

Abstract
Attempted syntheses of (smif)2Ti (smif = 1,3-di-(2-pyridyl)-2-azaallyl) based on metatheses of TiClnLm (n = 2–4) with M(smif) (M = Li, Na), in the presence of a reducing agent (Na/Hg) when necessary, failed, but several apparent Ti(II) species were identified by X-ray crystallography and multidimensional NMR spectroscopy: (smif){Li(smif-smif)}Ti (1, X-ray), [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), (smif)Ti(κ3-N,N(py)2-smif,(smif)H) (3), and (smif)Ti(dpma) (4). NMR spectroscopy and K-edge XAS showed that each compound possesses ligands that are redox non-innnocent, such that d1 Ti(III) centers AF-couple to ligand radicals: (smif){Li(smif-smif)2−}TiIII (1), [(smif2−)TiIII]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), [(smif2−)TiIII](κ3-N,N(py)2-smif,(smif)H) (3), and (smif2−)TiIII(dpma) (4). The instability of the (smif)2Ti relative to its C-C coupled dimer, 2, is rationalized via the complementary nature of the amide and smif radical anion ligands, which are also common to 3 and 4. Calculations support this contention.
Introduction
During the course of examining polydentate chelates that could effect strong fields about first row transition elements,1–3 a ligand degradation occurred that enabled the synthesis of (smif)CrN(TMS)2 (smif = 1,3-di-(2-pyridyl)-2-azaallyl),1 as shown in Scheme 1. A condensation route was then employed to prepare (smif)H (1,3-di-(2-pyridyl)-2-azapropene), which could be deprotonated by standard basic reagents to afford (smif)M′ (M′ = Li, Na, etc.). The neutral and anionic precursors were then utilized to synthesize a new series of Werner complexes, (smif)2M (M = V, Cr, Mn, Fe, Co, Ni, Zn), via metathetical procedures, in addition to other derivatives.4,5 The compounds have unusual optical properties due to intraligand (IL) transitions emanating from the transfer of charge from non-bonding allyl-type orbitals (CNCnb), located principally on the azaallyl, to Π*-orbitals on the pyridine rings. Conspicuously absent in the application to the first row transition elements are the metals titanium and copper.
Scheme 1.

Calculations on (smif)2M, and redox chemistry of related azaallyl compounds, revealed that the relative energy of the CNCnb orbitals vs. those of the metal are critical with regard to the stability of smif-containing complexes.4–6 For the case of hypothetical “(smif)2Cu”, it is likely that the copper dz2 and dx2-y2 orbitals are below those of the smif anion, a situation where the metal internally oxidizes the ligand, thereby causing decomposition. In support of this contention, all attempts at generating “(smif)2Cu” generated degradation products.
The situation for titanium is different. Calculations suggest that the energies of the d-orbitals are well above those of the CNCnb orbitals that constitute the HOMOs of the smif anion, but the Π*-orbitals of smif are likely to be close, raising the possibility that smif may be redox non-innocent.6 In (smif)2Cr, spectroscopic and calculational investigations provided evidence that the compound was best formulated as (smif(−))(smif(2−))CrIII, and related studies of (smif)2V suggested that the HOMO of the compound possessed roughly even amounts of ligand and metal character (i.e., “V(2.5+)”).5
In the quest for (smif)2Ti, the possibility that smif may be redox non-innocent6 is consistent with the limited number of Ti(II) coordination compounds that have been characterized with confidence. Divalent titanium species typically contain organometallic strong field ligands, such as cyclopentadienyl,7–9 pentadienyl,10,11 cyclohexadienyl,12,13 cyclooctadienyl,14 and ligands capable of significant p-backbonding,15 which stabilize the low oxidation state by dispersing excess electron density. The coordination compounds that can legitimately be described as Ti(II) are limited to trans-(tmeda)2TiCl2,16–18 Girolami’s bis-dmpe complexes (i.e., trans-(dmpe)2TiX2; X = Cl, BH4, Me, OPh)19–22 and related derivatives (e.g., (dmpe)TiMe2(diene)23),24,25 (porphyrin)Ti(alkynes),26 and the recently prepared bis-(tris-pyrazolylborate)Ti.27 In the context of applications to organic synthesis,28–31 calixarene dianion Ti(II) arene and alkyne complexes have also been explored.32,33
Herein is described the quest for (smif)2Ti and the actual compounds generated, which were identified within mixtures by modern multidimensional NMR spectroscopic methods. In addition, K-edge spectroscopy is utilized as a means to validate oxidation state assessments based on NMR spectroscopic findings.
Results
Synthesis of (smif)Ti Compounds
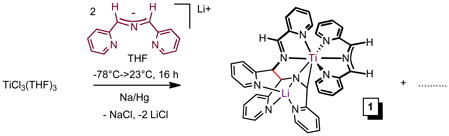
Analogous to the synthesis of (smif)2V,5 treatment of TiCl3(THF)334 with 2 equiv of Li(smif) – generated in situ from LiN(TMS)2 and (smif)H – and one equiv Na/Hg in THF led to a deep purple solution containing a mixture of diamagnetic products, which was later determined to be comprised of four main compounds (1–4, eq 1, Scheme 2). Crystals of (smif){Li(smif-smif)}Ti (1) were deposited upon concentration of a benzene solution, and its structure was subsequently determined by X-ray crystallography. The compound possesses one isolated smif anion, but the remaining two smif ligands are connected by a new C-C bond (shown in red),4,35 and this coupled-smif ligand contains a pocket in which the lithium resides, rendering it an anion. Complex 1 is drawn such that an anionic metalaaziridene unit is bound to a 7-coordinate titanium center, consistent with a formal Ti(II) oxidation state; either the amide or its adjacent carbon can be considered bound to Li, and the other an anion with respect to titanium.
Scheme 2.

 |
(1) |
 |
(2) |
The purification of one component of the mixture (1 in 46%) obtained from eq 1 prompted efforts to vary synthetic conditions and reagents toward additional isolations. Ti-containing starting materials TiCl3, TiCl3(THF)3,34 TiCl4(THF)2, and TiCl2(tmeda)2,16 were exposed to Li(smif), Na(smif), (smif)2Zn5 and (smif)2Mg (eq 2, 68%) in various solvents under varied conditions, and no clean route to any of the products was obtained. The attempts are compiled in the Supplemental Material and reveal that in certain cases only one or two of the products that were ultimately identified are present, but in these cases additional unidentified substances hampered purification. For example, direct treatment of TiCl2(tmeda)2 with two anionic smif equiv obviated the generation of (smif){Li(smif-smif)}Ti (1), but additional species appeared. Furthermore, the sporadic nature of the product distributions was consistent with highly sensitive species.
When (smif){Li(smif-smif)}Ti (1) was redissolved in C6D6, degradation occurred to afford significant quantities of compound 2 within 10 min, and similar amounts of compounds 3 and 4 within 44 h after heating at 80°C (Scheme 2). Given the similar characteristics of the products as judged by 1H NMR spectroscopy, it seemed plausible that modern multidimensional NMR spectroscopic methods could differentiate the products, despite the difficulties encountered in attempting to purify the mixture.
X-ray Crystal Structure of (smif){Li(smif-smif)}Ti (1)
Selected crystallographic and refinement data for (smif){Li(smif-smif)}Ti (1) are given in Table 1, while important metric parameters are listed in the caption of Fig. 1, which provides a view of the molecule. The coordination geometry of 1 is pseudo-octahedral, counting the metalaaziridine as a single site opposite a pyridine of the Li(smif-smif) unit. In contrast to the (smif)2Mn (n = 0, M = V, Cr, Fe, Co, and Ni; n = +1, M = Cr, Co) series,5 which possess smif bite angles from ~77–84°, ∠N1-Ti-N2 = 73.41(4)° and ∠N3-Ti-N2 = 72.63(4)°, concomitant with an elongated titanium-aza distance (d(Ti-N2) = 2.1956(12) Å) expected for the low valent early metal (rcov(Ti) = 1.32). The corresponding titanium-nitrogen distances of the smif pyridines are also longer, with d(Ti-N1) = 2.1693(11) Å and d(Ti-N3) = 2.1678(13) Å, and the CNC “backbone” distances of d(N2-C6) = 1.3437(18) Å and d(N2-C7) = 1.3234(19) Å are consistent with a delocalized anion.
Table 1.
Selected crystallographic and refinement data for (smif){Li(smif-smif)}Ti (1).
| 1a | |
|---|---|
| formula | C45H39N9LiTi |
| formula wt | 760.69 |
| space group | P1bar |
| Z | 2 |
| a, Å | 10.5641(5) |
| b, Å | 10.7536(5) |
| c, Å | 17.1299(9) |
| α, deg | 88.685(2) |
| β, deg | 78.420(2) |
| γ, deg | 82.808(2) |
| V, Å3 | 1891.38(16) |
| ρcalc, g·cm−3 | 1.336 |
| μ, mm−1 | 0.273 |
| temp, K | 173(2) |
| λ(Å) | 0.71073 |
| R indices [I > 2σ(I)]b,c |
R1 = 0.0416 wR2 = 0.0987 |
| R indices (all data)b,c |
R1 = 0.0625 wR2 = 0.1084 |
| GOFd | 1.089 |
The asymmetric unit contains 1 and 1.5 molecules of C6H6.
R1 = Σ||Fo| − |Fc||/Σ|Fo|.
wR2 = [Σw(|Fo| − |Fc|)2/ΣwFo2]1/2.
GOF (all data) = [Σw(|Fo| − |Fc|)2/(n − p)]1/2, n = number of independent reflections, p = number of parameters.
Fig. 1.

Molecular view of (smif){Li(smif-smif)}Ti (1). Selected bond distances (Å) and angles (°): Ti-N1, 2.1693(11); Ti-N2, 2.1956(12); Ti-N3, 2.1678(12); Ti-N4, 2.1120(12); Ti-N5, 2.0231(12); Ti-N8, 1.9722(12); Ti-C31, 2.3260(14); Li-N6, 2.106(3); Li-N7, 2.095(3); Li-N8, 2.005(3); Li-N9, 2.056(3); N2-C6, 1.344(2); N2-C7, 1.323(2); N4-C17, 1.411(2); N5-C18, 1.379(2); C17-C18, 1.367(2); N5-C19, 1.466(2); C19-C25, 1.547(2); N8-C25, 1.453(2); N8-C31, 1.390(2); C31-C32, 1.424(2); N1-Ti-N2, 73.41(4); N1-Ti-N3, 145.73(5); N1-Ti-N4, 90.18(4); N1-Ti-N5, 98.99(5); N1-Ti-N8, 102.80(5); N1-Ti-C31, 84.90(5); N2-Ti-N3, 72.63(4); N2-Ti-N4, 93.56(5); N2-Ti-N5, 165.87(5); N2-Ti-N8, 117.99(5); N2-Ti-C31, 82.46(5); N3-Ti-N4, 87.47(4); N3-Ti-N5, 113.15(5); N3-Ti-N8, 96.95(5); N3-Ti-C31, 95.07(5); N4-Ti-N5, 74.29(5); N4-Ti-N8, 148.05(5); N4-Ti-C31, 174.39(5); N5-Ti-N8, 74.94(5); N5-Ti-C31, 109.12(5); N8-Ti-C31, 36.59(5); C6-N2-C7, 125.07(13); C18-N5-C19, 118.19(12); C25-N8-C31, 124.99(11); N6-Li-N7, 89.64(11); N6-Li-N8, 93.07(11); N6-Li-N9, 153.60(14); N7-Li-N8, 84.76(10); N7-Li-N9, 116.45(12); N8-Li-N9, 85.79(10).
The Li(smif-smif) ligand contains a reduced pyridine-imine component, with an elongated d(N5-C18) of 1.3789(18) Å adjacent to a N5-C19 single bond of 1.4656(18) Å, a shortened C17–C18 distance of 1.367(2) Å, and a long pyridine N4-C17 distance of 1.4105(18) Å. In the context of Wieghardt’s and Chirik’s analyses,36–38 the pyridine-imine is at least mono-reduced, and a case can be made for the dianionic form. A normal titanium-pyridine d(Ti-N4) of 2.1120(12) Å, and a titanium-imine d(Ti-N5) of 2.0231(12) Å, are also observed, and the latter is considerably shorter than the aforementioned Ti-Naza linkage of the smif ligand. The remaining (CN)Ti interaction is best construed as an anionic metalaaziridine unit, where an elongated imine (d(N8-C31) = 1.3903(17) Å) is bound to the titanium via a short d(Ti-N8) of 1.9722(12) Å, and a long d(Ti-C31) of 2.3260(14) Å. The adjacent N8-C25 and C31-C32 bond lengths of 1.4528(18) and 1.424(2) Å, respectively, clearly represent single bonds, and a new carbon-carbon single bond is formed from the coupling of Li(smif) and smif (d(C19-C25) = 1.5472(19) Å). The remaining three pyridine rings associated with the coupled ligand and N8 are ligated to Li in a highly distorted trigonal monoprismatic geometry: ∠N8-Li1-N6 = 93.07(11)°, ∠N8-Li1-N7 = 84.76(10)°, ∠N8-Li1-N9 = 85.79(10)°, ∠N6-Li1-N7 = 89.64(11)°, ∠N6-Li1-N9 = 153.60(14) °, ∠N7-Li1-N9 = 116.45(12) °.
The structure of (smif){Li(smif-smif)}Ti (1) exhibits three anionic ligand or ligand fragments: the smif anion, the reduced pyridine-imine considered as a radical anion,36 and the anionic aziridine of the coupled Li(smif-smif) ligand. From the structure and its diamagnetism, it is proposed that 1 contains a d1 Ti(III) center antiferromagnetically coupled to the pyridine-imine radical anion.
NMR Spectroscopic Characterization
1. Structure Elucidation
With the crystal structure of (smif){Li(smif-smif)}Ti (1) determined, NMR spectroscopy was used to investigate 1 in solution to see if its solid state conformation was retained. The spectral assessment proved more challenging than anticipated when degradation compounds – all of which had been observed in the aforementioned synthetic efforts – appeared during data acquisition. As Scheme 2 reveals, 1 degraded to give substantial quantities of 2 within 30 min, and further decomposition to 3 and 4 occurred over extended periods. A rough mixture of 1-4 was conveniently obtained for spectroscopic purposes upon heating 1 at 80°C for 44 h.
In order to elucidate the identity of the four compounds in solution, a series of 2D NMR spectroscopic experiments were employed, the details of which are given in the Supplemental Material. Initial efforts focused on 2D correlation spectroscopy (COSY), which facilitated identification of protons that were spin coupled to one another, specifically adjacent protons on pyridine rings. From these correlations, the gCOSY (gradient selected) spectrum provided an estimate of 15 different pyridine rings, and identified the most downfield chemical shifts as mostly o-CH resonances. Unfortunately, due to overlapping cross peaks and weak correlations, all 15 rings were not solely assembled via gCOSY experiments. Adiabatic heteronuclear single quantum coherence (HSQCAD) 2D spectroscopy displayed correlations between 75 protons and their corresponding carbons, and permitted identification of backbone (-CHNCH-) and pyridine ring protons. Diverse 13C-chemical shifts aided in differentiating overlapping proton resonances, thereby helping to elucidate 6 pyridine rings. Additionally, HSQCAD revealed new 1H-13C correlations that went undetected in the COSY experiments because they were weak, and a total of 15 pyridine rings were ultimately identified. The quaternary carbons of the pyridine rings were observed via strong correlations with the o-CH positions obtained from 2D gHMBCAD (adiabatic heteronuclear multiple bond correlation) experiments. Cross peaks were observed that proved useful towards assigning the protons around the pyridine rings, as each displayed the same correlation patterns and 13C chemical shift patterns. Ligand fragments were established by correlations observed between pyridine rings and their adjacent methine or methylene backbone resonances.
In order to finish assigning the 15 pyridine rings, total correlation spectroscopy (TOCSY) was utilized since all protons within a specific spin system produced a series of cross peaks. Each pyridine ring possessed its own spin system, and all protons corresponding to one ring appeared in a line, subsequently verifying assignments made from other 2D experiments. Rotating-frame Overhauser effect spectroscopy (ROESY) was used to determine which ligand environments were associated with a specific titanium center, and provided information regarding the spatial orientation of the ligands. The complete assignments of compounds 1-4 are given in Scheme 2.
The solution structure of (smif){Li(smif-smif)}Ti (1) was found to be the same as in the solid state, with coupling of smif and Li(smif) units generating a complicated tetradentate ligand comprised of an imino-pyridine group connected to an anionic metalaaziridine via a C-C bond.4,35 Compounds 2-4 were clearly differentiated by o-pyridine resonances of δ 13.88, 13.99, and 13.61 that corresponded to bound pyridines linked through a –C(sp3)-N-C(sp3)- amide bridge. Compound 2 initially appeared to be “(smif)2Ti” due to the presence of two disconnected smif ligands, but only 5 proton resonances would be expected for a D2d arrangement, yet 15 were observed. Furthermore, one smif possessed mirror symmetry, and backbone 1H and 13C resonances (1H, δ 2.19; 13C, δ 74.93) more consistent with an sp3 carbon center. A structure that accommodates the presence of an asymmetric smif ligand and sp3-carbons in the backbone is [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), where dimerization of “(smif)2Ti” has occurred via the formation of two C-C bonds4,34 between the CNC backbones of the ligands. In the C2h dimer, a 180° rotation axis splits the two C-C bonds of the coupled smifs, and renders the uncoupled smif ligands asymmetric, as both lie in the horizontal mirror plane. A related coupling of smif ligands was observed in the structurally characterized [(TMS)2NFe]2(μ-κ3,κ3-N,N(py)2-smif,smif).4
Compound 3 also possessed an asymmetric smif ligand and two symmetric smif components with sp3-carbon backbone spectral signatures (1H (δ), 13C (δ): 4.38, 73.85; 2.80, 66.39). A key finding was an NH resonance that suggested a structure related to [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), but with one titanium-smif moiety replaced by a proton. A Cs symmetric structure, (smif)Ti(κ3-N,N(py)2-smif,(smif)H) (3), possesses an uncoupled smif ligand in the plane of symmetry, and a smif(smif)H ligand that manifests mirror symmetry. Methylene backbone resonances (1H, δ 3.29; 13C, δ 61.83) consistent with a symmetric di-2-pyridylmethyl-amide (dpma) ligand,4 in addition to a smif ligand, characterized compound 4, which was thus formulated as (smif)Ti(dpma) (4).
2. NMR Evidence of Redox Non-innocence
The X-ray structure determination of 1 led to the conclusion that the metal center was best considered (smif){Li(smif-smif)2−}TiIII (1); the smif anion, a pyridine-imine radical anion, and an anionic metalaaziridene comprised the three formally negative entities. The structural evidence and diamagnetism of 1 is satisfied by a model whereby a d1 Ti(III) center is antiferromagnetically coupled to the radical anion of the pyridine imine unit. Consider the structures proposed for 2-4. All of the titanium centers contain nominally two anionic ligands, one smif and one amide, rendering the metals formally Ti(II), a situation that appears remarkable in view of the paucity of Ti(II) coordination compounds and the potential for the redox non-innocence of smif.5
Since the smif in (smif){Li(smif-smif)}Ti (1) appears to be a standard, closed-shell anion, its chemical shifts can be compared with those in compounds 2-4 to check for differences indicative of redox non-innocence. The –CHNCH- backbone proton resonances of 1 are δ 6.34 and 6.42 (1H), whereas the related signals on 2-4 average δ 3.29(16). The 13C chemical shifts of 1 are δ 110.72 and 112.87, which are close but high relative to the average shift value (δ 108.52(19)) for the related carbons on 2-4.
Pyridine proton chemical shifts for compounds 2-4 were dramatically lower than those observed for (smif){Li(smif-smif)}Ti (1), lending credence to the possibility that all smif ligands in these species were radical dianions.5 The o-CH resonances of δ 7.17 and 8.15 for 1 were ~3 ppm greater than the average o-CH chemical shift for 2-4 (δ 4.28(60)), and the p-CH proton chemical shifts of 1 (δ 6.59, 6.62) were about 2 ppm greater than the corresponding average shift (δ 4.39(20)) for 2-4. The m-CH nearest the quaternary center changed the least, but the average shift from species 2-4 of δ 4.83(22) was still over 1 ppm lower than the corresponding values for 1 (δ 5.95, 6.40). The remaining meta-position -- opposite the bridge -- had an average shift of δ 3.10(33) for compounds 2-4, roughly 3 ppm below those of compound 1 (δ 5.78, 6.02).
While the proton chemical shifts provided a clear indication that the smif ligands in 2-4 were electronically different than the model smif of (smif){Li(smif-smif)}Ti (1), comparison of the 13C resonances afforded a subtler differentiation. The quaternary centers and o-C 13C resonances were statistically similar for all four complexes, but significant deviations were observed for the meta- and para-resonances. The average p-C 13C chemical shift of δ 125.66(33) for 2-4 was ~8 ppm lower that those of 1, while the average resonance (δ 122.81(29)) of the m-C adjacent to the quaternary center was ~5 ppm greater than the corresponding shifts of 1, and the remaining m-C average chemical shift of δ 100.34(34) was ~11 ppm lower than those of 1. Overall, the statistical similarities of the resonances pertaining to compounds 2-4, and their clear differences over those of 1, provide a compelling case for an electronically differentiated ligand. In accord with these observations, compounds 2-4 are taken to be [(smif2−)TiIII]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), [(smif2−)TiIII](κ3-N,N(py)2-smif,(smif)H) (3), and (smif2−)TiIII(dpma) (4). The smif radical dianion is considered to be antiferromagnetically coupled to Ti(III) d1 centers in compounds 2-4.
X-Ray Absorption Spectroscopy
Corroboration for the NMR analysis that suggested the presence of a smif dianion radical in compounds 2-4 was sought from K-edge X-ray Absorption Spectroscopy.5,39–41 Fig. 2 shows normalized K-edge titanium spectra for (smif){Li(smif-smif)}Ti (1) and [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), which was generated in ~90% purity via removal of 1 via crystallizations. For comparison, TiCl3(THF)3 and TiO2 were also examined as references for Ti(III) and Ti(IV) formal oxidation states, respectively. An expanded view of the pre-edge region is also given, and it shows that the lowest features for 1, 2 and TiCl3(THF)3 are shifted to lower energies relative to TiO2 by roughly 1 eV, consistent with a Ti(III) oxidation state assignment. Furthermore, the two features that comprise the leading edge of 1 and 2, presumably the 1s->3d(Π) and 1s->3d(σ*) transitions, are separated by >2 eV, consistent with the expected 17,000–20,000 cm−1 field strength for a smif dianion and dipyridyl-amide ligand set.5
Fig. 2.
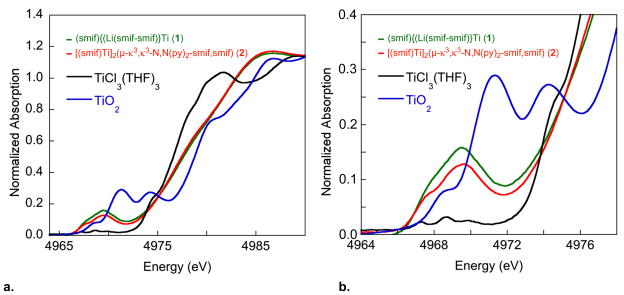
a. Normalized Ti K-edge spectra (10 K) for (smif){Li(smif-smif)}Ti (1), a sample of [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2) that was assayed as ~90% pure by 1H NMR spectroscopy, TiCl3(THF)3 as a reference Ti(III) sample, and TiO2 as a reference for Ti(IV). b. Expansion of the pre-edge region.
Discussion
Nature of (smif)2Ti
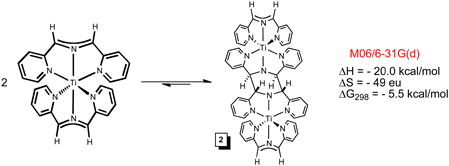
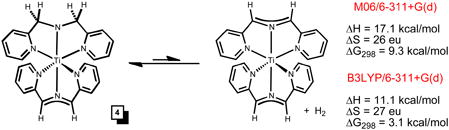
While the quest for (smif)2Ti failed, it was the intrinsic instability of the compound that prevented isolation, as the NMR spectroscopic identification of [(smif)Ti]2(μ-κ3,κ3- N,N(py)2-smif,smif) (2) revealed. Complex 2 is essentially “(smif)2Ti” trans-coupled to another “(smif)2Ti” via two C-C bonds linking the CNC backbones.4,35 At the M06/6-31G(d) level of calculation, the free energy for dimerization at 298K of −5.5 kcal/mol suggests that (smif)2Ti may be thermally accessed via 2, as eq 3 indicates; B3LYP calculations yielded substantially worse agreement.
 |
(3) |
Nature of (smif)Ti Complexes
Although (smif){Li(smif-smif)}Ti (1), [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), (smif)Ti(κ3-N,N(py)2-smif,(smif)H) (3), and (smif)Ti(dpma) (4) may typically and more easily be viewed as Ti(II) species with closed-shell ligands, the structural characterization of 1, and the XAS and NMR spectroscopic investigations of 2-4 suggest otherwise. The metric parameters of 1 indicate the presence of a pyridine-imine radical anion, rendering the complex best described as Ti(III), i.e., (smif){Li(smif-smif)2−}TiIII (1), with the d1 metal center anti-ferromagnetically (AF) coupled to the ligand radical anion. Likewise, the spectral evidence is persuasive for a smif radical dianion AF-coupled to the d1 Ti(III) centers of compounds 2-4, affording [(smif2−)TiIII]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), [(smif2−)TiIII](κ3-N,N(py)2-smif,(smif)H) (3), and (smif2−)TiIII(dpma) (4) as better descriptions.
The portrayals of 1-4 as Ti(III) d1 metal centers AF-coupled to ligand radical anions is not surprising in view of the paucity of non-organometallic Ti(II) species of unquestionable formal oxidation state. One wonders whether bis-(tris-pyrazolylborate)Ti27 and (porphyrin)Ti(alkynes)26 actually have related electronic features that would better fit a Ti(III) description, although the latter compounds have metalacyclopropene character, and are thus “oxidized” by p-backbonding.
 |
(4) |
One surprise from this study and analysis is the finding that the combination of dipyridyl-amide (dpma) and smif act as a pair of tridentate ligands that clearly renders the Ti(III) centers stable. The stability was calculationally assayed via the dehydrogenation of (smif)Ti(dpma) illustrated in eq 4, and the numbers essentially run counter to those calculated for the dehydrogenation of (smif)Fe(dpma) to (smif)2Fe and H2 (ΔH = −2.9; ΔS = 8 eu; ΔG298 = −5.2). Viewing (smif)Ti(dpma) (4) in C2v such that the tridentate ligands are in the xz and yz planes, an electronic configuration of (dxy)1(ligand Π*)1 is expected. The N(pΠ)->Ti(dyz) donation is coupled with the slight σ*-character of the dyz-orbital that derives from the bite angle of smif being less than 180°. A smif radical dianion might be expected to have shorter d(Ti-N), than the monoanion, and a correspondingly greater bite angle. This stronger interaction, presumably electrostatic in origin, would attenuate the σ*-character of the dyz-orbital, thereby permitting a stronger N(pΠ)->Ti(dyz) donation. In this fashion, the tridentate amide ligand and smif radical dianion are complementary in their interactions with the Ti(III) center. In contrast, transient (smif)2Ti may not be metrically and electronically disposed toward a Ti(III) electronic configuration, but in dimerizing, an amide-smif ligand set is achieved along with the higher oxidation state of Ti(III).
Molecular Orbital Diagram of (smif)Ti(dpma) (4)
A truncated molecular orbital diagram of (smif)Ti(dpma) (4) is given in Fig. 3, and while the calculations support the above contention regarding complementary binding of amide and smif, significant covalency in the metal-ligand interactions provides a greater perspective on why these complexes are stable. Central to the figure is the CNCnb smif Π* orbital that shows greater delocalization than in the previous set of (smif)2M calculations.5,6 For M = Fe, Co, Ni, the “t2g” orbitals are below the CNCnb pair, whereas for Mn, Cr, V and Ti, they are above. As in the calculations of hypothetical (smif)2Ti, the d-orbitals are 1.5–2.0 eV above the CNCnb orbital in 4, allowing for significant mixing with Π*-orbitals on the dpma and smif ligands.
Fig. 3.

Truncated molecular orbital diagram of (smif)Ti(dpma) (4; the dpma is in the xz plane, the smif is in the yz plane) illustrating orbitals near the HOMO/LUMO gap. The left side of the diagram features orbitals with dpma character, while the right side pictures those with smif character. The SOMOs whose electrons are AF-coupled to afford the ground state singlet are at ~−2.0 eV; the left orbital is ~50% dxy while the right orbital is ~67% smif Π*, with some dxz admixed. The remaining orbitals with predominantly d-character are not shown, but dxz and dyz are ~ −0.17 and −0.40 eV, respectively, while the σ*-orbitals (dz2 and dx2-y2) are at ~ +1.4 and +2.0 eV.
The AF-coupled α and β electrons reside in orbitals that have considerable ligand character. As anticipated, the lowest “d-orbital” SOMO is dxy, but it is only ~50% titanium, and it manifests subtle Π-bonding with a dpma Π* orbital. The remainder of dxy is spread among two higher orbitals with mostly smif Π* character. The other SOMO is ~67% smif Π* that is bonding with respect to dxz (~33%); again the majority of dxz is at ~−0.17 (not shown) where it interacts with a different smif Π* orbital. The dyz orbital at ~ −0.40 is decidedly antibonding with respect to the N(pΠ), as predicted. While a “back of the envelope” depiction of a Ti(III) d1 complex would surely be (dxy)1, the covalency in the complex reveals the metal d-character to be smeared between dxy and dxz; the other electron is localized on the smif, rendering it a dianion. Given the disparate nature of dpma and smif, it is not surprising that very few of the MOs show a delocalization about both ligands in C2v symmetry.
Mechanism of (smif){Li(smif-smif)}Ti (1) Degradation
Scheme 3 illustrates a potential process by which highly sensitive (smif){Li(smif-smif)}Ti (1) degrades into various products. Reversible formation of the C-C bond (in red) that can be considered to form via nucleophilic attack of Li(smif) on a bound (smif)Ti entity permits generation of (smif)2Ti, which can then dimerize (eq 3) to give [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2). Coupling of Li(smif) and (smif)2Ti via two C-C bonds could lead to 3-(-H)-Li, and adventitious protonation of this entity could generate (smif)Ti(κ3-N,N(py)2-smif,(smif)H) (3). Alternatively, protonolysis or hydrolysis of 2 can also afford 3, and oxidation of “(smif)TiOH” or related species with water can generate the additional dihydrogen needed to covert H(smif) to H(dpma), thereby permitting amide amine exchange of 2 or 3 to afford (smif)Ti(dpma) (4).
Scheme 3.

Conclusion
Metathesis and reduction processes have allowed the synthesis of various titanium compounds, (smif){Li(smif-smif)}Ti (1), [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), (smif)Ti(κ3-N,N(py)2-smif,(smif)H) (3), and (smif)Ti(dpma) (4), whose solution structures have been determined from multidimensional NMR spectroscopy. NMR spectroscopy and K-edge XAS have shown that each compound possesses ligands that are redox non-innocent.5,6,35–48 Consequently, the best electronic descriptions of the complexes are d1 Ti(III) centers AF-coupled to ligand radicals: (smif){Li(smif-smif)2−}TiIII (1), [(smif2−)TiIII]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), [(smif2−)TiIII](κ3-N,N(py)2-smif,(smif)H) (3), and (smif2−)TiIII(dpma) (4). Calculations of 4 show considerable covalency, consistent with the proximity of the titanium d-orbitals with the extended Π*-orbitals of dpma and smif.
Experimental
General Considerations
All manipulations were performed using either glovebox or high vacuum line techniques. All glassware was oven dried. THF and ether were distilled under nitrogen from purple sodium benzophenone ketyl and vacuum transferred from the same prior to use. Hydrocarbon solvents were treated in the same manner with the addition of 1–2 mL/L tetraglyme. Benzene-d6 and toluene-d8 were dried over sodium, vacuum transferred and stored over activated 4Å molecular sieves. THF-d8 was dried over sodium and vacuum transferred from sodium benzophenone ketyl prior to use. Cl2Ti(tmeda)2,16 Li(smif), Na(smif), (smif)2Zn,5 and 1,3-(2-pyridyl)-2-azapropene (smifH)49 were prepared according to literature procedures. Lithium bis(trimethylsilyl)amide was purchased from Aldrich and recrystallized from hexanes prior to use. All other chemicals were commercially available and used as received.
NMR spectra were obtained using an INOVA 400 MHz and 600 MHz spectrometers. Chemical shifts are reported relative to benzene-d6 (1H δ 7.16; 13C{1H} δ 128.39), toluene-d8 (1H δ 2.09; 13C{1H} δ 20.4), and THF-d8 (1H δ 3.58; 13C{1H} δ 67.57). All multidimensional techniques were conducted using INOVA software affiliated with the spectrometers.
Synthesis
1. (smif)2Mg (6-Mg)
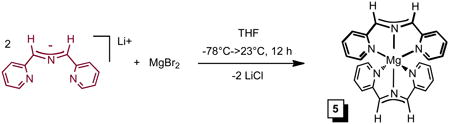
To a 25 mL round bottom flask charged with MgBr2 (0.091g, 0.49 mmol) and Li(smif) (0.200 g, 0.98 mmol) was vacuum transferred 10 mL THF at −78 °C resulting in a deep magenta solution which was slowly warmed to 23 °C. After the solution stirred for 12 h, the volatiles were removed in vacuo. The solid was triturated with Et2O (3 × 5 mL) and filtered to yield (smif)2Mg as a microcrystalline gold solid (0.140 g, 68%). 1H NMR (C6D6, 400 MHz): δ 5.82 (dd, py-C5H, 1 H, J = 6.3, 1.0 Hz), 6.46 (d, py-C3H, 1 H, J = 8.3 Hz), 6.72 (dd, py-C4H, 1 H, J = 7.7, 1.8 Hz), 7.07 (s, CH, 1 H), 7.76 (d, py-C6H, 1 H, J = 5.1 Hz). 13C{1H} NMR (C6D6, 100 MHz): δ 112.20 (CH), 114.14 (py-C3H), 117.61 (py-C5H), 137.04 (py-C4H), 147.90 (py-C6H), 158.46 (py-C2).
2. Typical Approach to “(smif)2Ti”; TiCl3(THF)3, Li(smif) and Na/Hg to 1-4
To a 50 mL 3-neck flask charged with lithium bis(trimethylsilyl)amide (0.340 g, 2.04 mmol) and 0.95 % sodium amalgam (0.024 g Na, 1.06 mmol) was vacuum transferred 15 mL THF at −78 °C. A solution of smifH (0.400 g, 2.02 mmol) in THF (20 mL) was slowly added to the 3-neck flask via a dropping funnel under argon. The solution immediately turned magenta and stirred at −78 °C for 2 h prior to the addition of TiCl3(THF)3 (0.376 g, 2.25 mmol). The reaction mixture turned dark purple after slowly warming to 23 °C and was stirred for 16 h. The volatiles were removed in vacuo, and the resulting dark black-purple solid was filtered in toluene yielding a mixture of compounds 1-4 (0.290 g).
3. Optimized Synthesis of (smif){Li(smif-smif)}Ti (1)
To a 100 mL 3-neck flask charged with lithium smif (0.412 g, 2.04 mmol) and 0.95 % sodium amalgam (0.024 g Na, 1.06 mmol) was vacuum transferred 40 mL THF at −78 °C. The solution immediately turned magenta prior to the addition of TiCl3(THF)3 (0.376 g, 1.01 mmol). The reaction mixture turned dark purple after slowly warming to 23 °C and was stirred for 24 h. The solution was filtered and washed with THF (3 × 10 mL) and removed of all volatiles in vacuo, to give a dark purple crystalline solid (0.300g) in 46% yield. Recrystallization from benzene afforded X-ray quality crystals.
NMR Spectral Assignments
4. Lithium[(k-C,Nam,Nim,Npy3-1,2-bis(pyridin-2-yl)-2-(pyridin-2-ylmethyleneamino)ethyl)(pyri-din-2-ylmethideyl)amido)Ti(smif)] or (smif){Li(smif-smif)}Ti (1)
1H NMR (C6D6, 600 MHz): δ 4.15 (s, backboneC-CH, 1 H), 4.82 (d, backbone-CH, 1 H, J = 5.9 Hz), 5.07 (t, pyim-C5H, 1 H, J = 6.3, 1.2 Hz), 5.30 (d, backbone-CH, 1 H, J = 6.0 Hz), 5.78 (t, pyasym′-C5H, 1 H, J = 6.3 Hz), 5.95 (d, pyasym′-C3H, 1 H, J = 8.5 Hz), 5.97 (t, pyim-C4H, 1 H, J = 5.9, 1.2 Hz), 6.02 (t, pyasym-C5H, 1 H, J = 6.3, 1.0 Hz), 6.11 (t, py‴-C5H, 1 H, J = 6.3, 1.1 Hz), 6.16 (t, py″-C5H, 1 H, J = 6.3, 1.1 Hz), 6.31 (t, py′-C5H, 1 H, J = 6.0 Hz), 6.34 (s, azaasym′-CH, 1 H), 6.40 (d, pyasym-C3H, 1 H, J = 7.7 Hz), 6.41 (d, py‴-C3H, 1 H, J = 9.0 Hz), 6.42 (s, azasym-CH, 1 H), 6.58 (d, py″-C3H, 1 H, J = 7.2 Hz), 6.59 (t, pyasym′-C4H, 1 H, J = 8.0 Hz), 6.62 (dd, pyasym-C4H, 1 H, J = 8.1, 1.4 Hz), 6.70 (t, py″-C4H, 1 H, J = 6.3, 1.8 Hz), 6.70 (s, im-CH, 1 H), 6.73 (d, py′-C3H, 1 H, J = 7.7 Hz), 6.74 (d, pyim-C3H, 1 H, J = 9.0 Hz), 6.82 (t, py′-C4H, 1 H, J = 7.7, 1.8 Hz), 6.97 (t, py‴-C4H, 1 H, J = 7.7 Hz), 7.17 (d, pyasym′-C6H, 1 H, J = 4.4 Hz), 7.51 (d, py‴-C6H, 1 H, J = 4.7 Hz), 7.65 (d, py′-C6H, 1 H, J = 4.7 Hz), 7.69 (d, py″-C6H, 1 H, J = 4.8 Hz), 8.15 (d, pyasym-C6H, 1 H, J =5.5 Hz), 8.42 (d, pyim-C6H, 1 H, J = 6.6 Hz). 13C{1H} (C6D6, 150 MHz): δ 76.50 (backbone-CH), 78.88 (backbone-CH), 87.72 (backboneC-CH), 99.68 (pyim-C5H), 109.18 (py‴-C5H), 110.72 (azasym-CH), 111.58 (pyasym-C5H), 111.75 (pyasym′-C5H), 112.87 (azaasym′-CH), 115.38 (im-CH), 114.95 (py‴-C3H), 117.60 (pyasym-C3H), 117.62 (pyasym′-C3H), 121.43 (py″-C5H), 121.59 (py′-C5H), 121.68 (pyim-C4H), 122.93 (py″-C3H), 123.34 (pyim-C3H), 124.88 (py′-C3H), 133.60 (pyasym′-C4H), 133.82 (py‴-C4H), 134.39 (pyasym-C4H), 135.78 (py″-C4H), 136.90 (py′-C4H), 142.74 (pyim-C2), 146.83 (py‴-C6H), 147.40 (pyasym′-C6H), 147.45 (pyim-C6H), 147.67 (py′-C6H), 147.87 (py″-C6H), 149.74 (pyasym-C6H), 156.80 (pyasym′-C2), 156.93 (pyasym-C2), 164.33 (py′-C2), 164.52 (py″-C2), 164.82 (py‴-C2).
5. [(smif)Ti]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2)
1H NMR (C6D6, 600 MHz): δ 2.19 (s, azasym-CH, 2 H), 2.60 (td, pyasym′-C5H, 1 H, J = 6.6, 1.1 Hz), 3.05 (s, azaasym′-CH, 1 H), 3.18 (t, pyasym-C4H, 1 H, J = 8.6 Hz), 3.35 (s, azaasym-CH, 1 H), 3.49 (d, pyasym′-C6H, 1 H, J = 7.3 Hz), 4.09 (t, pyasym′-C4H, 1 H, J = 7.8 Hz), 4.40 (dd, pyasym-C5H, 1 H, J = 9.3, 6.0 Hz), 4.53 (d, pyasym′-C3H, 1 H, J = 9.3 Hz), 4.71 (d, pyasym-C6H, 1 H, J = 7.0 Hz), 4.98 (d, pyasym-C3H, 1 H, J = 9.7 Hz), 6.44 (d, pysym-C3H, 2 H, J = 8.1 Hz), 7.36 (t, pysym-C4H, 2 H, J = 8.6 Hz), 8.08 (t, pysym-C5H, 2 H, J = 6.6 Hz), 13.88 (d, pysym-C6H, 2 H, J = 4.6 Hz). 13C{1H} (C6D6, 150 MHz): δ 74.93 (azasym-CH), 100.00 (pyasym-C4H), 100.59 (pyasym′-C5H), 108.46 (azaasym′-CH), 108.76 (azaasym-CH), 122.37 (pyasym′-C3H), 123.00 (pyasym-C3H), 124.63 (pysym-C5H), 125.54 (pyasym-C5H), 125.65 (pysym-C3H), 126.19 (pyasym′-C4H), 136.92 (pysym-C4H), 146.22 (pyasym′-C6H), 147.17 (pyasym-C6H), 150.78 (pysym-C2), 154.25 (pysym-C6H), 156.67 (pyasym-C2), 156.74 (pyasym′-C2).
6. (κ-Nam,Npy2-2,3,5,6-tetrakis(pyridin-2-yl)piperazin-1-yl)(smif)Ti or (smif)Ti(κ3-N,N(py)2-smif,(smif)H) (3)
1H NMR (C6D6, 600 MHz): δ 2.80 (t, backbonesymUC-CH, 2 H, J = 9.8 Hz), 3.01 (t, NH, 1 H, J = 9.8 Hz), 3.02 (t, pyasym-C5H, 1 H, J = 7.6 Hz), 3.20 (s, azaasym-CH, 1 H), 3.41 (s, azaasym′-CH, 1 H), 3.52 (t, pyasym′-C4H, 1 H, J = 6.5 Hz), 4.12 (d, pyasym-C6H, 1 H, J = 6.8 Hz), 4.33 (dd, pyasym-C4H, 1 H, J = 9.3, 6.0 Hz), 4.38 (d, backbonesymC-CH, 2 H, J = 8.7 Hz), 4.63 (dd, pyasym′-C5H, 1 H, J = 9.0, 5.8 Hz), 4.68 (d, pyasym′-C3H, 1 H, J = 9.1 Hz), 5.02 (d, pyasym′-C6H, 1 H, J = 6.9 Hz), 5.04 (d, pyasym′-C3H, 1 H, J = 9.0 Hz), 5.99 (d, pysymC-C3H, 2 H, J = 5.8 Hz), 6.30 (d, pysymUC-C3H, 2 H, J = 7.7 Hz), 6.56 (t, pysymUC-C5H, 2 H, J = 6.8 Hz), 6.87 (t, pysymUC-C4H, 2 H, J = 7.7 Hz), 7.09 (t, pysymC-C4H, 2 H, J = 7.7, 1.4 Hz), 7.92 (t, pysymC-C5H, 2 H, J = 6.3 Hz), 8.38 (d, pysymUC-C6H, 2 H, J = 4.4 Hz), 13.99 (d, pysymC-C6H, 2 H, J = 4.8 Hz). 13C{1H} (C6D6, 150 MHz): δ 66.39 (backbonesymUC-CH), 73.85 (backbonesymC-CH), 100.20 (pyasym′-C4H), 100.79 (pyasym-C5H), 108.28 (azaasym′-CH), 108.66 (azaasym-CH), 122.47 (pysymUC-C5H), 122.80 (pyasym′-C3H), 123.12 (pyasym′-C3H), 123.38 (pysymC-C3H), 124.55 (pysymC-C5H), 124.69 (pysymUC-C3H), 125.30 (pyasym′-C5H), 125.71 (pyasym-C4H), 135.87 (pysymUC-C4H), 137.11 (pysymC-C4H), 146.81 (pyasym-C6H), 147.65 (pyasym′-C6H), 149.92 (pysymUC-C6H), 152.74 (pysymC-C2), 154.23 (pysymC-C6H), 156.11 (pyasym-C2), 156.21 (pyasym′-C2),159.96 (pysymUC-C2).
7. (dpma)Ti(smif) (4)
1H NMR (C6D6, 600 MHz): δ 3.19 (t, pysmif-C5H, 1 H, J = 8.6 Hz), 3.29 (s, dpma-CH2, 2 H), 3.43 (s, aza-CH, 1 H), 4.08 (d, pysmif-C6H, 1 H, J = 7.1 Hz), 4.51 (t, pysmif-C4H, 1 H, J = 9.3, 6.0, 1.2 Hz), 4.91 (d, pysmif-C3H, 1 H, J = 9.3 Hz), 6.86 (d, pydpma-C3H, 1 H, J = 9.4 Hz), 7.36 (t, pydpma-C4H, 1 H, J = 6.0 Hz), 7.84 (t, pydpma-C5H, 1 H, J = 6.6 Hz), 13.61 (d, pydpma-C6H, 1 H, J = 5.0 Hz). 13C{1H} (C6D6, 150 MHz): δ 61.83 (dpma-CH2),100.11 (pysmif-C5H), 108.43 (aza-CH), 121.68 (pydpma-C3H), 122.76 (pysmif-C3H), 124.13 (pydpma-C5H), 125.54 (pysmif-C4H), 138.17 (pydpma-C4H), 146.08 (pysmif-C6H), 153.12 (pydpma-C6H), 153.88 (pydpma-C2), 155.72 (pysmif-C2).
XAS Spectroscopy
XAS data were measured at the Stanford Synchrotron Radiation Lightsource using focused beam line 7-3, under ring conditions of 3 GeV and 60–100 mA. A Si(220) double-crystal monochromator was used for energy selection and a Rh-coated mirror (set to an energy cutoff of 9 keV) was utilized in combination with 30% detuning for rejection of higher harmonics. All samples were prepared as dilutions in BN and measured as transmission spectra. Samples were maintained at 10K using an Oxford continuous flow. To check for reproducibility, 2–3 scans were measured for all samples. The energy was calibrated from Ti foil spectra, with the first inflection set to 4966.0 eV. A step size of 0.11 eV was used over the edge region. Data were averaged, and a smooth background was removed from all spectra by fitting a polynomial to the pre-edge region and subtracting this polynomial from the entire spectrum. Normalization of the data was accomplished by fitting a flattened polynomial or straight line to the post-edge region and normalizing the edge jump to 1.0 at 5000 eV.
Single Crystal X-Ray Diffraction Study of (smif){Li(smif-smif)}Ti (1)
A metallic gold-green block (0.35 × 0.25 × 0.15 mm) of (smif){Li(smif-smif)}Ti (1) was obtained from benzene at 23 °C. Upon isolation, the crystals were covered in polyisobutenes and placed under a 173 K N2 stream on the goniometer head of a Siemens P4 SMART CCD area detector (graphite-monochromated MoKα radiation, λ = 0.71073 Å). The structures were solved by direct methods (SHELXS). All non-hydrogen atoms were refined anisotropically unless stated, and hydrogen atoms were treated as idealized contributions (Riding model). A total of 29,322 reflections were collected with 8,504 determined to be symmetry independent (Rint = 0.0356), and 6,256 were greater than 2σ(I). A semi-empirical absorption correction from equivalents was applied, and the refinement utilized w−1 = σ2(Fo2) + (0.0461p)2 + 0.3555p, where p = ((Fo2 + 2Fc2)/3).
Computational Methods
B3LYP50–52 geometry optimization utilized the Gaussian03 suite of programs; the 6–31G(d) basis set was employed. Tests with the larger 6-311+G(d) basis set did not reveal significant differences in the optimized geometries. No symmetry constraints were employed in geometry optimizations, which were started from a pseudo-C2v structure. Calculation of the energy Hessian was performed to confirm species as minima on their respective potential energy surfaces at this level of theory.
Supplementary Material
Synopsis.
Multidimensional NMR spectroscopy, X-ray crystallography, and K-edge XAS show that the best electronic descriptions of 1 and its degradation products are d1 Ti(III) centers antiferromagnetically coupled to ligand radicals: (smif){Li(smif-smif)2−}TiIII (1), [(smif2−)TiIII]2(μ-κ3,κ3-N,N(py)2-smif,smif) (2), [(smif2−)TiIII](κ3-N,N(py)2-smif,(smif)H) (3), and (smif2−)TiIII(dpma) (4; dpma = di-2-pyridylmethyl-amine).
Acknowledgments
PTW thanks the NSF (CHE-0718030) and Cornell University, and TRC the NSF (CHE-1057758) for financial support. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209).
Footnotes
Supporting Information. CIF file for (smif){Li(smif-smif)}Ti (1), descriptions of NMR spectroscopic structure elucidations of 1–4, and a listing of synthetic attempts at “(smif)2Ti” are given. This material is available via the Internet at http://pubs.acs.org.
References
- 1.Frazier BA, Wolczanski PT, Lobkovsky EB. Inorg Chem. 2009;48:11576–11585. doi: 10.1021/ic901329z. [DOI] [PubMed] [Google Scholar]
- 2.Volpe EC, Wolczanski PT, Lobkovsky EB. Organometallics. 2010;29:364–377. [Google Scholar]
- 3.Volpe EC, Manke DR, Bartholomew ER, Wolczanski PT, Lobkovsky EB. Organometallics. 2010;29:6642–6652. [Google Scholar]
- 4.Frazier BA, Wolczanski PT, Lobkovsky EB, Cundari TR. J Am Chem Soc. 2009;131:3428–3429. doi: 10.1021/ja8089747. [DOI] [PubMed] [Google Scholar]
- 5.Frazier BA, Bartholomew ER, Wolczanski PT, DeBeer S, Santiago-Berrios M, Abruña HD, Lobkovsky EB, Bart SC, Mossin S, Meyer K, Cundari TR. Inorg Chem. 2011;50:12414–12436. doi: 10.1021/ic200376f. [DOI] [PubMed] [Google Scholar]
- 6.Hachmann J, Frazier BA, Wolczanski PT, Chan GKL. Chem Phys Chem. 2011;12:3236–3244. doi: 10.1002/cphc.201100286. [DOI] [PubMed] [Google Scholar]
- 7.Chirik PJ. Organometallics. 2010;29:1500–1517. [Google Scholar]
- 8.(a) You Y, Girolami GS. Organometallics. 2008;27:3172–3180. [Google Scholar]; (b) You Y, Wilson SR, Girolami GS. Organometallics. 1994;13:4655–4657. [Google Scholar]
- 9.Devore DD, Timmers FJ, Hasha DL, Rosen RK, Marks TJ, Deck PA, Stern CL. Organometallics. 1995;14:3132–3134. [Google Scholar]
- 10.(a) Wilson AM, Rheingold AL, Waldman TE, Klein M, West FG, Ernst RD. J Organomet Chem. 2009;694:1112–1121. [Google Scholar]; (b) Harvey BG, Mayne CL, Arif AM, Ernst RD. J Am Chem Soc. 2005;127:16426–16435. doi: 10.1021/ja052287b. [DOI] [PubMed] [Google Scholar]; (c) Hylakryspin I, Waldman TE, Melendez E, Trankarnpruk W, Arif AM, Ziegler ML, Ernst RD, Gleiter R. Organometallics. 1995;14:5030–5040. [Google Scholar]; (d) Gedridge RW, Arif AM, Ernst RD. J Organomet Chem. 1995;501:95–100. [Google Scholar]; (e) Ernst RD, Freeman JW, Stahl L, Wilson DR, Arif AM, Nuber B, Ziegler ML. J Am Chem Soc. 1995;117:5075–5081. [Google Scholar]
- 11.Varga V, Polášek M, Hiller J, Thewalt U, Sedmera P, Mach K. Organometallics. 1996;15:1268–1274. [Google Scholar]
- 12.(a) Basta R, Harvey BG, Arif AM, Ernst RD. J Am Chem Soc. 2005;127:11924–11925. doi: 10.1021/ja052214s. [DOI] [PubMed] [Google Scholar]; (b) Arif AM, Basta R, Ernst RD. Polyhedron. 2006;25:876–880. [Google Scholar]; (c) Basta R, Arif AM, Ernst RD. Organometallics. 2005;24:3982–3936. [Google Scholar]; (d) Wilson AM, West FG, Rheingold AL, Ernst RD. Inorg Chim Acta. 2000;300:65–72. [Google Scholar]; (e) Wilson AM, West FG, Arif AM, Ernst RD. J Am Chem Soc. 1995;117:8490–8491. [Google Scholar]
- 13.DiMauro PT, Wolczanski PT. Organometallics. 1987;6:1947–1954. [Google Scholar]
- 14.(a) Kulsomphob V, Tomaszewski R, Rheingold AL, Arif AM, Ernst RD. J Organomet Chem. 2002;655:158–166. [Google Scholar]; (b) Kulsomphob V, Tomaszewski R, Yap GPA, Liable-Sands LM, Rheingold AL, Ernst RD. J Chem Soc Dalton Trans. 1999:3995–4001. [Google Scholar]
- 15.Cloke FGN, Hanks JR, Hitchcock PB, Nixon JF. Chem Commun. 1999:1731–1732. [Google Scholar]
- 16.Edema JJH, Duchateau R, Gambarotta S, Hynes R, Gabe E. Inorg Chem. 1991;30:154–156. [Google Scholar]
- 17.Fowles GWA, Lester TE. Chem Commun. 1967;1:47–48. [Google Scholar]
- 18.Fowles GWA, Lester TE, Walton RA. J Chem Soc A, Inorg Phys Theor. 1968:1081–1085. [Google Scholar]
- 19.Girolami GS, Wilkinson G, Galas AMR, Thornton-Pett M, Hursthouse MB. J Chem Soc, Dalton Trans. 1985:1339–1348. [Google Scholar]
- 20.Morris RJ, Girolami GS. Inorg Chem. 1990;29:4167–4169. [Google Scholar]
- 21.Jensen JA, Girolami GS. Inorg Chem. 1989;28:2107–2113. [Google Scholar]
- 22.Jensen JA, Wilson SR, Schultz AJ, Girolami GS. J Am Chem Soc. 1987;109:8094–8096. [Google Scholar]
- 23.Spencer MD, Wilson SR, Girolami GS. Organometallics. 1997;16:3055–3067. [Google Scholar]
- 24.Goedde DM, Girolami GS. Inorg Chem. 2006;45:1380–1388. doi: 10.1021/ic051556w. [DOI] [PubMed] [Google Scholar]
- 25.Spencer MD, Morse PM, Wilson SR, Girolami GS. J Am Chem Soc. 1993;115:2057–2059. [Google Scholar]
- 26.(a) Thorman JL, Young VG, Jr, Boyd PDW, Guzei IA, Woo LK. Inorg Chem. 2001;40:499–506. doi: 10.1021/ic0003426. [DOI] [PubMed] [Google Scholar]; (b) Woo LK, Hays JA, Jacobson RA, Day CL. Organometallics. 1991;10:2102–2104. [Google Scholar]; (c) Woo LK, Hays JA, Young VG, Day CL, Caron C, D’Souza F, Kadish KM. Inorg Chem. 1993;32:4186–4192. [Google Scholar]
- 27.Kayal A, Kuncheria J, Lee SC. Chem Commun. 2001:2482–2483. doi: 10.1039/b108115b. [DOI] [PubMed] [Google Scholar]
- 28.Sato F, Urabe H, Okamoto S. Chem Rev. 2000;100:2835–2886. doi: 10.1021/cr990277l. [DOI] [PubMed] [Google Scholar]
- 29.Kulinkovich OG, de Meijere A. Chem Rev. 2000;100:2789–2834. doi: 10.1021/cr980046z. [DOI] [PubMed] [Google Scholar]
- 30.Wong HNC. Acc Chem Res. 1989;22:145–152. [Google Scholar]
- 31.Hanamoto T, Yamada K. J Org Chem. 2009;74:7559–7561. doi: 10.1021/jo901392n. [DOI] [PubMed] [Google Scholar]
- 32.Ladipo FT. Comm Inorg Chem. 2006;27:73–102. [Google Scholar]
- 33.(a) Kingston JV, Ozerov OV, Parkin S, Brock CP, Ladipo FT. J Am Chem Soc. 2002;124:12217–12224. doi: 10.1021/ja0271577. [DOI] [PubMed] [Google Scholar]; (b) Ozerov OV, Parkin S, Brock CP, Ladipo FT. Organometallics. 2000;19:4187–4190. [Google Scholar]; (c) Ozerov OV, Patrick BO, Ladipo FT. J Am Chem Soc. 2000;122:6423–6431. [Google Scholar]
- 34.Manzer LE. Inorg Synth. 1982;21:135–140. [Google Scholar]
- 35.Hulley EB, Wolczanski PT, Lobkovsky EB. J Am Chem Soc. 2011;133:18058–18061. doi: 10.1021/ja207638h. [DOI] [PubMed] [Google Scholar]
- 36.(a) Lu CC, Bill E, Weyhermüller T, Bothe E, Wieghardt K. J Am Chem Soc. 2008;130:3181–3197. doi: 10.1021/ja710663n. [DOI] [PubMed] [Google Scholar]; (b) Mondal A, Weyhermüller T, Wieghardt K. Chem Commun. 2009:6098–6100. doi: 10.1039/b912604j. [DOI] [PubMed] [Google Scholar]
- 37.de Bruin B, Bill E, Bothe E, Weyhermüller T, Wieghardt K. Inorg Chem. 2000;39:2936. doi: 10.1021/ic000113j. [DOI] [PubMed] [Google Scholar]
- 38.(a) Bart SC, Chlopek K, Bill E, Bouwkamp MW, Lobkovsky E, Neese F, Wieghardt K, Chirik PJ. J Am Chem Soc. 2006;128:13901. doi: 10.1021/ja064557b. [DOI] [PubMed] [Google Scholar]; (b) Bouwkamp MW, Bowman AC, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2006;128:13340–13341. doi: 10.1021/ja064711u. [DOI] [PubMed] [Google Scholar]; (c) Russell SK, Milsmann C, Lobkovsky E, Weyhermüller T, Chirik PJ. Inorg Chem. 2011;50:3159–3169. doi: 10.1021/ic102186q. [DOI] [PubMed] [Google Scholar]; (d) Tondreau AM, Milsmann C, Patrick AD, Hoyt HM, Lobkovsky E, Wieghardt K, Chirik PJ. J Am Chem Soc. 2010;132:15046–15059. doi: 10.1021/ja106575b. [DOI] [PubMed] [Google Scholar]
- 39.Sarangi R, DeBeer George S, Jackson Rudd D, Szilagyi RK, Ribas X, Rovira C, Holm RH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2007;129:2316–2326. doi: 10.1021/ja0665949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapre R, Bothe E, Weyhermüller T, DeBeer George S, Muresan N, Wieghardt K. Inorg Chem. 2007;46:7827–7839. doi: 10.1021/ic7008607. [DOI] [PubMed] [Google Scholar]
- 41.Banerjee P, Sproules S, Weyhermüller T, DeBeer George S, Wieghardt K. Inorg Chem. 2009;48:5829–5847. doi: 10.1021/ic900154v. [DOI] [PubMed] [Google Scholar]
- 42.(a) Ray K, Petrenko T, Wieghardt K, Neese F. Dalton Trans. 2007:1552–1556. doi: 10.1039/b700096k. [DOI] [PubMed] [Google Scholar]; (b) Ray K, DeBeer George S, Solomon EI, Wieghardt K, Neese F. Chem Eur J. 2007;13:2783–2797. doi: 10.1002/chem.200601425. [DOI] [PubMed] [Google Scholar]; (c) Ray K, Weyhermüller, Neese F, Wieghardt K. Inorg Chem. 2007;44:5345–5360. doi: 10.1021/ic0507565. [DOI] [PubMed] [Google Scholar]; (d) Petrenko T, Ray K, Wieghardt KE, Neese F. J Am Chem Soc. 2006;128:4422–4436. doi: 10.1021/ja0578451. [DOI] [PubMed] [Google Scholar]; (e) Herebian D, Wieghardt KE, Neese F. J Am Chem Soc. 2003;125:10997–11005. doi: 10.1021/ja030124m. [DOI] [PubMed] [Google Scholar]; (f) (a) Spikes GH, Bill E, Weyhermüller T, Wieghardt K. Angew Chem Int Ed. 2008;47:2973–2977. doi: 10.1002/anie.200705410. [DOI] [PubMed] [Google Scholar]
- 43.(a) Scarborough CC, Sproules S, Weyhermüller T, DeBeer S, Wieghardt K. Inorg Chem. 2011;50:12446–12462. doi: 10.1021/ic201123x. [DOI] [PubMed] [Google Scholar]; (b) Sproules S, Weyhermüller T, Goddard R, Wieghardt K. Inorg Chem. 2011;50:12623–12631. doi: 10.1021/ic2016534. [DOI] [PubMed] [Google Scholar]
- 44.(a) Nguyen AI, Zarkesh RA, Lacy DC, Thorson MK, Heyduk AF. Chem Sci. 2011;2:166–169. [Google Scholar]; (b) Nguyen AI, Blackmore KJ, Carter SM, Zarkesh RA, Heyduk AF. J Am Chem Soc. 2009;131:3307–3316. doi: 10.1021/ja808542j. [DOI] [PubMed] [Google Scholar]; (c) Blackmore KJ, Lal N, Ziller JW, Heyduk AG. Eur J Inorg Chem. 2009:735–743. [Google Scholar]; (d) Ketterer NA, Fan H, Blackmore KJ, Yang X, Ziller JW, Baik MH, Heyduk AF. J Am Chem Soc. 2008;130:4364–4374. doi: 10.1021/ja077337m. [DOI] [PubMed] [Google Scholar]; (e) Zaresh RA, Ziller JW, Heyduk AF. Angew Chem Int Ed. 2008;47:4715–4718. doi: 10.1002/anie.200800812. [DOI] [PubMed] [Google Scholar]
- 45.Stanciu C, Jones ME, Fanwick PE, Abu-Omar MM. J Am Chem Soc. 2007;129:12400–12401. doi: 10.1021/ja075396u. [DOI] [PubMed] [Google Scholar]
- 46.Smith AL, Hardcastle KI, Soper JD. J Am Chem Soc. 2010;132:14358–14360. doi: 10.1021/ja106212w. [DOI] [PubMed] [Google Scholar]
- 47.(a) Pierpont CG. Coord Chem Rev. 2001;219:415–433. [Google Scholar]; (b) Pierpont CG. Coord Chem Rev. 2001;216:99–125. [Google Scholar]
- 48.(a) Sproules S, Wieghardt K. Coord Chem Rev. 2011;255:837–860. [Google Scholar]; (b) Sproules S, Wieghardt K. Coord Chem Rev. 2010;254:1358–1382. [Google Scholar]
- 49.Incarvito C, Lam M, Rhatigan B, Rheingold AL, Qin CJ, Gavrilova AL, Bosnich B. J Chem Soc, Dalton Trans. 2001:3478–3488. [Google Scholar]
- 50.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 51.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 52.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN’03, Revision C.02. Gaussian, Inc; Carnegie Office Park, Building 6, Suite 230, Carnegie, PA 15106: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.