) Association between JAK2 mRNA expression and JAK2 copy number determined by real-time PCR. (C) Association of JAK2 protein expression and JAK2 copy number. Correlation coefficient and p-value was determined by Spearman's method. (D) Mutations of JAK1, JAK2, JAK3, and TYK2 in lung cancers from COSMIC database. SCLC: small cell lung cancer (n=63, 166, 62, 61, respectively), SCC: squamous cell carcinoma (n=269, 254, 269, 247, respectively), Adeno: adenocarcinoma (n=467, 462, 467, 441, respectively), others: other lung cancers (n=108, 930, 53, 30, respectively).
Inhibition of Janus kinases decreases proliferation of a subset of SCLC cells
To explore the biologic importance of JAKs in SCLC, we treated SCLC cells with the pan-JAKs inhibitor AZD1480 (29). AZD1480 inhibited SCLC cell proliferation with an IC50 between 0.7 to 3.1 μM in 6 out of 13 (46%) SCLC cell lines tested (Figure 2A); no cytotoxic effect was reported in cancer cell lines of many solid tumors at these drug concentration (29). To test whether the inhibition of proliferation is AZD1480 specific, we treated N592 and H82 cells with INCB16562, a janus kinase 1/2 inhibitor (35). The IC50 to INCB16562 was 2.8μM for NCI-N592 cells and 3.0μM for NCI-H82 cells (Figure 2A), suggesting that proliferation of a portion of SCLC cells were abrogated by janus kinase inhibitors. The sensitivity to AZD1480 was unrelated to the protein expression of JAK1 (r=0.18, p=0.55 by Spearman's method), JAK2 (r=−0.20, p=0.50), or STAT3 (r= 0.10, p=0.73) (Figure S1). Upon AZD1480 treatment, STAT3 phosphorylation was inhibited in the AZD1480 sensitive GLC4 and NCI-N592 cells at a dose dependent manner (Figure 2C). For AZD1480 resistant cells, STAT3 phosphorylation was inhibited in the NCI-H128 cells but not in the NCI-H187 cells (Figure 2B).
Figure 2. Proliferation of SCLC cells after JAK inhibition.

(A) Cell viability of SCLC cell lines upon AZD1480 or INCB15652 treatments (B) STAT3 phosphorylation in the GLC4, N592, H128, and H187 cells upon AZD1480 treatment. (C) Proliferation of H82 cells upon knockdown of JAK2 by 2 siRNAs. *p=0.03 by t-test for both siRNAs. (D) Proliferation of GLC4 cells upon knockdown of JAK1 by 2 siRNAs. * p=0.02, ** p<0.01 by t-test.
We further explored whether inhibition of JAKs attenuates proliferation of SCLC cells directly. Proliferation of NCI-H82 cells, which express JAK2 but little JAK1 proteins (Figure 1B), was reduced upon treatment of the cells with two JAK2 siRNAs, compared to control siRNA (Figure 2C). JAK1 siRNA did not influence expression of JAK2 protein in the NCI-H82 cells (Figure S2A), and JAK1 siRNA resulted in less inhibition of the proliferation of the cells in comparison (Figure S2B).
In GLC4 cells, which express JAK1 but little JAK2 proteins (Figure 1B), STAT3 was dephosphorylated upon JAK1 knockdown; proliferation of GLC4 cells was reduced upon treatment of cells with two JAK1 siRNAs compared with control siRNA (Figure 2D). JAK2 siRNA did not influence expression of JAK1 protein as well as phospho-STAT3 protein in the GLC4 cells (Figure S2C), and it resulted in less inhibition of the proliferation of the cells (Figure S2D).
AZD1480 induces G2/M cell cycle arrest and apoptosis in SCLC cells
AZD1480 caused significant dose-dependent accumulation of 4N DNA contents in NCI-N592, GLC4, and NCI-H82 cells (Figure 3A), indicating G2/M arrest. Noticeably, a small portion of G2/M cells was in mitosis as exemplified by the dose-dependent increase of phospho-Histone 3 serine 10 in N592 cells, as demonstrated by western blot (Figure 3B) and immunofluorescence (Figure 3C). Interestingly, whereas we did not observe changes in the cell cycle when we knocked-down JAK1 by siRNA in the GLC4 cells and JAK2 in the NCI-H82 cells, we observed increase of G2/M portion of the cells upon knocking-down both JAK1 and JAK2 simultaneously in the cells (Figure S2E and S2F).
Figure 3. G2/M cell cycle arrest in SCLC cells upon AZD1480 treatment.

(A) Cell cycle analysis of N592, GLC4 and H82 cells upon AZD1480 treatment (percentages of each cell cycle stage were averages of triplicate experiments). The images are representative cell cycle analysis of N592 cells upon AZD1480 treatment. (B) Western blot of Histone 3 serine 10 (H3S10) phosphorylation in N592 cells upon AZD1480 treatment. (C) Phospho-H3S10 positive N592 cells, determined by immunofluorescence upon AZD1480 treatment (100 cells were analyzed per each low-power field, and 6 low-power fields were analyzed per each concentration, vertical bar: standard deviation). p=0.004 among groups by one-way ANOVA. (D) Combination index in GLC4 cells upon treatment with cisplatin plus AZD1480 and etoposide plus AZD1480.
Since the cells arrested in G2/M by AZD1480 may survive from the acute drug treatment, we tested whether the combination with cisplatin or etoposide may show any synergistic inhibitory effect on SCLC cells. We observed that most combination indexes (CI) values were less than 1.0 when AZD1480 was combined with either cisplatin or etoposide in GLC4 cells (Figure 3D), suggesting a synergistic effect.
After AZD1480 treatment for 48 hours, we observed decreased expression of Mcl-1, a downstream molecule of STAT3, in N592 and GLC4 cells (Figure 4A). We also demonstrated a dose-dependent decrease of uncleaved PARP expression as well as an increase of cleaved-Caspase 3 and PARP in NCI-N592 and GLC4 cells upon AZD1480 treatment, and an increase of cleaved-Caspase 3 in the NCI-H82 cells (Figure 4B). In line with this result, an increase of Annexin-V positive cells was also evident in NCI-N592 cells upon AZD1480 treatment (Figure 4C and 4D). Taken together, these results suggest that AZD1480 induced apoptosis in SCLC cells.
Figure 4. Apoptosis in SCLC cells upon AZD1480 treatment.

(A) MCL1 expression in GLC4 and N592 cells upon AZD1480 treatment. (B) Expression of cleaved Caspase 3 (c-Caspase 3), uncleaved-PARP (uc-PARP), and cleaved-PARP (c-PARP) in SCLC cells upon AZD1480 treatment for 48 hours. (C) Flow cytometry studies of 7-AAD and Annexin V-stained N592 cells upon AZD1480 treatment. (D) Percentage of early apoptotic (Annexin-V positive and 7-AAD negative) N592 cells and late apoptotic (Annexin-V positive and 7-AAD positive) N592 cells upon AZD1480 treatment for 48 hours. * p= 0.03 for early apoptosis and 0.001 for late apoptosis by one-way ANOVA. Values were average of experiments performed in triplicate; vertical bars represent standard deviation.
AZD1480 attenuated the growth of SCLC xenografts
We evaluated the effect of AZD1480 in SCLC cells in vivo. We observed a decrease of xenograft growth upon AZD1480 treatment, in NCI-H82 cells (Figure 5A) and GLC4 cells (Figure 5B), compared to vehicle treatment. AZD1480 inhibited STAT3 phosphorylation in GLC4 xenografts (Figure 5C).
Figure 5. SCLC xenografts upon AZD1480 treatment.

(A) Growth curves of H82 xenografts upon AZD1480 60mg/kg/day or vehicle treatment (n=5 in each group). *p<0.001 by t-test on day 21 of treatment; vertical bar: standard error. Images are representative tumors of H82 xenografts. (B) Growth curves of GLC4 xenografts upon AZD1480 60mg/kg/day or vehicle treatment (n=7 in each group). * p=0.02 by t-test on day 15 of treatment; vertical bar: standard error. Images are representative tumors of GLC4 xenografts. (C) Phospho-STAT3 activities in GLC4 xenografts upon AZD1480 or vehicle treatments.
On H&E staining, we observed trends of more necrotic cells in GLC4 as well as NCI-H82 xenografts treated with AZD1480 than vehicle. (Figure 6A). We did not observe difference of Ki-67 staining between vehicle and AZD1480 treated SCLC xenografts (43.6% and 39.2% in average in vehicle treated and AZD1480 treated, respectively, GLC4 xenografts, p=0.34; 55% and 52% in NCI-H82 xenografts, p=0.39) (Figure 6B). We also detected stronger signals of cleaved caspase 3/7 in AZD1480 treated SCLC xenografts, suggesting more apoptic cells in AZD1480 treated xenografts (p=0.001 for GLC4 xenografts and p=0.004 for NCI-H82 xenografts) (Figure 6C). We further detected decrease of CD31 positive endothelial cells in AZD1480 treated SCLC xenografts (p=0.06 for GLC4 xenografts and p<0.001 for NCI-H82 xenografts) (Figure 6D); our findings are consistent with a previous report of anti-angiogenic effect of AZD1480 in vivo (36).
Figure 6. Immunohistochemistry study of GLC4 xenografts.

(A) Frequencies of necrotic cells in SCLC xenografts treated with vehicle or AZD1480 (n=5–7 for each group). p=0.23 for GLC4 xenografts and p=0.37 for NCI-H82 xenografts. Images are representative images of H&E staining of GLC4 xenografts upon vehicle and AZD1480 treatment. *area of necrosis. Arrows indicated nuclear pycnosis. (B) Representative images of Ki-67 staining of GLC4 xenografts upon vehicle and AZD1480 treatment. Ki-67 positive cells are stained in brown. (C)Singals of cleaved caspase 3/7 in SCLC xenografts treated with vehicle or AZD1480 (n=4 for each group). p=0.001 for GLC4 xenografts and p=0.004 for NCI-H82 xenografts. (D) Frequencies of CD31 positive endothelial cells per 200X fields in NCI-H82 SCLC xenografts treated with vehicle or AZD1480 (n=15–22 200 X fields for each group). p=0.06 for GLC4 xenografts and p<0.001 for NCI-H82 xenografts. Images are representative images of the CD31 staining of NCI-H82 xenografts upon vehicle and AZD1480 treatment. Arrows indicate CD31 positive endothelial cells.
Discussion
Here we provide pre-clinical evidence of a potential value of JAKs as therapeutic targets in SCLC. AZD1480 was cytotoxic to SCLC cells at sub-micromolar concentrations. Incubation of SCLC cells with AZD1480 resulted in G2/M cell cycle arrest and apoptosis. Finally, AZD1480 inhibited growth in vivo in two SCLC xenograft models. We also observed more necrosis and apoptosis in AZD1480 treated SCLC xenografts.
SCLC is characterized by a high number of genomic alterations and somatic gene mutations (14, 15). The mutation rate in SCLCs was 5.5 to 7.4 protein-changing mutations per million base pairs, which is higher than other solid tumors such as breast cancer, ovarian cancer, prostate cancer, and renal cancer (14, 15). Even though some candidate oncogenic drivers have recently been identified (14, 15), most of the mutated genes are not druggable. We searched the COSMIC database and found that the frequencies of mutations in JAK1, JAK2, and JAK3 genes were very low: 4.8%, 0%, and 1.6%, respectively (Figure 1D) (37), and we did not detect JAK2 V617F mutation, a druggable mutation, in the 14 SCLC cells. Using next-generation sequencing techniques, Peifer et al. (14) and Rudin et al. (15) reported non-synonymous mutations of the JAK1 gene in 2 out of 29 and 1 out of 42 SCLC specimens, respectively; no non-synonymous mutation of the JAK2, JAK3, or TYK2 gene was detected in the two reports. Little is known on how these mutations influence the function of the proteins. High level amplifications of JAK1 and JAK2 were reported in 2.5% and 5% of SCLC in the Tumorscape website, respectively (17), and we previously showed high copy amplification of the JAK2 gene in one out of 33 (3.3%) SCLC tumors (16). As we failed to observe correlations between sensitivity to AZD1480 and the expression of JAK1 or JAK2 proteins in SCLC cells, the factor(s) or target(s) predicting sensitivity to AZD1480 in SCLC remain unknown and warrant further studies.
We demonstrated that AZD1480 inhibits SCLC viability in vitro and xenograft growth in vivo. It has been reported that AZD1480 inhibited xenograft growth of multiple solid tumor cell lines at least partly through its anti-angiogenic effect (29, 36), whereas only a few reports suggested direct cytotoxicity of AZD1480 in cancer cells. AZD1480 induced apoptosis in multiple myeloma cells by inhibiting JAK2 and FGFR3 (38). In solid tumors, McFarland et al. demonstrated that 10μM AZD1480 resulted in apoptosis of U251-MG glioblastoma cells (39). Here we showed that AZD1480 induced G2/M cell cycle arrest and apoptosis in SCLC cells at sub-micromolar concentrations (Figure 3 and Figure 4). SCLC is a neuroendocrine tumor (40) and is rather distinct from other carcinomas for several reasons. Genetically SCLC tumors carry more mutations than other carcinomas and displays typical cigarette-related G:C→T:A transversions (15). Our study provides the first preclinical evidence that inhibition of Janus kinases may be a valid strategy to explore for the treatment of SCLCs.
We observed that AZD1480 slowed SCLC xenograft growth but did not induce regression of the xenografts (Figure 5). Similar findings were reported in other molecular targeted therapies to treat SCLC tumors when used alone. Shoemaker et al. demonstrated that the Bcl-2 family inhibitor navitoclax (ABT-263) induced xenograft shrinkage of H146, H889, and H1963 SCLC cells and inhibited tumor growth rate in 8 other SCLC cell lines (41). Whereas the Hedgehog inhibitor NVP-LDE225 only slowed xenograft growth of LX22 SCLC cells when given alone, it significantly enhanced anti-tumor activity of carboplatin and etoposide (42). We also observed synergistic effects between AZD1480 and chemotherapeutic drugs such as cisplatin or etoposide in GLC4 cells (Figure 3D). As SCLC tumors tend to grow rapidly, decreased growth rate of xenografts by AZD1480 monotherapy may not be translated into marked clinical benefit of SCLC patients. The anti-tumor effect of AZD1480 in combination with traditional chemotherapies on the other hand warrants further investigation.
AZD1480 is a multi-kinase inhibitor with potent activities against TrkA, JAK2, Aurora-A, and Flt4 (29). To exclude the anti-cancer effect is AZD1480 specific, we showed that INCB16562, another janus kinases inhibitor, inhibited proliferation of SCLC cells at micromolar level. We showed that AZD1480 resulted in increased activities of caspase 3/7 and decreased number of CD31 positive endothelial cells in SCLC xenografts (Figure 6D), suggesting that both direct cytotoxicity and anti-angiogenesis may contribute to the effect of AZD1480 on SCLC xenografts (36). We also demonstrated that knocking-down JAK1 and/or JAK2 in SCLC cells inhibited cell proliferation (Figure 2C–2D) as well as G2/M arrest of the cells (Figure S2E and S2F). Considering the potential off-target effects of siRNAs, we do not exclude the possibility that targets in addition to janus kinases contribute to the effect of AZD1480 in SCLC cells.
In conclusion, janus kinases inhibitor AZD1480 attenuated SCLC growth in vitro and in vivo. Clinical development of anti-JAK therapies, including AZD1480, in SCLC is warranted.
Supplementary Material
Statement of translational relevance.
Small cell lung cancer (SCLC) is characterized by rapid growth and high lethality. There has been only a very limited progress in the treatment of SCLC during the past two decades, and this has been mainly due to introduction of radiation to the chest and brain. Chemotherapy with cisplatin and etoposide remains the standard first-line treatment and topotecan is the standard second-line treatment. Here we demonstrated that inhibition of janus kinases (JAKs) by either the small molecule inhibitor AZD1480 or siRNAs attenuated growth of SCLC cells. AZD1480 induced apoptosis of SCLC cells in vitro and attenuated growth of SCLC xenografts in vivo. Given the limited therapeutic options for advanced SCLC, our findings may influence clinical development of medical treatment of small cell lung cancer.
Acknowledgments
The study was sponsored by the intramural grant of the National Institutes of Health, USA
Footnotes
The authors declare no conflict-of-interest
References
- 1.van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378:1741–55. doi: 10.1016/S0140-6736(11)60165-7. [DOI] [PubMed] [Google Scholar]
- 2.Oze I, Hotta K, Kiura K, Ochi N, Takigawa N, Fujiwara Y, et al. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: disappointing results. PLoS One. 2009;4:e7835. doi: 10.1371/journal.pone.0007835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC. Small cell lung cancer: have we made any progress over the last 25 years? Oncologist. 2007;12:1096–104. doi: 10.1634/theoncologist.12-9-1096. [DOI] [PubMed] [Google Scholar]
- 4.Lara PN, Jr., Natale R, Crowley J, Lenz HJ, Redman MW, Carleton JE, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–5. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jotte R, Von Pawel J, Spigel DR, Socinski MA, O'Brien M, Paschold EH, et al. Randomized phase III trial of amrubicin versus topotecan (Topo) as second-line treatment for small cell lung cancer (SCLC) Journal of Clinical Oncology. 2011;29:7000. doi: 10.1200/JCO.2013.54.5392. [DOI] [PubMed] [Google Scholar]
- 6.Gitlitz BJ, Moon J, Glisson BS, Reimers HJ, Bury MJ, Floyd JD, et al. Sorafenib in platinum-treated patients with extensive stage small cell lung cancer: a Southwest Oncology Group (SWOG 0435) phase II trial. J Thorac Oncol. 2010;5:1835–40. doi: 10.1097/JTO.0b013e3181f0bd78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heymach JV, Johnson DH, Khuri FR, Safran H, Schlabach LL, Yunus F, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with sensitive relapse small-cell lung cancer. Ann Oncol. 2004;15:1187–93. doi: 10.1093/annonc/mdh315. [DOI] [PubMed] [Google Scholar]
- 8.Pujol JL, Breton JL, Gervais R, Tanguy ML, Quoix E, David P, et al. Phase III double-blind, placebo-controlled study of thalidomide in extensive-disease small-cell lung cancer after response to chemotherapy: an intergroup study FNCLCC cleo04 IFCT 00-01. J Clin Oncol. 2007;25:3945–51. doi: 10.1200/JCO.2007.11.8109. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paik PK, Rudin CM, Pietanza MC, Brown A, Rizvi NA, Takebe N, et al. A phase II study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in relapsed small cell lung cancer. Lung Cancer. 2011;74:481–5. doi: 10.1016/j.lungcan.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 12.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 13.Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–90. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature genetics. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature genetics. 2012;44:1111–6. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voortman J, Lee JH, Killian JK, Suuriniemi M, Wang Y, Lucchi M, et al. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl Acad Sci U S A. 2010;107:13040–5. doi: 10.1073/pnas.1008132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeiffer M, Hartmann TN, Leick M, Catusse J, Schmitt-Graeff A, Burger M. Alternative implication of CXCR4 in JAK2/STAT3 activation in small cell lung cancer. Br J Cancer. 2009;100:1949–56. doi: 10.1038/sj.bjc.6605068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang CL, Liu YY, Ma YG, Xue YX, Liu DG, Ren Y, et al. Curcumin Blocks Small Cell Lung Cancer Cells Migration, Invasion, Angiogenesis, Cell Cycle and Neoplasia through Janus Kinase-STAT3 Signalling Pathway. PLoS One. 2012;7:e37960. doi: 10.1371/journal.pone.0037960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renauld JC. Class II cytokine receptors and their ligands: key antiviral and inflammatory modulators. Nat Rev Immunol. 2003;3:667–76. doi: 10.1038/nri1153. [DOI] [PubMed] [Google Scholar]
- 21.Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11:69–74. doi: 10.1016/s0168-9525(00)89000-9. [DOI] [PubMed] [Google Scholar]
- 22.Constantinescu SN, Girardot M, Pecquet C. Mining for JAK-STAT mutations in cancer. Trends Biochem Sci. 2008;33:122–31. doi: 10.1016/j.tibs.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 23.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 24.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 25.Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–17. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quintas-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nature reviews Drug discovery. 2011;10:127–40. doi: 10.1038/nrd3264. [DOI] [PubMed] [Google Scholar]
- 27.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–98. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 29.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–97. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Voortman J, Dingemans AM, Voeller DM, Pham T, Wang Y, et al. MicroRNA expression and clinical outcome of small cell lung cancer. PLoS One. 2011;6:e21300. doi: 10.1371/journal.pone.0021300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 32.Murugesan G, Aboudola S, Szpurka H, Verbic MA, Maciejewski JP, Tubbs RR, et al. Identification of the JAK2 V617F mutation in chronic myeloproliferative disorders using FRET probes and melting curve analysis. Am J Clin Pathol. 2006;125:625–33. doi: 10.1309/TK0X-L917-XK2V-LRPQ. [DOI] [PubMed] [Google Scholar]
- 33.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrini I, Wang Y, Zucali PA, Lee HS, Pham T, Voeller D, et al. Copy number aberrations of genes regulating normal thymus development in thymic epithelial tumors. Clin Cancer Res. 2013;19:1960–71. doi: 10.1158/1078-0432.CCR-12-3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Favata M, Kelley JA, Caulder E, Thomas B, Wen X, et al. INCB16562, a JAK1/2 selective inhibitor, is efficacious against multiple myeloma cells and reverses the protective effects of cytokine and stromal cell support. Neoplasia. 2010;12:28–38. doi: 10.1593/neo.91192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601–10. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic acids research. 2010;38:D652–7. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scuto A, Krejci P, Popplewell L, Wu J, Wang Y, Kujawski M, et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia. 2011;25:538–50. doi: 10.1038/leu.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10:2384–93. doi: 10.1158/1535-7163.MCT-11-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertino EM, Confer PD, Colonna JE, Ross P, Otterson GA. Pulmonary neuroendocrine/carcinoid tumors: a review article. Cancer. 2009;115:4434–41. doi: 10.1002/cncr.24498. [DOI] [PubMed] [Google Scholar]
- 41.Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–77. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- 42.Park KS, Martelotto LG, Peifer M, Sos ML, Karnezis AN, Mahjoub MR, et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat Med. 2011;17:1504–8. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.