

Abstract
Ischemic revascularization involves extensive structural adaptation of the vasculature, including both angiogenesis and arteriogenesis. Previous studies suggest that fibroblast growth factor (FGF)-2 participates in both angiogenesis and arteriogenesis. Despite this, the specific role of endogenous FGF-2 in vascular adaptation during ischemic revascularization is unknown. Therefore, we used femoral artery ligation in Fgf 2+/+ and Fgf 2−/− mice to test the hypothesis that endogenous FGF-2 is an important regulator of angiogenesis and arteriogenesis in the setting of hindlimb ischemia. Femoral ligation increased capillary and arteriole density in the ischemic calf in both Fgf 2+/+ and Fgf 2−/− mice. The level of angiographically visible arteries in the thigh was increased in the ischemic hindlimb in all mice, and no significant differences were observed between Fgf 2+/+ and Fgf 2−/− mice. Additionally, limb perfusion progressively improved to peak values at day 35 postsurgery in both genotypes. Given the equivalent responses observed in Fgf 2+/+ and Fgf 2−/− mice, we demonstrate that endogenous FGF-2 is not required for revascularization in the setting of peripheral ischemia. Vascular adaptation, including both angiogenesis and arteriogenesis, was not affected by the absence of FGF-2 in this model.
Keywords: revascularization, angiogenesis, basic fibroblast growth factor, arteriogenesis, collateralization
The prevalence of ischemic disease in the heart and limbs has led to extensive investigation in the area of therapeutic blood vessel growth (11). Much of the research in this area has focused on the use of growth factors to induce or augment the body’s naturally occurring revascularization process (24). Despite recent work in this field, much remains unanswered in regard to the basic biological events of vessel growth in the adult organism. Importantly, the specific roles of endogenously produced growth factors in ischemic revascularization, the very same molecules used therapeutically, remain largely unknown.
Spontaneously occurring revascularization, as a consequence of arterial occlusion, is characterized by significant vascular adaptation, including both angiogenesis and arteriogenesis (9, 12). Angiogenesis refers to the growth of capillaries via endothelial cell sprouting, whereas arteriogenesis refers to the growth and remodeling of preexistent arterial vessels into functional collateral arteries (12). The endogenous mediators of these processes are only beginning to be identified. Vascular endothelial growth factor appears to be a major regulator of ischemia-induced angiogenesis (7), and endothelial nitric oxide synthase (eNOS) may be required for both angiogenesis and arteriogenesis during peripheral revascularization (16, 25). In addition, a host of other molecules have been implicated in angiogenesis and arteriogenesis, including fibroblast growth factor (FGF)-2 or basic FGF, tumor necrosis factor-α, monocyte chemoattractant protein-1, and transforming growth factor-β1 (1, 10, 27, 28, 31).
In particular, FGF-2 upregulation parallels vessel growth and improved blood flow in models of hindlimb arterial occlusion (1, 5) and myocardial ischemia (6). Increased expression of FGF-2 colocalizes to growing and newly formed microvascular segments in or around ischemic tissues (4, 28). Additionally, exogenous FGF-2, a potent vascular cell mitogen, enhances angiogenesis and arteriogenesis in animal models of peripheral arterial occlusion (2, 4, 31). Lastly, in experimental models of angiogenesis, addition of FGF-2 upregulated vascular endothelial growth factor expression in capillary vascular cells (23) and modulated remodeling of the microvascular tree (19).
Given these observations, it is often assumed that endogenous FGF-2 is a required mediator of vascular growth and adaptation during ischemic revascularization. However, these studies do not definitively identify FGF-2 as the natural mediator of these processes in vivo because they are based largely on application of exogenous FGF-2 or monitoring of endogenous FGF-2 expression. Thus we used a model of hindlimb ischemia to compare the responses of mice lacking FGF-2 (Fgf 2−/−) and wild-type mice (Fgf 2+/+) to directly examine the importance of FGF-2 during revascularization. We evaluated angiogenesis, arteriogenesis, and the recovery of hindlimb perfusion to determine the specific role of endogenous FGF-2 in this model.
MATERIALS AND METHODS
Animals
Male and female Fgf 2+/+ and Fgf 2−/− mice (33) (8–12 wk of age, 50% Black Swiss and 50% 129 SV) were used for all experiments according to the University of Arizona Institute Animal Care and Use Committee-approved procedures. All mice were genotyped by PCR by using primers specific for the Fgf 2 wild-type allele (forward, 5′-GCTGTACACTCAAGGGGCTC-3′; reverse, 5′-CGCCGT-TCTTGCAGTAGAG-3′) and the Fgf 2 knockout allele (forward, 5′-TCCAAAGCCTGACTTGATCC-3′; reverse, 5′-CTG-ACTAGGGGAGGAGTAGAAGG-3′), after collection of genomic DNA from tail clips. Note that equal numbers of male and female mice were included in each experimental group. Mice were anesthetized with 2.5% Avertin (2.5% 2,2,2-tribromo-ethanol, 2.5% tert-amyl alcohol in PBS; Aldrich) at a dose of 0.15 ml/10 g body wt ip.
Hindlimb ischemia
The model used to produce hindlimb ischemia has been described previously (16). To reduce flow to the left hindlimb, we ligated and excised a portion of the left femoral circulation (both artery and vein). The proximal femoral ligation was made upstream to the medial circumflex femoral artery and the popliteal artery branches. The distal ligation was made in the saphenous artery and vein midway between the ankle and knee. Care was taken to leave the femoral nerve undamaged. Skin incisions were closed with 7.5-mm Michel suture clips and 7.0-mm prolene nonabsorbable suture (Ethicon).
Fgf 2 transcript levels in ischemic hindlimb (RT-PCR)
Tissue (~100 mg) from the calf of ischemic and contralateral (nonischemic) hindlimbs of Fgf 2+/+ and Fgf 2−/− mice was harvested at days 3, 7, and 14 after surgery (n = 2 for each time point). The tissue was immediately homogenized in RNAzol B (Tel-Test) for RNA extraction per manufacturer’s instructions. Equal amounts (1 μg) of total RNA from each sample were reverse transcribed into first-strand cDNA by using Superscript reverse transcriptase (GIBCO BRL). Fgf 2 and Gapdh transcripts were amplified by PCR in separate 100-μl reactions (33). Aliquots of 20 μl were withdrawn at sequential cycles during the PCR reaction for evaluation.
Laser Doppler perfusion imaging
Hindlimb perfusion was measured by using a laser Doppler perfusion imager (LDPI; PIM II, Lisca AB) (7, 29). LDPI values scale linearly with the product of red blood cell velocity and the number of blood cells within the scanned tissue, thus providing a unitless perfusion value (29). Perfusion was evaluated before (control) and immediately after surgery (day 0) as well as serially at days 3, 7, 14, 21, 28, and 35 after induction of ischemia (Fgf 2+/+, n = 4; Fgf 2−/−, n = 4). For LDPI measurements, mice were kept on a heating pad maintained at 37°C for 10 min before scanning, as well as during scanning, to minimize temperature variations during perfusion scans. A perfusion ratio was calculated by dividing the mean perfusion value of the ischemic hindlimb (dorsal side of calf and foot) by the mean perfusion value of an identical region in the nonischemic hindlimb from the same scan. As a positive control, male eNOS knockout mice (eNOS−/−; n = 4) and eNOS wild-type mice (eNOS+/+; n = 4) were evaluated by using LDPI after femoral ligation. eNOS−/− mice have been previously shown to have impaired hindlimb revascularization (16, 25). eNOS mice on a C57BL/6J background were obtained from The Jackson Laboratory (Bar Harbor, ME).
Histology and vessel densities
Animals were killed without surgery (control) and after hindlimb surgery at days 7, 14, 21, and 35 for histology (Fgf 2+/+, n = 4; Fgf 2−/−, n = 4). Whole animals were perfuse fixed with Histochoice (Amresco) at constant pressure (90–100 Torr), and hindlimbs were placed in fixative overnight. The entire hindlimb between the knee and ankle was isolated, and the tibia bone was carefully removed. Microvessels were identified on 6-μm paraffin sections cut from the middle portion of the calf by using standard histochemistry techniques. Capillaries were identified as GS1 (Griffonia simplicifolia I lectin; EY-Labs)-positive vessels smaller than 10 μm in outer diameter. Arterioles were identified as vessels having an outer diameter between 10 and 30 μm that were stained positive for α-smooth muscle actin (SMA; monoclonal antibody against α-SMA; Sigma Chemical, St. Louis, MO) around the entire vessel cross section. Capillaries and arterioles were counted per 20 randomly selected high-power fields (162 × 162-μm field) on two whole limb transverse sections from each hindlimb. Vessel densities were expressed as the number of vessels per square millimeter.
Average muscle fiber cross-sectional area
To calculate cross-sectional area per muscle fiber, digitized images were captured by using a charge-coupled device digital camera (Sony DKC-5000) attached to a microscope (Nikon Optiphot) and then analyzed with image analysis software (Scion Image 4.0). For each animal, a total of three images (420 × 320-μm field) was taken of the same generalized region of the calf (medial gastrocnemius). Perimeters were measured, and cross-sectional area was calculated for all muscle fibers completely within the image frame. Also, the number of muscle fibers per image was recorded. These measurements were performed on control, day 7, and day 35 animals (n = 4 per genotype for each time point).
Cell proliferation
Animals were injected with bromodeoxyuridine (BrdU; 30 mg/kg body wt; Sigma Chemical) intraperitonially at 24 and 12 h before death. BrdU incorporation into the nuclei of proliferating cells was identified on 6-μm sections by using a peroxidase-conjugated sheep anti-BrdU antibody (Biodesign International) as described previously (7). BrdU-positive nuclei were counted per 20 randomly selected high-power fields (162 × 162-μm field) on two whole limb transverse sections from each hindlimb. Proliferation is expressed as the number of BrdU-positive nuclei per square millimeter. Serially cut sections were used to identify the same microscopic fields to colocalize GS1 stained capillaries and BrdU-positive nuclei to determine the identity of the proliferating cells. It has been previously shown (7) that endothelial cells comprise the predominant proliferative cell type in the mouse ischemic hindlimb. Analysis of cell proliferation in the hindlimb was performed on control (nonischemic) and day 7 ischemic limbs based on the previous report of peak proliferation at day 7 in this mouse model (7).
Microangiography
Collateral artery growth (arteriogenesis) was evaluated at days 14 and 35 by using microangiography (Fgf 2+/+, n = 4; Fgf 2−/−, n = 4). Animals were anesthetized and subsequently overdosed with 2.5% Avertin after exposure of the abdominal aorta. Hindlimbs were perfused (90–100 Torr) with PBS containing 1 × 10−5 M sodium nitroprusside through a tapered polyethylene catheter (PE-50) inserted into the aorta, above the iliac branches, to induce maximal vasodilation and to ensure complete filling with contrast agent (barium sulfate 210% wt/vol, Liqui-Coat, Lafayette Pharmaceuticals). In nonischemic limbs, a ligature was placed around the ankle to occlude arteriovenous shunts present in the foot (data not shown) and to limit filling to arteries only. High-definition angiograms (Faxitron Systems) were generated and scanned into a computer for quantitative evaluation by using image analysis software (Scion Image). Vessel area was calculated from inverted images for the entire thigh region between the ligation site and the knee and below the femur by thresholding out nonfilled structures. Next, vessel area, measured from thresholded images, was divided by the total area of the region examined (normalized vessel area). Lastly, an angiographic score was obtained by dividing the normalized vessel area for the left limb (ischemic) by the value obtained for the right limb (nonischemic).
Statistical analysis
Values are presented as means ± SE. Comparison between two means was done by using Student’s unpaired t-test. Multiple groups were compared by one-way ANOVA with a Student-Newman-Keuls test. Statistical significance was set at P < 0.05.
RESULTS
Fgf 2 expression is increased in ischemic hindlimbs of Fgf 2+/+ mice
We used semiquantitative RT-PCR (33) to confirm that Fgf 2 mRNA expression is increased in the hindlimb after ischemia in this model. Fgf 2 transcript levels increased in the ischemic hindlimb of Fgf 2+/+ mice by approximately twofold at day 3 relative to the contralateral, nonischemic hindlimb. This difference was greater than fourfold by day 7 and returned to control levels by day 14 (Fig. 1). As expected, no Fgf 2 transcript was detected in hindlimb tissue from Fgf 2−/− mice (Fig. 1).
Fig. 1.

A: relative Fgf 2 mRNA expression by RT-PCR in ischemic and nonischemic (contralateral) hindlimbs of Fgf 2+/+ mice. Shown are representative results at 3, 7, and 14 days after left femoral artery ligation. Aliquots of the amplification reaction were removed during sequential cycles and gel electrophoresed. Gapdh serves as control for cDNA template quantity and linear amplification. B: no Fgf 2 mRNA was detected in tissues from Fgf 2−/− mice with RT-PCR.
Recovery of resting hindlimb perfusion
Serial perfusion measurements, by LDPI in the same animal over 5 wk, were used to evaluate the temporal recovery of resting perfusion in the ischemic hindlimb. In both Fgf 2+/+ and Fgf 2−/− mice, the hindlimb perfusion ratio (ischemic to nonischemic) was reduced immediately after surgery (Fig. 2). Perfusion progressively improved after induction of ischemia to near normal by day 35 for both Fgf 2+/+ and Fgf 2−/− mice. No significant differences were observed between the perfusion ratios of Fgf 2+/+ and Fgf 2−/− mice at any of the time points examined. As previously shown (16, 25), recovery of hindlimb perfusion was significantly impaired in eNOS−/− mice. These mice were studied to confirm that the LDPI methods used to evaluate the Fgf 2 mice would in fact detect any differences if present. Importantly, the lack of revascularization in the eNOS−/− mice was associated with a progressive necrosis of the ischemic hindlimb. In fact, the majority of eNOS−/− mice studied lost some portion of the distal hindlimb to necrosis as early as day 7. By contrast, Fgf 2−/− mice (as well as Fgf 2+/+ and eNOS+/+) had no signs of necrosis or tissue loss in the hindlimb after femoral ligation. It should be noted that LDPI measurements in our study were done under resting conditions, and resting perfusion values typically reflect only a small portion of the entire perfusion capacity in the hind-limb. However, others (21) report that warming of the mice during LDPI (as performed in our study) induces some degree of vasodilation in the hindlimb. Thus perfusion measurements performed in this manner may represent a larger fraction of total perfusion capacity than actual resting perfusion values.
Fig. 2.

A: representative laser Doppler perfusion imager (LDPI) scans of Fgf 2+/+ and Fgf 2−/− mice before (Pre) and serially after left femoral ligation. Red tones represent highest perfusion, and blue indicates lowest perfusion. B: time course of perfusion recovery after femoral ligation measured from LDPI scans. Perfusion ratio is the average left (ischemic) hindlimb perfusion value divided by the average right (nonischemic) perfusion value. Comparisons between Fgf 2+/+ and Fgf 2−/− were not significantly different. *Significant difference between eNOS+/+ and eNOS−/−, P < 0.05.
Ischemia-induced changes in microvessel density and muscle fiber area
We measured capillary and arteriole density in the calf skeletal muscle of nonischemic and revascularized hindlimbs to assess angiogenesis and microvascular remodeling. Capillary density (per mm2) in the ischemic calves of both Fgf 2+/+ and Fgf 2−/− mice increased significantly (P < 0.05) by day 7 to nearly 1.5 times the respective contralateral, nonischemic hindlimb densities (Fig. 3). Capillary density remained elevated in the ischemic hindlimb in all mice through day 35. No such changes in capillary density were detected in the thighs of ischemic hindlimbs for either genotype (data not shown). No significant differences in capillary density were observed in revascularizing hindlimbs between Fgf 2+/+ and Fgf 2−/− mice. Arteriole density (per mm2) in day 7 ischemic hind-limbs of both Fgf 2+/+ and Fgf 2−/− mice was elevated nearly twofold relative to nonischemic hindlimbs (P < 0.05 vs. control; Fig. 3). As with capillary density, arteriole density remained elevated in the ischemic hindlimbs at day 35. No significant differences in arteriole density were observed between Fgf 2+/+ and Fgf 2−/− mice. We evaluated muscle fiber cross-sectional area, given that changes in fiber area can affect capillary density independent of angiogenesis (20). As shown in Fig. 4, fiber area was not different between the genotypes at the various time points examined. Thus comparison of vessel density between Fgf 2+/+ and Fgf 2−/− mice is not being affected by differential changes in fiber area in one particular genotype. However, we did measure a significantly reduced fiber area at day 7 in all mice. This suggests that capillary density changes measured at this early time point might reflect both changes in fiber area and vascular growth (angiogenesis). Based on previous data (20), the fiber area decrease observed in our study would not completely explain the increase in capillary density measured at day 7 (~300 capillaries/mm2). Nevertheless, fiber area returned to control values when measured at day 35. Thus changes in capillary density in the ischemic limb measured at day 35 appear to be due to angiogenesis rather than to changes in muscle fiber area.
Fig. 3.

A: representative photomicrographs of muscle cross sections from day 35 postischemic hindlimbs of Fgf 2+/+ (WT) and Fgf 2−/− (KO) that were stained with Griffonia simplicifolia I (GSI) lectin to identify capillaries (left) or antibody against α-smooth muscle actin to identify arterioles (right). Sections are counterstained with hematoxylin. B: capillary density in hindlimb muscle without femoral ligation (control) and 7, 14, 21, and 35 days after ligation. C: arteriole density in hindlimb muscle without femoral ligation (control) and 7 and 35 days after ligation. *P < 0.05 vs. Fgf 2+/+ control. #P < 0.05 vs. Fgf 2−/− control. Fgf 2+/+ vs. Fgf 2−/−, not significantly different.
Fig. 4.
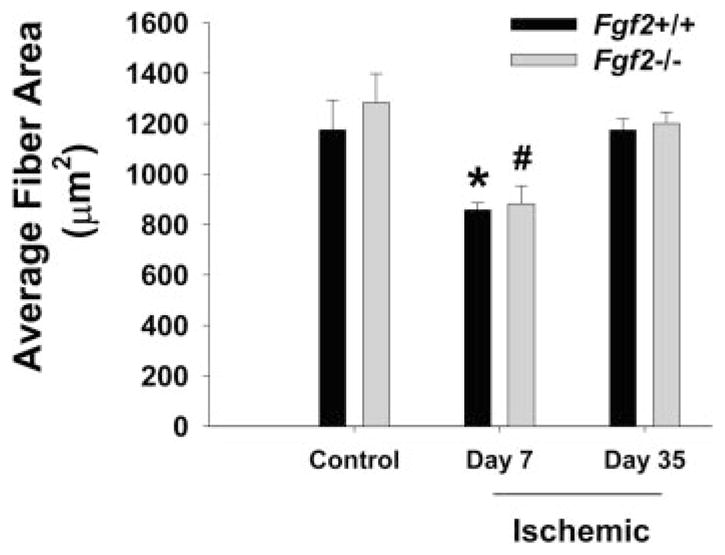
Average muscle fiber area (cross sectional) in hindlimb muscle without femoral ligation (control) and days 7 and 35 after ligation. *P < 0.05 vs. Fgf 2+/+ control (day 35). #P < 0.05 vs. Fgf 2−/− control (day 35). Fgf 2+/+ vs. Fgf 2−/−, not significantly different.
Ischemia-induced changes in cell proliferation
Numerous proliferating cells were detected in the ischemic limbs of both genotypes at day 7 in the ischemic limb (Fig. 5). Very few BrdU-positive cells were seen in nonischemic hindlimbs. Staining of serial sections showed rather consistent colocalization of BrdU-positive cells with GS1-labeling. This is consistent with previous data showing that endothelial cells are the predominant cell type undergoing proliferation at day 7 in the ischemic mouse hindlimb (7). Finally, there was no difference in cell proliferation between Fgf 2+/+ and Fgf 2−/− mice in the day 7 ischemic limbs (Fig. 5).
Fig. 5.

A: representative photomicrographs of muscle cross sections from day 7 postischemic hindlimbs of Fgf 2+/+ (WT) and Fgf 2−/− (KO) that are stained with bromodeoxyuridine (BrdU; blackish-brown stained nuclei) to identify proliferating cells. B: serial sections (6 μm each) from hindlimb stained with BrdU or GS1 to identify proliferating endothelial cells. C: quantification of cell proliferation in hindlimb muscle without femoral ligation (control) and 7 days after ligation.*P < 0.05 vs. respective control. Fgf 2+/+ vs. Fgf 2−/−, not significantly different.
Arteriogenesis in the ischemic mouse hindlimb
We evaluated arteriogenesis (collateral artery development) in the ischemic hindlimbs by using high-resolution microangiography. This technique allowed us to visualize vessels with inner diameters of approximately ≥50 μm. Representative angiograms from Fgf 2+/+ and Fgf 2−/− mice are shown in Fig. 6. Angiograms of nonischemic hindlimbs showed no architectural differences in the vasculature between Fgf 2+/+ and Fgf 2−/− mice. Few angiographically visible collateral vessels were evident immediately after femoral ligation (day 0). At days 14 and 35 postsurgery, collateral arteries (with typical corkscrew patterns) were visible, spanning from the lateral circumflex femoral and deep femoral arteries to the genual arteries (near the knee) and saphenous artery branches. We observed the same general pattern of collateral vessel growth for both Fgf 2+/+ and Fgf 2−/− mice at all time points examined. We used an angiographic score to quantify visible vascular area in the hindlimb. This score was expressed as a ratio of normalized vessel area in the affected (left) limb divided by normalized vessel area in the unaffected (right) limb. As expected, the scores were ~1.0 for nonischemic (control) mice. Immediately after ligation (day 0), scores decreased significantly (P < 0.05) in both the Fgf 2+/+ and Fgf 2−/− mice. At day 14, scores for Fgf 2+/+ and Fgf 2−/− mice increased significantly (P < 0.05) above presurgery values and remained elevated in the revascularized hindlimb through day 35 (Fig. 6). The amount of angiographically visible arteries in the hindlimb did not differ between Fgf 2+/+ and Fgf 2−/− mice.
Fig. 6.

A: representative angiograms in left hindlimbs of Fgf 2+/+ and Fgf 2−/− mice without ligation (control), immediately after ligation (day 0), 14 days after surgery, and 35 days after surgery. B: quantification of angiographically visible arteries in hindlimb. Angiographic score is expressed as left hindlimb vessel area divided by right hindlimb vessel area. *P < 0.05 vs. control and vs. day 0. #P < 0.05 vs. day 0. Fgf 2+/+ vs. Fgf 2−/−, not significantly different.
DISCUSSION
To our knowledge, this is the first study to directly examine the importance of endogenous FGF-2 during revascularization in the setting of peripheral ischemia. The absence of FGF-2 did not appear to affect vascular growth in this model. We did not detect any differences in the vessel density, including capillaries, arterioles, and angiographically visible arteries, when comparing the ischemic limbs of Fgf 2+/+ and Fgf 2−/− mice. Consistent with the above findings, recovery of resting hindlimb perfusion was similar between the FGF-2 wild-type and knockout animals. Furthermore, the absence of any tissue necrosis or gross abnormality in the ischemic limbs of Fgf 2−/− mice provides additional evidence that the revascularization process does not require FGF-2.
These results are somewhat surprising in view of the extensive collection of research implicating FGF-2 in the revascularization of ischemic tissue (1, 4, 5, 28). In animal models, FGF-2 expression has been identified around newly formed microvessels during hindlimb ischemia (4), and strong FGF-2 staining was detected in monocytes accumulating in growing collateral arteries (1). In other studies, FGF-2 is upregulated more globally in the revascularizing regions (5, 6, 28). Similarly, in our study, Fgf 2 expression was elevated in the ischemic hindlimb of wild-type mice at days 3 and 7. During ischemia, the temporal change in FGF-2 expression and its localization to growing collateral arteries and capillaries strongly implicates FGF-2 in the revascularization process. Importantly, Walgenbach et al. (28) used anti-FGF-2 antibody therapy to significantly reduce angiogenesis in ischemic skeletal muscle with the use of a rabbit model. In a study examining injury-induced revascularization in mice (13), antibody neutralization of endogenous FGF-2 significantly reduced the capillary density measured in ischemic muscle of the hindlimb. In contrast, there was no evidence that angiogenesis was altered in the ischemic limbs of Fgf 2−/− mice in our study. Even the amount of cell proliferation (presumably endothelial cells) within ischemic tissues was not different between Fgf 2+/+ and Fgf 2−/− mice, further indicating that angiogenesis was not affected by the absence of FGF-2. Our data are consistent with other results using Fgf 2−/− mice that show that angiogenesis during ischemic retinopathy (18) and ocular choroidal injury (26) does not require endogenous FGF-2. Perhaps the most novel finding in the present study is that arteriogenesis appeared unaffected by the lack of FGF-2. The previous studies cited above examined vascular adaptation at the level of microcirculation rather than adaptation and growth of larger conductance arteries. Our evaluation of angiographically visible arteries, tissue perfusion, and hindlimb viability strongly suggests that collateral artery growth is not impaired in Fgf 2−/− mice. In contrast to Fgf 2−/− and wild-type mice, eNOS−/− animals had reduced limb perfusion and no angiographically visible collateral vessels (data not shown), which was associated with progressive tissue necrosis.
The conflicting results between our study and the various studies using antibody neutralization of FGF-2 could be because of several possibilities. Neutralization studies that identify FGF-2 as an essential mediator of angiogenesis may be overestimating the role of FGF-2 due to inhibition of other FGF proteins. There are at least 22 FGF family members, and these proteins bind to a common group of receptors, although with differing affinities (3, 30). Alternatively, chronic gene loss (e.g., gene knockout) and acute protein loss (e.g., antibody neutralization) may result in distinct responses to the same stimulus. Lastly, differences in the animal models used to examine ischemia and angiogenesis might also account for the contradictory observations.
The apparently normal revascularization response observed in Fgf 2−/− mice may reflect compensation for the loss of FGF-2 by another gene product. Given the large number of FGF proteins, it is possible that there is some degree of redundancy among FGF family members. Genetic ablation of two or more genes in a single animal (e.g., double knockout) is one approach to directly test for compensation. Recently, a double knockout of FGF-1 and FGF-2 was shown to have the same phenotype as Fgf 2−/− mice (14). This suggests that FGF-1, the most closely related FGF family member to FGF-2, is not compensating for the loss of FGF-2 in situations of vascular growth. However, we cannot rule out that FGF-1 or other proteins are acting to functionally replace FGF-2 in our model. Alternatively, it is possible that there is not compensation and that other growth factors or molecules may be the actual mediators of biological events currently ascribed to FGF-2. In this regard, increased FGF-2 expression observed in ischemic tissue may be mediating some other process during revascularization (e.g., vessel responsiveness) that is either unrelated to or not critical for structural adaptation of the vasculature. Of course, we cannot exclude that subtle differences may have been present in the vessels of the ischemic limb of Fgf 2−/− mice that were simply not detected in our study.
To date, three independently generated Fgf 2−/− mouse lines have all been shown to be fertile and to grow to maturity (8, 17, 33). However, numerous phenotypes have been identified in these Fgf 2−/− mice, including thrombocytosis, altered blood pressure regulation, decreased vein vascular smooth muscle activity, impaired cerebral cortex development, and decreased bone mass (8, 15, 17, 33). Studies of adult Fgf 2−/− mice in pathological situations have identified an indispensable and uncompensated role for FGF-2 in wound healing, pressure-induced cardiac hypertrophy, and neurogenesis after brain injury (17, 22, 32). Nevertheless, the present study demonstrates that endogenous FGF-2 is not required for vascular adaptation during ischemic revascularization in the mouse hindlimb. Overall, the lack of a detectable deficit in vascular growth, either developmental or pathological, in Fgf 2−/− mice suggests that we may have to reconsider the importance of endogenous FGF-2 in angiogenesis and arteriogenesis.
Acknowledgments
This work was supported by an American Heart Association, Desert/Mountain Affiliate Predoctoral Fellowship no. 9910147Z (to C. J. Sullivan) and National Heart, Lung, and Blood Institute Grants HL-63732 (to J. B. Hoying) and HL-58511 (to T. Doetschman).
References
- 1.Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101:40–50. doi: 10.1172/JCI119877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baffour R, Berman J, Garb JL, Rhee SW, Kaufman J, Friedmann P. Enhanced angiogenesis and growth of collaterals by in vivo administration of recombinant basic fibroblast growth factor in a rabbit model of acute lower limb ischemia: dose-response effect of basic fibroblast growth factor. J Vasc Surg. 1992;16:181–191. [PubMed] [Google Scholar]
- 3.Bikfalvi A, Klein S, Pintucci G, Rifkin D. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- 4.Bush R, Pevec W, Ndoye A, Cheung A, Sasse J, Pearson D. Regulation of new blood vessel growth into ischemic skeletal muscle. J Vasc Surg. 1998;28:919–928. doi: 10.1016/s0741-5214(98)70070-9. [DOI] [PubMed] [Google Scholar]
- 5.Chleboun J, Martins R. The development and enhancement of the collateral circulation in an animal model of lower limb ischemia. Aust NZ J Surg. 1994;64:202–207. doi: 10.1111/j.1445-2197.1994.tb02179.x. [DOI] [PubMed] [Google Scholar]
- 6.Cohen M, Vernon J, Yaghdjian V, Hatcher V. Longitudinal changes in myocardial basic fibroblast growth factor (FGF-2) activity following coronary artery ligation in the dog. J Mol Cell Cardiol. 1994;26:683–690. doi: 10.1006/jmcc.1994.1081. [DOI] [PubMed] [Google Scholar]
- 7.Couffinhal T, Silver M, Zheng LP, Kearney M, Witzen-bichler B, Isner JM. Mouse model of angiogenesis. Am J Pathol. 1998;152:1667–1679. [PMC free article] [PubMed] [Google Scholar]
- 8.Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hershey JC, Baskin EP, Glass JD, Hartman HA, Gilberto DB, Rogers IT, Cook JJ. Revascularization in the rabbit hindlimb: dissociation between capillary sprouting and arteriogenesis. Cardiovasc Res. 2001;49:618–625. doi: 10.1016/s0008-6363(00)00232-7. [DOI] [PubMed] [Google Scholar]
- 10.Hoefer IE, van Royen N, Buschmann IR, Piek JJ, Schaper W. Time course of arteriogenesis following femoral artery occlusion in the rabbit. Cardiovasc Res. 2001;49:609–617. doi: 10.1016/s0008-6363(00)00243-1. [DOI] [PubMed] [Google Scholar]
- 11.Isner JM, Takayuki A. Therapeutic angiogenesis. Front Biosci. 1998;3:E49–E69. doi: 10.2741/a367. [DOI] [PubMed] [Google Scholar]
- 12.Ito WD, Arras M, Scholz D, Winkler B, Htun P, Schaper W. Angiogenesis but not collateral growth is associated with ischemia after femoral artery occlusion. Am J Physiol Heart Circ Physiol. 1997;273:H1255–H1265. doi: 10.1152/ajpheart.1997.273.3.H1255. [DOI] [PubMed] [Google Scholar]
- 13.Lefaucheur J, Gjata B, Lafont H, Sebille A. Angiogenic and inflammatory responses following skeletal muscle injury are altered by immune neutralization of endogenous basic fibroblast growth factor, insulin-like growth factor-1 and transforming growth factor β1. J Neuroimmunol. 1996;70:37–44. doi: 10.1016/s0165-5728(96)00099-9. [DOI] [PubMed] [Google Scholar]
- 14.Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260–2268. doi: 10.1128/mcb.20.6.2260-2268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, Doetschman T, Coffin JD, Hurley MM. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093. doi: 10.1172/JCI8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozaki H, Okamoto N, Ortega S, Chang M, Ozaki K, Sadda S, Vinores MA, Derevjanik N, Zack DJ, Basilico C, Campochiaro PA. Basic fibroblast growth factor is neither necessary nor sufficient for the development of retinal neovascularization [see comments] Am J Pathol. 1998;153:757–765. doi: 10.1016/S0002-9440(10)65619-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons-Wingerter P, Elliott KE, Clark JI, Farr AG. Fibroblast growth factor-2 selectively stimulates angiogenesis of small vessels in arterial tree. Arterioscler Thromb Vasc Biol. 2000;20:1250–1256. doi: 10.1161/01.atv.20.5.1250. [DOI] [PubMed] [Google Scholar]
- 20.Sarelius IH, Maxwell LC, Gray SD, Duling BR. Capillarity and fiber types in the cremaster muscle of rat and hamster. Am J Physiol Heart Circ Physiol. 1983;245:H368–H374. doi: 10.1152/ajpheart.1983.245.2.H368. [DOI] [PubMed] [Google Scholar]
- 21.Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, Schaper W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34:775–787. doi: 10.1006/jmcc.2002.2013. [DOI] [PubMed] [Google Scholar]
- 22.Schultz JE, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, Pawlowski SA, Lorenz JN, Kimball TR, Doetschman T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest. 1999;104:709–719. doi: 10.1172/JCI7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seghezzi G, Patel S, Ren C, Guanlandris A, Pintucci G, Robbins E, Shapiro R, Galloway A, Rifkin D, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, Laham RJ, Li W, Pike M, Sellke FW, Stegmann TJ, Udelson JE, Rosengart TK. Clinical trials in coronary angiogenesis: issues, problems, consensus: an expert panel summary. Circulation. 2000;102:E73–E86. doi: 10.1161/01.cir.102.11.e73. [DOI] [PubMed] [Google Scholar]
- 25.Tamarat R, Silvestre JS, Kubis N, Benessiano J, Duriez M, deGasparo M, Henrion D, Levy BI. Endothelial nitric oxide synthase lies downstream from angiotensin II-induced angiogenesis in ischemic hindlimb. Hypertension. 2002;39:830–835. doi: 10.1161/hy0302.104671. [DOI] [PubMed] [Google Scholar]
- 26.Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, Vinores SA, Basilico C, Campochiaro PA. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol. 1998;153:1641–1646. doi: 10.1016/S0002-9440(10)65753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Royen N, Hoefer I, Buschmann I, Heil M, Kostin S, Deindl E, Vogel S, Korff T, Augustin H, Bode C, Piek JJ, Schaper W. Exogenous application of transforming growth factor beta 1 stimulates arteriogenesis in the peripheral circulation. FASEB J. 2002;16:432–434. doi: 10.1096/fj.01-0563fje. [DOI] [PubMed] [Google Scholar]
- 28.Walgenbach K, Gratas C, Shestak K, Becker D. Ischaemia-induced expression of bFGF in normal skeletal muscle: a potential paracrine mechanism for mediating angiogenesis in ischemic skeletal muscle. Nat Med. 1995;1:453–459. doi: 10.1038/nm0595-453. [DOI] [PubMed] [Google Scholar]
- 29.Wardell K, Jakobsson A, Nilsson GE. Laser Doppler perfusion imaging by dynamic light scattering. IEEE Trans Biomed Eng. 1993;40:309–316. doi: 10.1109/10.222322. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 31.Yang H, Deschenes M, Ogilvie R, Terjung R. Basic fibroblast growth factor increases collateral blood flow in rats with femoral arterial ligation. Circ Res. 1996;79:62–69. doi: 10.1161/01.res.79.1.62. [DOI] [PubMed] [Google Scholar]
- 32.Yoshimura S, Takagi Y, Harada J, Teramoto T, Thomas SS, Waeber C, Bakowska JC, Breakefield XO, Moskowitz MA. FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc Natl Acad Sci USA. 2001;98:5874–5879. doi: 10.1073/pnas.101034998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 control of vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]