

The current clinical standards in cancer chemotherapy are limited by shortcomings such as poor water solubility, short circulation half-life, high toxicity and cellular resistance. Targeted nanoparticle drug delivery systems have the potential to overcome a number of these challenges [1-4]. Nanodiamonds (NDs) have shown great promise in the fields of biomedical imaging and drug delivery and have been functionalized with a variety of small molecule therapeutics, contrast agents, proteins, polymers and nucleic acids[1, 2, 5-16]. Although NDs are both safe[2, 17-20] and effective at delivering contrast agents[2, 15] and chemotherapeutics[2, 9, 19], in vivo tumor localization has been primarily reliant on passive mechanisms. Previous studies have reported the ability to target nanoparticles to specific subpopulations of cells through the covalent attachment of a protein ligand or antibody [9, 21-24]. For this reason we have developed self-assembled nanodiamond-lipid hybrid particles (NDLPs) that harness the potent interaction between the nanodiamond (ND)-surface and small molecules, while providing a mechanism for cell-targeted delivery of imaging or therapeutic payloads.
The identification of key extracellular receptors and signaling pathways in cancer provides an opportunity to develop targeted drug delivery strategies capable of enhancing efficacy and reducing toxicity. Receptor targeted therapies, such as Her2 inhibitory antibodies and selective estrogen-receptor modulators (SERMs), are now routinely incorporated into breast cancer chemotherapy regimens [25]. However, in order to benefit from these hormone-receptor targeted therapies, the cancer cells must express the appropriate receptors. Triple negative breast cancers (TNBCs) are named as such because they do not express the estrogen receptor (ER), progesterone receptor (PR) and Her2, all of which are commonly targeted with breast cancer therapies [26-28]. TNBCs are among the most aggressive breast cancer subtypes and are associated with poor prognosis, enhanced metastasis and greater rates or recurrence after conventional treatment [29, 30]. Although TNBCs fail to express many typical breast cancer receptors they do typically overexpress epidermal growth factor receptor (EGFR), which could be harnessed as a therapeutic target for improved drug delivery [27, 31, 32].
Herein we report the synthesis, characterization and evaluation of novel nanodiamondlipid hybrid particles (NDLPs) targeted to EGFR. We demonstrate that NDLPs can be readily self-assembled from a variety of modified-NDs and used to specifically deliver imaging or therapeutic molecules to TNBC cells (MDA-MB-231) cells in vitro and in vivo. We show that NDLP-mediated epirubicin delivery improves chemotherapeutic tolerance by mitigating drug-induced mortality and markedly increases treatment efficacy compared to epirubicin alone. Finally, we demonstrate that both NDs and NDLPs appear to be non-toxic in vivo. NDLPs are a scalable, biocompatible and modular platform for the delivery of a wide variety of biological agents, representing an important advance in targeted chemotherapeutic delivery.
NDLPs were generated by the rehydration of lipid thin films containing phosphatidylcholine, cholesterol and distearoylphosphoethanolamine-polyethyleneglycol-biotin with concentrated solutions of ND-complexes. The resulting hybrid particles were then targeted by attaching biotinylated antibodies to biotin-modified lipids with streptavidin cross-bridges (Scheme 1). This system facilitated active targeting of ND-drug complexes without disrupting or limiting the potent ND-drug interaction that enhances therapeutic tolerance and efficacy. NDLPs were synthesized from a variety of functionalized ND complexes, such as dye-labeled and drug-loaded NDs, which suggests that hybrid particle formation is dependent on the ND rather than the complexed molecules. NDLP sizes ranged from 40 to 110 nm in diameter and were strongly dependent on the size of the foundation ND-conjugate (Figure 1a). For the dyeconjugated NDs, the particles in the NDLP suspension are slightly larger than those in the original ND solution, which is likely due to the addition of a lipid monolayer or bilayer to the particle surface. In contrast, NDLP-epirubicin is slightly smaller than ND-epirubicin, which may be due to the lipids stabilizing smaller ND clusters and/or preventing charge-based aggregation. The size of the NDLP-epirubicin increased appropriately with the addition of streptavidin and then the biotinylated antibody, which suggests that the protein additions did not alter complex stability (Figure S1, Supporting Information). In contrast, NDLP zeta-potential was nearly neutral in all cases regardless of the starting ND type (Figure 1b). This is consistent with studies of liposomes composed of phosphatidylcholine (PC) and polyethylene glycol functionalized distearoyl-phosphoethanolamine (PEG-DSPE) [33]. Furthermore, NDLPs were exceptionally stable even in salt solutions, staying in suspension over the course of 72 hours without agitation (Figure S2).
Scheme 1.
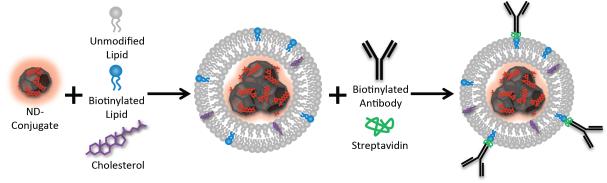
NDLPs are synthesized by rehydration of lipid thin films containing cholesterol and biotinylated lipids with concentrated ND solutions. Hybrid particles are then targeted using biotinylated antibodies and streptavidin crossbridges
Figure 1.

(a) NDLP hydrodynamic radius was strongly dependent on the foundation ND-complex size 488 – AlexaFluor 488, 750 – XenoLights CF750, Epi – Epirubicin. (b) NDLP zeta-potential was nearly neutral for all complexes. (c) Confocal microscopy of NDLPs prior to sizing demonstrated strong colocalization of ND (AlexaFluor 555) and lipid (DID) fluorescent labels. (d) Flow cytometry shows the presence of a dual positive NDLP population in addition to free NDs (AlexaFluor 488 only) and empty liposomes (DID only) (quantified in Table 1). (e) Size exclusion chromatography, nearly all fractions obtained from a size exclusion column contained the fluorescence signature of both the NDs and lipid bilayer in varying ratios. (f) Indirect ELISA using the fluorescence signature of ND-AlexaFluo 488 confirmed successful antibody conjugation to NDLPs. (g) Protein gel electrophoresis of tangential flow filtrate showed bands at approximately160 kDa (black arrowhead), 100kDa and 55kDa. (h) Quantification of the 160 kDa band demonstrated loading of 7.74 μg antibody/mg ND. (i) Epirubicin loading into ND clusters was quantified using absorbance at 485 nm of the supernatant after centrifugation showed consistent drug loading of greater than 85% of the stock solution or 17.3% (wt).
In order to characterize the NDLPs, NDs were labeled with visible range fluorophores and lipid-bilayers were labeled with the lipophilic dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DID). Confocal microscopy of NDLPs prior to sizing demonstrated strong co-localization of the fluorescence signatures of AlexaFluor-555 modified NDs and DID labeled lipid-layers (Figure 1c). Additionally, the NDLP suspension was analyzed by flow cytometry after sizing by probe sonication (Figure 1d). Particles that were positive for both fluorophores – DID and AlexaFluor 488 – were identified as NDLPs. Prior to filtration the suspension was composed of 17.90% NDLPs, 4.47% free NDs and 75.80% non-ND lipid particles (Table 1). Filtration through a 0.2 μm polyethersulfone (PES) filter membrane served two purposes: sterilization and removal of free NDs. Although the particles in solution were smaller than the 200 nm pores of the PES membrane, the remarkable adsorptive capacity that NDs have for polymers in solution makes their affinity for a polymer membrane unsurprising [11, 34]. Additionally, the PES structure contains both aromatic and polarizable residues, which would provide the NDs with ample opportunity for non-covalent interaction by pi-pi stacking or dipole interactions [35]. Post-filtration the solution was composed of 6.40% NDLPs and only 0.59% free NDs, suggesting that any therapeutic effects would be related to ND-drug complexes in NDLPs and not free ND-drug conjugates.
Table 1.
Quantification of NDLPs analyzed by flow cytometry (Figure 1d) before and after filtration through 0.2μm membranes.
| NDLP | ND+ / Lipid − | ND − / Lipid + | |
|---|---|---|---|
| ND Only | 0.03% | 41.60% | 0.12% |
| Lipid Only | 0.67% | 0.51% | 98.60% |
| Unfiltered NDLP | 17.90% | 4.47% | 75.80% |
| Filtered NDLP | 6.40% | 0.59% | 92.90% |
Cryogenic transmission electron microscopy of NDLPs showed the presence of two particle subtypes within the solution. Some ND clusters appeared to be encapsulated within liposomes, while others appeared to form a ND-lipid hybrid particle (Figure S3). When the solution was fractionated using a sepharose CL-4B column after filtration all the fractions, except for 11 and 12, contained the fluorescence signatures of both the NDs and lipid-bilayer (Figure 1e). The lipid presence in each fraction was also confirmed by ICP-AES (Figure S4). The differing ratios of ND:lipid fluorescence in the various fractions are consistent with a solution containing liposome encapsulated NDs (Figure 1e, fractions 1-5), ND-lipid hybrid particles (fractions 6-10) and free NDs (fractions 11-12). This data demonstrates a strong association between the NDs and lipids, which is a key requirement for targeted ND-drug delivery using a lipid-based biotin-streptavidin cross-bridge strategy. Additionally, this data indicates that all fractions of the solution contain NDs, which suggests that the portion of the solution that was read as “lipid only” on flow cytometry may have contained low concentrations of ND as opposed to being without NDs altogether.
Confirmation of functional antibody attachment to NDLPs was initially obtained by indirect ELISA (Figure 1f). In order to more quantitatively determine the degree of antibody loading on the NLDPs the suspension was subjected to tangential flow filtration. Gel electrophoresis of the filtrates of antibody loaded and antibody free particles showed the presence of bands at approximately 160 kDa, 100 kDa and 55 kDa (Figure 1g), which likely correspond to antibody, bovine serum albumin (BSA) dimers and streptavidin respectively (BSA was a component of the antibody reconstitution buffer). As no 160 kDa band was evident in the antibody-free particle filtrate, the 160 kDa band was used to determine the degree of antibody loading (Figure 1h). Comparison to a standard curve generated with the same antibody indicated that the filtrate contained 4.92 μg of antibody. Thus, 60.6% of the antibody was successfully conjugated to NDLPs, resulting in 0.774 wt % loading. Of note, there were some higher molecular weight bands in the BSA control lanes, which likely correspond to BSA dimers and trimers. However the 160 kDa band is always more prominent in the antibody loaded lane, as compared to the paired control lane loaded with an equivalent amount of BSA. Epirubicin loading into ND clusters was also quantified using absorbance at 485 nm. Greater than 85% of the epirubicin stock was consistently loaded into the ND clusters, which resulted in an average of 17.3 wt % loading (Figure 1i). This result is consistent with previously published reports of doxorubicin loading onto NDs [2].
The targeting ability of the NDLPs was evaluated in two breast cancer cell lines: the EGFR-overexpressing MDA-MB-231 cells and MCF-7 cells, which express a lower level of EGFR proteins relative to the MDA-MB-231 cells [36]. NDLPs loaded with ND-AlexaFluor-488 bearing either an anti-EGFR antibody or a non-specific antibody were incubated with both cell lines for 24 hours prior to imaging by confocal microscopy. Increased uptake of NDLPs carrying the anti-EGFR antibody was observed in both cell lines (Figure 2a). The same effect was observed when both cell lines were incubated with varying doses of NDLPs functionalized with the anti-EGFR antibody or antibody-free NDLP (Figure 2b). The addition of the EGFR-targeting antibody to the NDLPs resulted in a 2.89-fold increase in fluorescence in the MDA-MB-231 cells as compared to a 2.23-fold increase in the MCF-7 cells. A similar effect was observed when Xenolight CF 750, a NIR fluorophore, labeled NDs were used as the NDLP foundation (Figure S5).
Figure 2.

(a) Confocal microscopy demonstrates anti-EGFR antibody targeting increases uptake by EGFR-overexpressing MDA-MB-231 cells as compared to non-EGFR-overexpressing MCF-7 cells and non-specific antibody targeting. (b) Increased uptake of EGFR-targeted NDLPs was observed across multiple concentration (*p<10−4, **p<10−5, ***p<10−6, 2-way ANOVA for targeted vs. untargeted, within MDA-MB-231 p=1.35×10-19, within MCF-7 p=3.36×10-16, 4 degrees of freedom). (c) Pre-treatment with EGF diminished uptake of EGFR-targeted NDLPs in a dose dependent fashion, indicating receptor dependent uptake (*p<0.005, **p<10−5, ***p<10- 6). (d) Treatment with anti-EGFR targeted, epirubicin-loaded NDLPs reduced cell viability in both cell lines, however the effect was only significant in the EGFR-overexpressing MDA-MB-231 cells (*p<0.05).
To determine if cells take up NDLPs in an EGFR-dependent fashion, MDA-MB-231 cells were pre-treated with epidermal growth factor (EGF), the natural ligand for the receptor. Cellular uptake of targeted Alexafluor 488-labeled NDLPs was inhibited in a dose dependent fashion by pre-treatment with EGF (Figure 2c), indicating that NDLP update was dependent on EGFR availability. In addition to the NDLPs synthesized from NDs covalently modified with a fluorophore, NDLPs were also made using drug-loaded NDs. ND-mediated delivery of anthracycline chemotherapeutics has been shown to improve therapeutic efficacy by reducing systemic drug toxicity, increasing circulation time and overcoming ABC transporter mediated drug resistance [2]. Epirubicin, an anthracycline chemotherapeutic commonly used to treat breast cancer [25], can be adsorbed onto NDs and targeted using NDLPs. When mammary carcinoma cells were treated for 24 hours, antibody targeting of epirubicin-loaded NDLPs reduced cell viability in both the EGFR-overexpressing and non-EGFR-overexpressing cell lines, however the effect was only significant in the EGFR-overexpressing MDA-MB-231 cells (Figure 2d). In contrast, there was no reduction in the viability of either cell line with the untargeted NDLP-epirubicin, which suggests that epirubicin was sequestered in NDLP clusters.
In order to evaluate the degree of tumor localization in vivo, targeted and untargeted NDLPs were synthesized from NDs conjugated to Xenolight CF 750. Mice bearing luciferase-expressing MDA-MB-231 (MDA-MB-231-luc-D3H2LN) tumor xenografts were administered targeted or untargeted NDLPs by tail vein injection and imaged daily for 10-days. Anti-EGFR antibody targeting enhanced the degree and duration of tumor localization of NDLPs (Figure 3a), with a significant difference in tumor localization at 48 and 72 hours (p=0.003, p=0.0001 respectively; Figure 3b) compared to untargeted NDLPs. Both targeted and untargeted NDLPs appeared to clear by day 7, which allowed for weekly dosing of therapeutic particles (Figure 3b).
Figure 3.

(a) Mice were injected with liposomal complexes 2 weeks after 1×106 MDA-MB-231 cells were implanted into the mammary fat pad of female Nod/scid mice. Luciferase and NIR signal were measured daily following injections. Representative images of MDA-MB-231 tumor-bearing mice injected by tail-vein injection with NDLP-750, anti-EGFR-NDLP-750 or PBS control. (b) Quantification of NIR signal of anti-EGFR-NDLP-750 (n=3) and NDLP-750 (n=3) treated mice (*p=0.003, **p=0.0001). (c) Mice were injected intravenously once a week with liposomal drug complexes and controls two weeks after 1×106 MDA-MB-231 cells were implanted into the mammary fat pad of female Nod/scid mice. Luciferase signal was measured weekly following injections. Representative images of MDA-MB-231 tumor-bearing mice injected by tail-vein injection with PBS control (n=3), epirubicin (Epi) (n=4), untargeted NDLP Epi (n=4) and anti-EGFR-NDLP-Epi (n=4) (d) Quantification of luciferase signal of tumor-bearing mice following treatment (*p=0.006, **p=0.0006)
EGFR targeting with NDLPs provides a method for enhancing delivery of ND-complexes to EGFR-overexpressing tumors. As such, we investigated the effects of NDLP-mediated delivery of therapeutic ND-complexes on tumor progression. Following 2 weeks of tumor growth, tumor-bearing mice were treated weekly with 150 μg of epirubicin or equivalent of targeted or untargeted epirubicin-loaded NDLPs. We elected to compare the targeted NDLPs to epirubicin in order to test their overall efficacy relative to the clinical standard. In addition, we also treated a group of mice with untargeted NDLPs to ascertain the degree of benefit added from targeting. Tumor volumes were measured weekly following treatment (Figure 3c). All mice treated with 150 μg of unmodified epirubicin suffered from drug-related mortality by week 4 (Figure 3d). In contrast, all mice treated with NDLP-epirubicin survived the 7-week study, indicating that NDLP-mediated delivery of epirubicin improves murine tolerance of chemotherapy (Figure 3d). In addition to improving tolerance of therapy, treatment with both anti-EGFR targeted and untargeted NDLP-epirubicin resulted in significant reductions in tumor volume when compared to phosphate buffered saline treated controls (Figure 3d). The most effective treatment was the anti-EGFR targeted NDLP-epirubicin, which resulted in a more than 50% reduction in luciferase signal relative to untargeted particles and nearly complete tumor regression by the end of the 7-week study. These results suggest that targeted delivery of epirubicin using NDLPs can enhance treatment efficacy through increased tumor localization and improve drug tolerance.
Although a number of different groups have evaluated the safety of NDs both in vitro and in vivo [2, 18-20], a comprehensive evaluation of both serum and hematological markers of their safety has, to our knowledge, not yet been reported. 150 μg of epirubicin (N=5), 150 μg equivalent of NDLP-epirubicin (targeted, N=3; untargeted, N=4), equivalent does of ND (833 μg, N=5) or vehicle control (N=5) were delivered to CD-1 mice by tail vein injection. Blood was sampled after 24 hours and subjected to serum chemistry analysis and blood counts. Following treatment, the serum chemistry analysis and blood counts for all of the groups were within the normal range (Figure 4). There was no elevation or suppression of white blood cell counts in any of the groups (Figure 4, WBC), no significant changes in platelet numbers (Figure 4, PLT) and no alternations in white blood cell differential (Figure 4, WBC Differential). When compared to the published normal range for 6-8 week old female CD-1 mice, the red blood cell counts were slightly higher (Figure 4, RBC) and hemoglobin (Figure 4, HGB) was slightly lower than expected. However, no significant differences were observed between groups. In contrast, hematocrit was within the normal range (Figure 4, HCT) and there was no significant perturbation in other red blood cell indices (Figure S6).
Figure 4.

CD-1 mice were treated with 150μg epirubicin (n=5), 833μg of ND (n=5), equivalent of untargeted NDLP (n=3), targeted NDLP (n=4) or vehicle control (n=5) for 24 hours prior to hematological and serum chemistry analysis. No significant differences between treatment and vehicle control groups were observed. Gray region indicates expected normal range for 6-8 week-old, female, CD-1 mice. WBC – white blood cells, PLT-platelets, BA-basophils, EO-eosinophils, MO – monocytes, LY – lymphocytes, NE – neutrophils, RBC – red blood cells, Hgb – hemoglobin, Hct – hematocrit, ALK – alkaline phosphatase, AST – aspartate aminotransferase, ALT – alanine aminotransferase, TPR – total protein, TBIL – total bilirubin, BUN – blood urea nitrogen.
In addition to the hematological analysis, the serum chemistry analysis also indicated no apparent toxicity for all groups. Specifically, we observed no increases in serum marker of inflammation alkaline phosphatase (Figure 4, ALK). There was also no induction of serum markers of hepatotoxicity alanine aminotransferase (Figure 4, ALT) or aspartate aminotransferase (Figure 4, AST), which are particularly important as some of the NDLPs localized to the liver (Figure 5a). There was also no apparent effect upon liver function as measured by total serum protein (Figure 4, TPR) and total bilirubin (Figure 4, TBIL). There was no elevation of blood urea nitrogen (Figure 4, BUN) or serum creatinine (Figure S7, CRE), which indicated good renal function. Finally, there were no major alterations in serum glucose, phosphate, calcium, cholesterol, triglycerides or albumin (Figure S7, GLU, PHOS, CAL, CHOL, TRG, ALB). In general, we observed some variation from the expected normal range published by the mouse supplier (Charles River Laboratories). As the mice were outbred we did expect to see more variation in all of the serum and hematological makers than in an inbred strain. However, there were no statistically significant changes between the vehicle control and any of the other groups. Overall these results indicate that both NDs and NDLPs appear to be biocompatible, which serves as a foundation for their continued preclinical evaluation.
The focus of this study was to develop a self-assembled, ND-based system for targeted delivery of therapeutic and imaging agents to TNBCs. Here we have demonstrated that NDLPs provide an effective method for targeting ND complexes, both for imaging and therapy, to EGFR-overexpressing breast cancer cells in vitro and in vivo. Targeted NDLP-mediated delivery enhanced EGFR-overexpressing tumor treatment in mice and induced tumor regression compared to the administration of the clinical standard while improving overall tolerance epirubicin. Furthermore, both NDs and NDLPs appeared to be non-toxic when systemically administered to mice.
In addition to their high degree of biocompatibility there are a number of features of the NLDPs, such as size and composition, which contribute to the overall success of the system. Size and charge of nanoparticle complexes are important characteristics to consider when designing a drug delivery system, particularly when applied to cancer imaging or therapy. Endothelial junctions in healthy tissue range from 4-8 nm while those in cancerous tissue are on average 40-80 nm, but can be as large as 1 μm [37]. In combination with defective lymphatic drainage near rapidly growing tumors, this irregular tumor vasculature promotes the enhanced permeation and retention of nanoparticles with diameters greater than 8 nm in tumors compared to healthy tissue [37-40]. While drug-delivery complexes larger than 8 nm are beneficial for cancer therapy, the larger particles are cleared at an increased rate by the reticuloendothelial system [41]. For instance, 400 nm diameter liposomes are cleared 7.5 times as quickly as 200 nm liposomes, which in turn are cleared 5 times as quickly as vesicles of approximately 65 nm [42]. Although the size effect on reticuloendothelial system clearance was somewhat mitigated by the addition of polyethylene glycol modified lipids – similar to those used in this study – 200 nm particles are still cleared 54% faster than their 100 nm counterparts [43]. As such, optimal drug-delivery complex size for tumor treatment likely resides between 8 nm and 100 nm. DLS analysis of NDLPs revealed that NDLPs fit closely to these size requirements for optimal drug delivery in cancer therapy. The complex size may be a contributing factor to enhanced drug efficacy and tolerance by both EGFR-targeted and untargeted NDLPs. Furthermore, previous studies with cationic and neutral lipid particles suggest that high cationic lipid content will cause severe aggregation of lipid particles in serum, which contributes to shorter circulation times and poorer distribution into tumor tissues as compared to neutral lipid particles [44]. Both the initial lipid content and zeta-potential analysis of the NDLPs indicates that the particles are nearly neutral, which may contribute to the enhanced tumor localization and drug-delivery of NDLPs by preventing the formation of larger lipid aggregates.
When considering the additional improvement in efficacy observed with the EGFR-targeted NDLPs as compared to untargeted NDLPs, the combination of imaging, efficacy, and safety studies that have been demonstrated in this work should be considered. Figures 3C and 3D clearly show that untargeted NDLP-drug delivery impaired tumor growth instead of inducing tumor regression. This was also recently observed with the untargeted, ND-Doxorubicin (NDX) complex[2]. Treatment with the targeted ND-drug complexes, however, resulted in tumor regression with virtually undetectable levels of luciferase expression (Figures 3C and 3D). The ND-drug interaction coupled with the lipid-antibody architecture that enables increased drug loading and potent ND-drug binding results in improved drug tolerance that is observable with both the untargeted and targeted ND-drug complexes and also with NDX. The enhanced specificity mediated by the antibody, however, appears to be critical to mediating tumor regression. This specificity is further evidenced by the targeted/untargeted imaging studies in figures 3A and 3B.
Part of the success of the NDLP drug delivery systems is due to improvements in drug tolerance. Our group has previously shown that NDs improve chemotherapeutic efficacy by improving tumor localization and increasing drug circulation half-life [2]. The improved drug tolerance seen with ND-mediated drug delivery is at due, in part, to functional drug sequestration within the ND clusters. Myelosuppression, or reduction in white blood cell count, is frequently the dose limiting side effect of chemotherapy as it renders the patient susceptible to opportunistic infections. When doxorubicin, an epimer of epirubicin, was delivered as a free acid, it caused a marked reduction in white blood cell count. However, when it was delivered in ND clusters, the potent but reversible ND-drug interaction eliminated the drug-induced myelosuppression while also improving drug effectiveness [2]. In the current study, premature drug release would have likely resulted in both decreased drug efficacy due to non-specific uptake by normal cells and drug efflux from tumor cells. Furthermore, administration of NDs alone likely does not affect tumor treatment efficacy or toxicity, as comprehensive serum marker in vivo biocompatibility assays in figures 4, S6, and S7 demonstrated every marker remains unchanged following ND-only administration. Therefore, the potent drug-ND binding was likely the basis for the improved drug tolerance observed with both targeted and untargeted NDLP delivery as compared to the administration of epirubicin alone.
Although enhanced drug tolerance is an important feature of the system, sufficient antibody loading is required for the specificity necessary to mediate tumor regression. Overall we were able to achieve loading of 0.774 wt % of antibody onto the NDLP clusters. Assessment of per-particle antibody loading was conducted based on the mass of the NDs in solution and the size of the ND-488 clusters. Utilizing an approximately spherical shape for the ND particles, an estimated 1.76×104 NDs reside in each cluster and approximately 8.42×1010 clusters/mg. If roughly the same concentration of clusters in the ND suspension is present within the NDLP solutions, then on average there were approximately 346 antibodies per particle. Previous studies on immunoliposomes have demonstrated effective cellular targeting in vitro with as few as 20 antibodies per liposome [45]. Therefore, although the antibody loading may vary due to the presence of free liposomes in solution, a 15-fold increase in the number of particles would still present a sufficient number of antibodies on each particle to mediate targeting.
While previous work performed by our laboratory suggests that NDs are cleared from the body, whole body clearance of NDs still remains in question. Yuan et al have previously demonstrated that approximately 60% of NDs remain in the body after 28 days [46]. The primary particles used in that study, however, were an order of magnitude larger than those used here. Furthermore, we observed virtually no fluorescence in the mice administered NDLP-750 10 days after injection. While ND clearance timeframes are a subject of continued study, the ability for whole-body clearance, coupled with the promising safety studies in this work serve as further evidence for the continued translation of NDs towards clinical applications.
Overall, we have presented a completely self-assembled and readily scalable drug delivery platform capable of targeting imaging and improving drug delivery. Furthermore, the platform we have developed appears to be non-toxic. Previous studies on biocompatibility of NDs have focused on in vitro assessment [19, 20], evaluation in nematodes [17] and specific organ toxicity in mice [2, 18]. Here we performed a comprehensive hematological and serum chemistry analysis looking for any evidence of an inflammatory or toxic response to both NDs and NDLPs. Our study demonstrated that there was no significant alteration in organ function with intravenous administration of plain NDs, targeted and untargeted NDLPs, indicating that NDs and NDLPs are well tolerated materials. This data is particularly important as it provides a key step forward towards the clinical translation of ND-based drug delivery.
In conclusion, NDLPs offer a highly scalable and readily adaptable platform for drug delivery and biomedical imaging. The system itself is entirely self-assembled under mild conditions and can serve as a vehicle for a broad array of targeting moieties, therapeutic compounds, and imaging agents. Here we chose to use an anti-EGFR antibody and epirubicin to treat triple negative breast cancers. There are a number of other cancers that could benefit from anti-EGFR targeted therapies, including esophageal, colorectal and lung carcinomas [47-49]. Additionally, the anti-EGFR antibody could be readily replaced by virtually any other biotinylated antibody to target other cancers subtypes, such as anti-CD20 to treat leukemias and lymphomas. Alternatively, CD20-targeted NDLPs could be used to treat B-cell mediated autoimmune diseases or assist in immunosuppression for organ transplant. Furthermore, the ND clusters used here can also be replaced by any number of different ND-types that have been previously described in the literature [2, 5, 11, 15, 50, 51]. This feature provides an opportunity to improve the imaging and treatment of a number of different disease states, ranging from cell-targeted magnetic resonance imaging to detect distant metastases to cell-specific gene delivery to treat inherited metabolic disorders. Furthermore, because NDLP mediated delivery enhances the stability of NDs in salt solutions this platform may also enhance the translational potential of a variety of different ND-complexes.
When compared to other nanoparticle systems both NDs and NDLPs offer a versatile platform for the improvement of drug delivery and biomedical imaging. The unique carbon surface provides the opportunity to employ a variety of different coupling methods, both covalent and adsorptive, while improving drug tolerance. NDs have been shown to improve the safety and efficacy of a variety of different therapeutic and imaging agents [1, 2, 9, 11, 15, 20]. Importantly, NDs also appear to be biocompatible. The combination of all these features in one nanoparticle is very promising for the future clinical translation of ND-based drug delivery platforms, including the NDLPs presented here.
Supplementary Material
Acknowledgements
We thank C. Newcomb and J. Wu for their assistance with CryoTEM and R. Ahn for his guidance in working with lipid particles. We also thank I. Kandela and the Tumor Biology Core for assistance with the mouse toxicity study. We also thank the Northwestern University Atomic and Nanoscale Characterization Experimental Center, High Throughput Analysis Lab, Biological Imaging Facility and Quantitative Bioelemental Imaging Center for the use of their facilities. D.H. gratefully acknowledges support from the National Science Foundation CAREER Award (CMMI-0846323), Center for Scalable and Integrated NanoManufacturing (DMI-0327077), CMMI-0856492, DMR-1105060, V Foundation for Cancer Research Scholars Award, Wallace H. Coulter Foundation Translational Research Award, Society for Laboratory Automation and Screening Endowed Fellowship, Beckman Coulter, and European Commission funding program FP7-KBBE-2009-3. D.H. and L.M. gratefully acknowledge support from National Cancer Institute grant U54CA151880 (The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health). L.M. also thanks the Katten Muchin Rosenman Travel Scholarship Program for their support. E.C. gratefully acknowledges support from the American Cancer Society Postdoctoral Fellowship (PF-08-196-01-MGO).
Footnotes
Experimental Experimental details can be found in the supplemental material.
Supporting Information is available online from Wiley InterScience or from the author.
Contributor Information
Laura K. Moore, Department of Biomedical Engineering Northwestern University Evanston, Illinois, 60208, USA
Edward K. Chow, George Williams Hooper Foundation University of California San Francisco San Francisco, California, 94143, USA
Dr. Eiji Osawa, NanoCarbon Research Institute Shinshu University Ueda, Nagano, Japan
J. Michael Bishop, Department of Microbiology and Immunology George Williams Hooper Foundation University of California San Francisco San Francisco, California, 94143, USA
Dean Ho, Department of Biomedical Engineering Northwestern University Evanston, Illinois, 60208, USA ;Mechanical Engineering Northwestern University Evanston, Illinois, 60208, USA; Institute for Bionanotechnology in Medicine (IBNAM) Robert H. Lurie Comprehensive Cancer Center Northwestern University Chicago. Illinois, 60611, USA.
References
- [1].Chen M, Pierstorff ED, Lam R, Li S-Y, Huang H, Osawa E, Ho D. ACS Nano. 2009;3:2016. doi: 10.1021/nn900480m. [DOI] [PubMed] [Google Scholar]
- [2].Chow EK, Zhang X-Q, Chen M, Lam R, Robinson E, Huang H, Schaffer D, Osawa E, Goga A, Ho D. Sci. Trans. Med. 2011;3:73ra21. doi: 10.1126/scitranslmed.3001713. [DOI] [PubMed] [Google Scholar]
- [3].W. K. L. Chou HH, Chen C a, Wei LH, Lai CH, Hsieh CY, Yang YC, Twu NF, Chang TC, Y. M. S. Gynecol. Oncol. 2006;101:423. doi: 10.1016/j.ygyno.2005.10.027. [DOI] [PubMed] [Google Scholar]
- [4].Gabizon A, Martin F. Drugs. 1997;54(Suppl 4):15. doi: 10.2165/00003495-199700544-00005. [DOI] [PubMed] [Google Scholar]
- [5].Liu KK, Zheng WW, Wang CC, Chiu YC, Cheng CL, Lo YS, Chen C, Chao JI. Nanotechnology. 2010;21:315106. doi: 10.1088/0957-4484/21/31/315106. [DOI] [PubMed] [Google Scholar]
- [6].Smith AH, Robinson EM, Zhang XQ, Chow EK, Lin Y, Osawa E, Xi J, Ho D. Nanoscale. 2011;3:2844. doi: 10.1039/c1nr10278h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wei L, Zhang W, Lu H, Yang P. Talanta. 2010;80:1298. doi: 10.1016/j.talanta.2009.09.029. [DOI] [PubMed] [Google Scholar]
- [8].Zhang Q, Mochalin VN, Neitzel I, Knoke IY, Han J, Klug CA, Zhou JG, Lelkes PI, Gogotsi Y. Biomaterials. 2011;32:87. doi: 10.1016/j.biomaterials.2010.08.090. [DOI] [PubMed] [Google Scholar]
- [9].Zhang X-Q, Lam R, Xu X, Chow EK, Kim H-J, Ho D. Adv. Mater. 2011;4770 doi: 10.1002/adma.201102263. [DOI] [PubMed] [Google Scholar]
- [10].Chen M, Zhang X-Q, Man HB, Lam R, Chow EK, Ho D. J. Phys. Chem. Lett. 2010;1:3167. [Google Scholar]
- [11].Zhang XQ, Chen M, Lam R, Xu X, Osawa E, Ho D. ACS Nano. 2009;3:2609. doi: 10.1021/nn900865g. [DOI] [PubMed] [Google Scholar]
- [12].Mochalin VN, Shenderova O, Ho D, Gogotsi Y. Nat. Nano. 2012;7:11. doi: 10.1038/nnano.2011.209. [DOI] [PubMed] [Google Scholar]
- [13].Gibson JD, Khanal BP, Zubarev ER. J. Am. Chem. Soc. 2007;129:11653. doi: 10.1021/ja075181k. [DOI] [PubMed] [Google Scholar]
- [14].Petráková V, Taylor A, Kratochvĺlová I, Fendrych F, Vacĺk J, Kučka J, Štursa J, Cĺgler P, Ledvina M, Fišerová A, Kneppo P, Nesládek M. Adv. Funct. Mater. 2012;22:812. [Google Scholar]
- [15].Manus LM, Mastarone DJ, Waters EA, Zhang XQ, Schultz-Sikma EA, Macrenaris KW, Ho D, Meade TJ. Nano Lett. 2010;10:484. doi: 10.1021/nl903264h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kruger A, Liang Y, Jarre G, Stegk J. J. Mater. Chem. 2006;16:2322. [Google Scholar]
- [17].Mohan N, Chen C-S, Hsieh H-H, Wu Y-C, Chang H-C. Nano Lett. 2010;10:3692. doi: 10.1021/nl1021909. [DOI] [PubMed] [Google Scholar]
- [18].Yuan Y, Wang X, Jia G, Liu J-H, Wang T, Gu Y, Yang S-T, Zhen S, Wang H, Liu Y. Diamond Relat. Mater. 2010;19:291. [Google Scholar]
- [19].Huang H, Pierstorff E, Osawa E, Ho D. Nano Lett. 2007;7:3305. doi: 10.1021/nl071521o. [DOI] [PubMed] [Google Scholar]
- [20].Zhang X, Hu W, Li J, Tao L, Wei Y. Toxicol. Res. 2012;1:62. [Google Scholar]
- [21].Sigot V, Arndt-Jovin DJ, Jovin TM. Bioconjug. Chem. 2010;21:1465. doi: 10.1021/bc100054c. [DOI] [PubMed] [Google Scholar]
- [22].Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nat. Mater. 2011;10:389. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Cancer Res. 2006;66:6732. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- [24].Mamot C, Drummond DC, Noble CO, Kallab V, Guo Z, Hong K, Kirpotin DB, Park JW. Cancer Res. 2005;65:11631. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- [25].Carlson RW. National Comprehensive Cancer Network. Vol. Version 2. Fort Washington, PA: 2012. [Google Scholar]
- [26].Rakha EA, Ellis IO. J. Clin. Pathol. 2007;60:1300. doi: 10.1136/jcp.2006.045377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Cancer. 2007;109:25. doi: 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- [28].Lippman ME. In: Harrison’s Principles of Internal Medicine. Fauci E. B. Anthony S., Kasper Dennis L., Hauser Stephen L., Longo Dan L., Jameson J. Larry, Loscalzo Joseph., editors. Vol. 17. McGraw Hill: 2008. [Google Scholar]
- [29].Perou CM, Sorlie T, Eisen MB, Rijn M. Van De, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale A-L, Brown PO, Botstein D. Nature. 2000;406:747. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- [30].Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, Rijn M. Van De, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Proc. Natl. Acad. Sci. 2001;98:10869. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Koletsa T, Kotoula V, Karayannopoulou G, Nenopoulou E, Karkavelas G, Papadimitriou CS, Kostopoulos I. Histol. Histopathol. 2010;25:1171. doi: 10.14670/HH-25.1171. [DOI] [PubMed] [Google Scholar]
- [32].Nogi H, Kobayashi T, Suzuki M, Tabei I, Kawase K, Toriumi Y, Fukushima H, Uchida K. Oncol. Rep. 2009;21:413. [PubMed] [Google Scholar]
- [33].Blume G, Cevc G. Biochim. Biophys. Acta Biomembr. 1990;1029:91. doi: 10.1016/0005-2736(90)90440-y. [DOI] [PubMed] [Google Scholar]
- [34].Barnard AS, Sternberg M. J. Mater. Chem. 2007;17:4811. [Google Scholar]
- [35].Adnan A, Lam R, Chen H, Lee J, Schaffer DJ, Barnard AS, Schatz GC, Ho D, Liu WK. Mol. Pharm. 2010;8:368. doi: 10.1021/mp1002398. [DOI] [PubMed] [Google Scholar]
- [36].Reilly RM, Kiarash R, Sandhu J, Lee YW, Cameron RG, Hendler A, Vallis K, Gariepy J. J Nucl Med. 2000;41:903. [PubMed] [Google Scholar]
- [37].Fox ME, Szoka FC, Frechet JM. Acc Chem Res. 2009;42:1141. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jain RK. Cancer Metastasis Rev. 1987;6:559. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- [39].Matsumura Y, Maeda H. Cancer Res. 1986;46:6387. [PubMed] [Google Scholar]
- [40].Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe B. Itty, Bawendi MG, Frangioni JV. Nat. Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Pharmacol Rev. 1999;51:691. [PubMed] [Google Scholar]
- [42].Senior J, Crawley JCW, Gregoriadis G. Biochim. Biophys. Acta Gen. Subj. 1985;839:1. doi: 10.1016/0304-4165(85)90174-6. [DOI] [PubMed] [Google Scholar]
- [43].Woodle MC, Matthay KK, Newman MS, Hidayat JE, Collins LR, Redemann C, Martin FJ, Papahadjopoulos D. Biochim. Biophys. Acta Biomembr. 1992;1105:193. doi: 10.1016/0005-2736(92)90194-q. [DOI] [PubMed] [Google Scholar]
- [44].Zhao W, Zhuang S, Qi XR. Int J Nanomedicine. 2011;6:3087. doi: 10.2147/IJN.S25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mizoue T, Horibe T, Maruyama K, Takizawa T, Iwatsuru M, Kono K, Yanagie H, Moriyasu F. Int. J. Pharm. 2002;237:129. doi: 10.1016/s0378-5173(02)00044-3. [DOI] [PubMed] [Google Scholar]
- [46].Y C. Y. Yuan, H Liu J, H Wang, Y L. Diamond Relat. Mater. 2009;18:95. [Google Scholar]
- [47].Okines A, Cunningham D, Chau I. Nat. Rev. Clin. Oncol. 2011;8:492. doi: 10.1038/nrclinonc.2011.45. [DOI] [PubMed] [Google Scholar]
- [48].Saif MW, Chu E. Cancer J. 2010;16:196. doi: 10.1097/PPO.0b013e3181e076af. [DOI] [PubMed] [Google Scholar]
- [49].Wen J, Fu J, Zhang W, Guo M. Mod. Pathol. 2011;24:932. doi: 10.1038/modpathol.2011.46. [DOI] [PubMed] [Google Scholar]
- [50].Mochalin VN, Gogotsi Y. J. Am. Chem. Soc. 2009;131:4594. doi: 10.1021/ja9004514. [DOI] [PubMed] [Google Scholar]
- [51].Igarashi R, Yoshinari Y, Yokota H, Sugi T, Sugihara F, Ikeda K, Sumiya H, Tsuji S, Mori I, Tochio H, Harada Y, Shirakawa M. Nano Lett. 2012;12:5726. doi: 10.1021/nl302979d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.