

Abstract
Tandem genetic duplications arise frequently between the seven directly repeated 5.5-kb rrn loci that encode ribosomal RNAs in Salmonella enterica. The closest rrn genes, rrnB and rrnE, flank a 40-kb region that includes the purHD operon. Duplications of purHD arise by exchanges between rrn loci and form at a high rate (10−3/cell/division) that remains high in strains blocked for early steps in recombination (recA, recB, and/or recF), but drops 30-fold in mutants blocked for later Holliday junction resolution (ruvC recG). The duplication defect of a ruvC recG mutant was fully corrected by an added mutation in any one of the recA, recB, or recF genes. To explain these results, we propose that early recombination defects activate an alternative single-strand annealing pathway for duplication formation. In wild-type cells, rrn duplications form primarily by the action of RecFORA on single-strand gaps. Double-strand breaks cannot initiate rrn duplications because rrn loci lack Chi sites, which are essential for recombination between two separated rrn sequences. A recA or recF mutation allows unrepaired gaps to accumulate such that different rrn loci can provide single-strand rrn sequences that lack the RecA coating that normally inhibits annealing. A recB mutation activates annealing by allowing double-strand ends within rrn to avoid digestion by RecBCD and provide a new source of rrn ends for use in annealing. The equivalent high rates of rrn duplication by recombination and annealing pathways may reflect a limiting economy of gaps and breaks arising in heavily transcribed, palindrome-rich rrn sequences.
Keywords: duplication, recombination, single-strand annealing, ribosomal RNA loci
THE most frequent tandem gene duplications in the Salmonella chromosome arise between directly repeated ribosomal RNA (rrn) loci (Anderson and Roth 1981; Reams et al. 2010). Duplications formed between such extensive repeats (5.5 kb) are generally thought to arise by unequal recombination between copies of the repeat in different sister chromosomes (Roth et al. 1996; Romero and Palacios 1997) (see Figure 1). Such exchanges are expected to depend heavily on the homologous recombination pathways that rely on the strand exchange enzyme RecA. In keeping with this expectation, duplications between chromosomal repeats of the lac operon form at high rates, comparable to those of rrn duplication, but depend heavily on RecA (A. Reams, M. Carter, and J. Roth unpublished results). In contrast, recent assays in two other situations suggest that RecA can be dispensable for formation of duplications between long repeats that are subject to frequent nicks or breaks (Reams et al. 2010, 2012). Duplication of regions not flanked by extensive repeats are also RecA independent, but arise at rates two to three orders of magnitude lower than those for rrn-mediated duplications (Reams and Neidle 2003, 2004; Kugelberg et al. 2006; Reams et al. 2010). Here we describe duplications arising by exchanges between directly repeated rrn loci that are separated by >40 kb and are normal residents of the Salmonella chromosome. The goal is to understand how these duplications arise at such high rates with or without RecA.
Figure 1.

Process of duplication formation and join-point identification. The duplications described here arise by genetic exchanges between different rrn loci in sister chromosomes and may occur by recombination or by single-strand annealing. Duplications were trapped, and their rrn junction sequences were amplified using PCR with primers that diverge in the parent sequence but converge at a duplication junction (circled). Only the rrnE/B duplication is diagrammed here. The diagram and the text discuss the critical exchange required for duplication without regard to reciprocality or to the ultimate fate of unjoined ends.
Seven rrn loci are scattered around the Salmonella enterica chromosome (Figure 2) and have nearly identical base sequences. Recombination between these loci generates a variety of chromosome rearrangements including duplications, inversions, and translocations (Anderson and Roth 1981; Liu and Sanderson 1998; Helm et al. 2003). Tandem duplications between rrn loci form at very high rates. For example, the argH gene, between rrnA and rrnB (see Figure 2), duplicates at 1.9 × 10−3/cell/division (Reams et al. 2010), and this rate is essentially unaltered in recA mutants (Reams et al. 2010). These duplications of argH are lost at a 10-fold higher rate (10−2/cell/division) by heavily RecA-dependent recombination between the extensive duplicated regions (>150 kb). Because of their high loss rate and fitness cost, argH duplications are carried as stable polymorphisms in unselected cultures and are maintained at a steady-state frequency of ∼1% (Reams et al. 2010). These high steady-state frequencies are likely to be a general property of all duplications in all organisms.
Figure 2.

Positions of rrn loci and nature of purHD duplication endpoints. The genetic map indicates the position of rrn loci on the circular S. enterica chromosome. The expanded top region shows the purHD region analyzed here. Map distances are not to scale. The distribution of duplications that include the purHD locus is based on 30 purH duplications isolated in a recA+ strain and 20 duplications (in parentheses) isolated in a recA mutant strain.
The surprising RecA independence of rrn-mediated duplication formation is examined here using the purHD locus. This region is flanked by the most closely spaced rrn cistrons, rrnB and rrnE (separated by 40 kb), and is held at the highest duplication steady-state frequency (∼3%) of any tested point in the chromosome (Anderson and Roth 1981). It is suggested that the high rate and apparent recombination independence of rrn duplications may reflect two features of rrn sequences. First, rrn cistrons are the most highly transcribed genes in the Salmonella and Escherichia coli chromosomes (Dennis 2004). Second, rrn sequences include many stem-loop structures that are responsible for folding of the ribosomal RNAs (16S, 23S, 5S) produced from each locus. The palindromic features of rrn DNA may allow untranscribed strands to form secondary structures that are subject to cutting or breakage. These unusual features may make rrn sequences prone to frequent gaps and breaks.
It is proposed here that blockage of early recombination steps (RecBCDA and RecFORA) can activate a single-strand annealing pathway of duplication formation that compensates for loss of recombination. When frequent DNA damage within rrn sequences remains unrepaired by recombination, these lesions can accumulate sufficiently that two different rrn loci can provide ends. Duplications can form by annealing when two single-strand ends are provided and neither strand is coated with inhibitory RecA protein. Activation of annealing pathways renders duplication formation independent of Holliday resolution activities (RuvC, RecG).
Materials and Methods
Strains and media
All strains were derivatives of S. enterica (Typhimurium) strain LT2 and are listed in Table 1. Rich medium was Luria broth (LB), used with antibiotics as described below.
Table 1. Strains used in this study.
| Strain | Genotype1 | Source |
|---|---|---|
| TR10000 | Wild-type S. enterica (serovar Typhimurium) LT2 | Lab collection |
| TT25620 | purH887::Tn10, tetA101::Kan(TcSKnR) DEL(Fels-2 Gifsy-1 Gifsy-2) sulA46::Spc lexA34::Rif(sw)/pSIM5(CmR) | This report |
| TT25621 | purH887::Tn10, tetA101::Kan(TcSKnR) DEL(Fels-2 Gifsy-1 Gifsy-2) sulA46::Spc lexA34::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT25671 | purH887::Tn10, tetA101::Kan(TcSKnR) DEL(Fels-2 Gifsy-1 Gifsy-2) sulA46::Spc lexA33:(Rif lexA3IND)/pSIM5(CmR) | This report |
| TT25672 | purH887::Tn10, tetA101::Kan(TcSKnR) DEL(Fels-2 Gifsy-1 Gifsy-2) sulA46::Spc lexA33:(Rif lexA3IND) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26376 | zja9229::tetRA(sw); tetA101::Kan (TcSKnR)/pSIM5(CmR) | This report |
| TT26377 | metA2382::tetRA(sw); tetA101::Kan (TcSKnR)/pSIM5(CmR) | This report |
| TT26379 | thrA537::tetRA(sw); tetA101::Kan (TcSKnR)/pSIM5(CmR) | This report |
| TT26380 | zja9229::tetRA(sw); tetA101::Kan (TcSKnR) rrnE::GntR(sw)/pSIM5(CmR) | This report |
| TT26381 | metA2382::tetRA(sw); tetA101::Kan (TcSKnR) rrnE::GntR(sw)/pSIM5(CmR) | This report |
| TT26382 | thrA537::tetRA(sw); tetA101::Kan (TcSKnR) rrnE::GntR (sw)/pSIM5(CmR) | This report |
| TT26402 | thrA537::tetRA(sw); tetA101::Kan (TcSKnR) rrnH::Spc (sw)/pSIM5(CmR) | This report |
| TT26403 | thrA537::tetRA(sw); tetA101::Kan (TcSKnR) rrnH::Spc(sw) rrnE::GntR(sw)/pSIM5(CmR) | This report |
| TT26404 | metA2382::tetRA(sw); tetA101::Kan (TcSKnR) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26405 | metA2382::tetRA(sw); tetA101::Kan (TcSKnR) rrnE::Gnt(sw) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26406 | zja9229::tetRA(sw); tetA101::Kan (TcSKnR) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26407 | zja9229::tetRA(sw); tetA101::Kan (TcSKnR) rrnE::Gnt(sw) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26438 | purH2411::tetAR; tetA101::Kan(TcSKnR)/pSIM5(CmR) | This report |
| TT26439 | purD2412::tetAR; tetA101::Kan(TcSKnR)/pSIM5(CmR) | This report |
| TT26441 | purD2412::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif/pSIM5(CmR) | This report |
| TT26442 | purH2411::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif/pSIM5(CmR) | This report |
| TT26443 | purD2412::tetAR; tetA101::Kan(kanR) ruvC4::Rif(sw)/pSIM5(CmR) | This report |
| TT26444 | purH2411::tetAR; tetA101::Kan(TcSKnR) ruvC4::Rif(sw)/pSIM5(CmR) | This report |
| TT26445 | purD2412::tetAR; tetA101::Kan(TcSKnR) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26446 | purD2412::tetAR; tetA101::Kan(TcSKnR) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26461 | purH2411::tetAR; tetA101::Kan(TcSKnR) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26462 | purH2411::tetAR; tetA101::Kan(TcSKnR) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26463 | purH2411::tetAR; tetA101::Kan(TcSKnR) recB10 his646(del:OGD) pro-47(del:AB)/pSIM5(CmR) | This report |
| TT26464 | purD2412::tetAR; tetA101::Kan(TcSKnR) recB10 his646(del:OGD) pro-47(del:AB)/pSIM5(CmR) | This report |
| TT26465 | purH2411::tetAR; tetA101::Kan(TcSKnR) DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26466 | purD2412::tetAR; tetA101::Kan(TcSKnR) DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26467 | purH2411::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif recB10 his646(del:OGD) pro-47(del:AB)/pSIM5(CmR) | This report |
| TT26468 | purD2412::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif recB10 his646(del:OGD) pro-47(del:AB)/pSIM5(CmR) | This report |
| TT26469 | purH2411::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26470 | purD2412::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26498 | purH2411::tetAR; tetA101::Kan(TcSKnR) ruvAB7::Spc(sw)/pSIM5(CmR) | This report |
| TT26500 | purH2411::tetAR; tetA101::Kan(TcSKnR) recN655::Spc(sw)/pSIM5(CmR) | This report |
| TT26502 | purD2412::tetAR; tetA101::Kan(TcSKnR) recN655::Spc(sw)/pSIM5(CmR) | This report |
| TT26507 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw)/pSIM5(CmR) | This report |
| TT26508 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw)/pSIM5(CmR) | This report |
| TT26522 | purH2411::tetAR; tetA101::Kan(TcSKnR) ruvAB7::Spc(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26528 | purD2412::tetAR; tetA101::Kan(kanR) ruvAB7::Spc(sw)/pSIM5(CmR) | This report |
| TT26530 | purD2412::tetAR; tetA101::Kan(KanR) ruvAB7::Spc(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26532 | purD2412::tetAR; tetA101::Kan(TcSKnR) DELmutS281/pSIM5(CmR) | This report |
| TT26534 | purD2412::tetAR; tetA101::Kan(kanR) ruvC4::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26536 | purH2411::tetAR; tetA101::Kan(TcSKnR) ruvC4::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26537 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw)/pSIM5(CmR) | This report |
| TT26538 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw)/pSIM5(CmR) | This report |
| TT26540 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26542 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26545 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26546 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26477 | pyrD2828::tetAR; tetA101::Kan(TcSKnR)/pSIM5(CmR) | This report |
| TT26479 | pyrD2828::tetAR; tetA101::Kan(TcSKnR) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26551 | purH2411::tetAR; tetA101::Kan(TcSKnR) rrnE::GntR(sw)/pSIM5(CmR) | This report |
| TT26552 | purH2411::tetAR; tetA101::Kan(TcSKnR) rrnE::GntR(sw) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26553 | purD2412::tetAR; tetA101::Kan(TcSKnR) rrnE::GntR(sw) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26556 | purH2411::tetAR; tetA101::Kan(TcSKnR) recN655::Spc(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26557 | purD2412::tetAR; tetA101::Kan(TcSKnR) recN655::Spc(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26559 | purH2411::tetAR; tetA101::Kan(TcSKnR) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26563 | purD2412::tetAR; tetA101::Kan(TcSKnR) rrnH::Spc(sw)/pSIM5(CmR) | This report |
| TT26567 | purH2411::tetAR; tetA101::Kan(TcSKnR) DEL1742(argA-recB) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26569 | purD2412::tetAR; tetA101::Kan(TcSKnR) DEL1742(argA-recB) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26571 | purH2411::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif DEL1742(argA-recB) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26573 | purD2412::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif DEL1742(argA-recB) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26578 | purD2412::tetAR; tetA101::Kan(TcSKnR) DEL(Fels-2) leuA414(UAG) sbcB1 recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26581 | purD2412::tetAR; tetA101::Kan(TcSKnR) sbcB::Zeo(sw)/pSIM5(CmR) | This report |
| TT26584 | purH2411::tetAR; tetA101::Kan(TcSKnR) sbcCD::Gnt(sw) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26587 | purD2412::tetAR; tetA101::Kan(TcSKnR) sbcCD::Gnt(sw) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26589 | purH2411::tetAR; tetA101::Kan(TcSKnR) recO656::GntR(sw)/pSIM5(CmR) | This report |
| TT26591 | purH2411::tetAR; tetA101::Kan(TcSKnR) recO656::GntR(sw) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26593 | purD2412::tetAR; tetA101::Kan(TcSKnR) recO656::GntR(sw)/pSIM5(CmR) | This report |
| TT26595 | purD2412::tetAR; tetA101::Kan(TcSKnR) recO656::GntR(sw) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26596 | purH2411::tetAR; tetA101::Kan(TcSKnR) sbcCD::Gnt(sw)/pSIM5(CmR) | This report |
| TT26597 | purD2412::tetAR; tetA101::Kan(TcSKnR) rrnE::GntR(sw)/pSIM5(CmR) | This report |
| TT26603 | purH2411::tetAR; tetA101::Kan(TcSKnR) recQ::Spc(sw) recA651::Rif(sw)/pSIM5(CmR) | This report |
| TT26605 | pyrD2828::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC6::Spc(sw)/pSIM5(CmR) | This report |
| TT26607 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) recF521::Tn5d-Rif DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26613 | purD2412::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26615 | purH2411::tetAR; tetA101::Kan(TcSKnR) recF521::Tn5d-Rif recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26618 | purD2412::tetAR; tetA101::Kan(TcSKnR) DEL(Fels-2) leuA414(UAG) sbcB1/pSIM5(CmR) | This report |
| TT26622 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26625 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26628 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) recF521::Tn5d-Rif DEL1742(argA-recB)/pSIM5(CmR) | This report |
| TT26632 | purH2411::tetAR; tetA101::Kan(TcSKnR) recJ657::Rif(sw)/pSIM5(CmR) | This report |
| TT26634 | purH2411::tetAR; tetA101::Kan(TcSKnR) recJ657::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26636 | purD2412::tetAR; tetA101::Kan(TcSKnR) recJ657::Rif(sw)/pSIM5(CmR) | This report |
| TT26638 | purD2412::tetAR; tetA101::Kan(TcSKnR) recJ657::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26640 | purH2411::tetAR; tetA101::Kan(TcSKnR) ligB::Rif(sw)/pSIM5(CmR) | This report |
| TT26642 | purH2411::tetAR; tetA101::Kan(TcSKnR) ligB::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26644 | purD2412::tetAR; tetA101::Kan(TcSKnR) ligB::Rif(sw)/pSIM5(CmR) | This report |
| TT26646 | purD2412::tetAR; tetA101::Kan(TcSKnR) ligB::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26657 | purD2412::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) recF521::Tn5d-Rif/pSIM5(CmR) | This report |
| TT26660 | purH2411::tetAR; tetA101::Kan(TcSKnR) recG646::Rif(sw) ruvC4::Spc(sw) recF521::Tn5d-Rif/pSIM5(CmR) | This report |
| TT26806 | purD2412::tetAR; tetA101::Kan(TcSKnR) DELmutS281 recA650::Gnt(sw)/pSIM5(CmR) | This report |
| TT26833 | pyrD2828::tetAR; tetA101::Kan(TcSKnR) ruvC6::Spc(sw) recG646::Rif(sw) recA650::Gnt(sw)/pSIM5(CmR) | This report |
“sw” designates alleles in which the normal sequence was replaced by a drug resistance determinant. “ tetA101::Kan (TcSKnR)“ refers to a kanamycin resistance determinant inserted into the middle of the tetA gene, rendering the strain sensitive to tetracycline and resistant to kanamycin. The “pSIM5(CmR)” plasmid carries the recombination genes (red) of phage lambda (Court et al. 2002).
Trapping duplications formed in overnight cultures
The drug-in-drug method for measuring duplication frequencies in unselected overnight cultures was described previously (Reams et al. 2010, 2012). For these assays, the strain to be tested carries the tetAR genes for tetracycline resistance (TcR) inserted as part of transposon Tn10 into the test locus (e.g., purH). These strains are then made sensitive to tetracycline by a kanamycin-resistance (KnR) determinant inserted into the tetA gene. The resulting strains are phenotypically Tcs KnR. Duplications are trapped by a Red-mediated recombineering cross in which a short single-strand fragment (80 bases) is introduced into the tested strain, which carries a pSIM5 plasmid and is grown at 30° (Court et al. 2002). This fragment, introduced at 42°, restores TcR by precisely excising the KnR determinant. Transformants are selected on LB–tetracycline agar plates. The majority of these transformants become TetR KanS. However, any recipient cells with a pre-existing duplication of the test locus become TetR, but retain their KanR phenotype and are detected by replica-printing to LB–tetracyline–kanamycin. Duplication frequencies are calculated by dividing the number of TcR KnR colonies by the total number of TcR colonies. Duplication of purHD by exchanges between rrn loci was verified by join-point PCR and whole-genome sequencing (see below).
The rate of duplication formation is defined here as “the duplication frequency attained by a culture during 33 generations of growth from a single cell without a duplication.” This frequency is below the steady-state level, and the nearly linear accumulation of duplications in this period is taken as the initial rate of duplication formation. The course of duplication accumulation was described previously (Reams et al. 2010). The forces that drive duplication frequency toward a steady state minimize the effects of Luria–Delbruck fluctuation. Low viability of some rec mutant strains does not interfere with comparison of rates to those in wild type because each rate is based on the fraction of viable tester cells that carry a duplication. The 33-generation period is assured by a single colony isolating the strain to be assayed and using an entire small colony to inoculate a liquid culture (4 ml) that is grown to stationary phase. The final titer (2 × 109 cells) is achieved after 33 generations of growth of the cell that initiated the colony.
Duplication join-point analysis
Trapped purHD duplications were analyzed for the nature of their rrn-mediated join points using a divergent primer PCR method described previously (Helm and Maloy 2001). Two divergent primers direct synthesis in opposite directions away from purHD. The primers anneal to regions immediately adjacent to various rrn locus (Figure 2). Since these primers direct divergent replication, a PCR product will be amplified only in strains with a specific rrn-mediated duplication junction. The following primer pairs were used to test for the presence of the various hybrid rrn combinations possible for purHD duplications: rrnE/B (TP2306+TP2311), rrnH/B (TP2306+TP2315), rrnE/H (TP2311+TP2316), rrnE/A (TP2304+TP2311), rrnH/A (TP2304+TP2315), rrnE/C (TP2308+TP2311), rrnH/C (TP2308+TP2315). The sequences of these primers are TP2304 (5′ TGCCTTCATTTTGCGGTGGTTAGAG 3′), TP2306 (5′ CCAGGCGCTCAGTAGTTGTTGTTCG 3′), TP2308 (5′ GCTGTTAGGGCACTTCACTTTGGCG 3′), TP2311 (5′ CGATAGGGGCGATGTGGTGCTGTTC 3′), TP2315 (5′ CCATCCGCAGGGCAGCATAGAAGAG 3′), and TP2316 (5′ CGGCAATAGCCTTTTCCATCAACGG 3′).
Prior to PCR, both the parent and various trapped purHD duplication strains were grown overnight in 2 ml LB media and diluted 10-fold. For each divergent primer pair, a PCR reaction was run on each strain. All PCR products were separated on a 0.7% agarose gel using standard electrophoresis procedures and visualized after staining with ethidium bromide. Strains carrying a duplication specific to the applied primers generated an ∼7-kb PCR product, whereas the parent strains showed no PCR product. Using this method, the specific rrn locus involved in forming the duplication was determined for all 30 of the tested purHD duplications. Whole-genome sequencing of several strains verified their PCR-determined rrn join points and demonstrated the presence of a simple duplication.
The crossover points of the exchanges that generated the hybrid rrn duplication join points were determined by sequencing the gel-purified 7-kb PCR products derived from junctions of independently isolated purHD duplications. PCR products were sequenced using 12 different primers that covered the entire rrn region. The sequences of hybrid junction elements were compared to those of participating parent rrn loci. The crossover event was inferred to have occurred in the region where the hybrid rrn sequence of one rrn sequence (e.g., rrnE) transitioned to that of another (e.g., rrnE).
Normalization of read-depth plots
Plots of Illumina read-depth data are often noisy due to uneven amplification in the PCR step of library preparation and from the effects of G+C differences on detection chemistry and DNA fragmentation. Since the profile is remarkably uniform between isogenic strains, the noise can be filtered out by comparing data from an isogenic reference strain (preferably from the same run). A normalization factor is determined at each position and used to correct the depth of the test genome, yielding a graph that clarifies the features of interest. An example is shown in Figure 3. The duplication strain tested was a population in which half the cells contained a large duplication. The duplication was unrecognized initially, but became clear following normalization.
Figure 3.

Identification of duplications by read-depth profiles. Full-genome sequences were determined (Illumina) for a haploid parent reference strain (top) and strain in which about half of the cells carried a duplication (middle). The duplication was clearer when the read-depth profile of the experimental strain was normalized to that of the parent (bottom).
Results
Duplications of the purHD operon arise between flanking rrn genes
Duplications were trapped as described previously (Reams et al. 2010), and junction points were classified by using PCR to determine which rrn loci recombined to form the duplication. For example, in identifying the rrnE/B junction (see Figure 1), one primer was directed clockwise toward rrnE and the other was directed counterclockwise toward rrnB as described previously (Helm and Maloy 2001). Cultures were diluted before DNA preparation to assure that the dominant structure was assayed and minimize contributions from any new rrn duplications appearing during growth. The presence of a rrnE/B duplication junction was demonstrated by production of a 7-kb PCR product using divergent primers (rrnE and rrnB). Other divergent primer pairs tested the presence of the various rrn join-point combinations studied here (e.g., rrnB/H, rrnA/E).
As seen in Figure 2, all 30 purHD duplications trapped in a recA+ strain showed hybrid rrn join points. Most (47%) involved the smallest interval (rrnE/B). Next most frequent (40%) were rrnH/B, and 13% involved rrnA/E (see Figure 2). None of the other rrn pairs flanking purHD were detected (rrnH/C, rrnH/A, rrnE/C). Several of the characterized rrn-mediated duplications were further analyzed by whole-genome sequencing. As seen in Figure 4, the Illumina read depth is elevated twofold for the genomic region between the corresponding rrn loci. This confirms that the trapped duplications carry two copies of the intervening region corresponding to the join point determined by PCR and no other obvious rearrangements.
Figure 4.
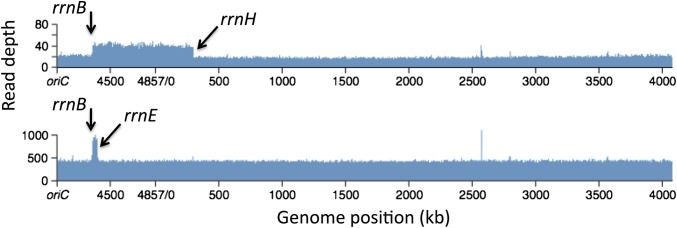
Genomic read-depth profile of a duplication strain. The read depth of a whole-genome sequence of a duplication strain (Illumina) was normalized to that of the parent strain. (Top) Results for a strain shown by PCR to have a rrnE/B junction. and (Bottom) A strain with an rrnH-rrnB junction. The few narrow spikes (bp 2500–3000) are not present in the raw data and are generated by the normalization process due to small deletion mutations carried by the standard strain.
Dependence of duplication rates on flanking rrn sequences
The duplication rate of the purHD operon is defined here as the duplication frequency attained during 33 generations of growth from a single cell lacking a duplication. During this period the frequency of purHD duplications reached 2.5%, a rate that corresponds to 3 × 10−3/cell/generation. This increase is nearly linear and is taken as an initial rate since it is far from the ultimate steady-state frequency.
The duplication rates of purHD and three nearby loci (zja, metA, and thrA) were assayed in strains lacking either or both of the rrnE and rrnH sequences. Positions of these loci are shown in Figure 2. Removal of both rrnE and rrnH caused a large decrease (170-fold) in the purHD duplication rate (Table 2). The duplication rates for the zja, metA, and thrA sites showed a similar drop after deletion of only rrnH. In all cases, the large drop in duplication rate resulted from removal of all the rrn loci from one side of the test site. The few residual duplications seen in these rrn-deficient backgrounds arose between diverse short sequence repeats and will be described elsewhere (A. Reams, E. Kofoid, and J. Roth, unpublished results).
Table 2. Effect of rrn deletions on rates of duplication formation.
| Duplication ratea of indicated sites with or without deletions of rrn loci (×100) |
||||
|---|---|---|---|---|
| Deleted rrn loci | purHD | xja | metA | thrA |
| None | 250 | 200 | 220 | 160 |
| rrnE | 210 | 100 | 200 | 100 |
| rrnH | 50 | 1.1 | 2.7 | 0.5 |
| rrnE and rrnH | 1.5 | 0.7 | 2.5 | 0.8 |
Duplication rates are expressed as the frequency of duplication-bearing cells reached during growth of a cell with no duplication for 33 generations.
As seen in Figure 2, roughly half of the purHD duplications arose by exchanges between rrnB and rrnE. Nevertheless, removal of rrnE alone had very little effect on the purHD duplication rate (Table 2), while removal of rrnH alone caused a fivefold reduction. The relatively large contribution of rrnH to the purHD duplication rate is not understood. We suggest that rrnH may be subject to more frequent chromosome gaps or breaks than rrnE, making rrnH/B duplications form more frequently than rrnE/B types. However, the larger (790 kb) rrnH/B duplication may have a higher fitness cost, leading to selective deletion of the region between rrnE and rrnH either before or after being trapped. Such remodeling would maintain the duplication state of purHD, but would leave a shorter (40 kb), lower-cost duplication with a rrnE/B join point.
Three sites in the chromosomal region between rrnE and rrnH (zja, metA, and thrA) were found to be duplicated at a slightly lower rate than purHD (see Table 2). Removal of rrnE had very little effect on the duplication rate of these loci, suggesting that most of these duplications form between rrnB and rrnH. The few duplications of this region that formed in strains without rrnH (for which rrn-mediated duplications are impossible) showed join points, suggesting that they arose by exchanges between diverse short sequences (A. Reams, E. Kofoid, and J. Roth, unpublished results).
Duplication rates are unaffected by defects in early steps of homologous recombination
The large size of the flanking rrn repeats and the high frequency of exchanges between them suggested that duplications of purHD are likely to form by homologous recombination. Strong RecA dependence has been seen in many situations in which duplications form between extensive perfect repeats (Roth et al. 1996; Romero and Palacios 1997). However, purHD duplication rates are essentially unaffected by mutations that eliminate enzymes important to standard pathways of recombination (see Figure 5). Furthermore, the distribution of junction types (rrnE/B, rrnH/B, and rrnE/A) for purHD duplications were the same in recA+ and recA mutant strains (see Figure 2, top). Taken at face value, these results suggest that RecA, RecB, or RecF enzymes are not involved in duplication formation. Despite these phenotypes, we suggest below that formation of these duplications in wild-type strains is heavily recombination dependent and occurs by single-strand annealing only in strains lacking these recombination functions.
Figure 5.

Duplication rates in several mutants defective for homologous recombination. Duplication rates were measured by determining the frequency of purHD duplication after 33 generations of growth from a single cell lacking a duplication. The frequency of duplications attained within this period is taken as the initial rate of duplication formation. The rates presented here are based on combined data for purD and the adjacent purH gene. Both sites individually gave essentially the same result.
The apparent RecA independence of rrn duplications suggested formation by single-strand annealing. Alternatively, RecA-independent events might be illegitimate exchanges mediated by topoisomerases that are likely to act within or near rrn loci in response to heavy transcription of these loci. The exchanges that produce duplications are not associated with formation of deletions or duplications within the rrn locus since the size of the rrn locus at the join point is as expected for a simple hybrid recombinant. To learn more about the nature of the exchanges, we analyzed hybrid rrn junction sequences to determine the approximate points within the rrn loci at which the genetic exchanges occurred. This is possible because the base sequences of different rrn loci are not identical. Each locus has sequence features that are unique, and every pair of rrn loci differs at several points. A tree of rrn sequence difference in the Appendix describes their differences. The aligned sequences of the three rrn loci involved in duplicating the purHD operon are diagrammed in Figure 6, where regions with many clustered sequence differences are boxed, and isolated single-base differences are indicated by colored triangles.
Figure 6.

Exchange points within rrn loci that generate a purHD duplication. Exchange points all fell in regions of identical sequence (I, II, or III) shared by rrnE and rrnB and by rrnH and rrnB. These regions are shaded and are defined either by blocks of different sequences (white regions) or by single-nucleotide differences (colored triangles). The number of exchanges found in each interval are indicated in the table above the map. IVS25 and IVS45 are intervening sequences (like introns) that are found in some rrn loci of Salmonella, but not E. coli (Mattatall and Sanderson 1996). The 25 and 45 refer to helices in the secondary structure of the 23S rRNA. The distribution of these sequences among rrn loci is described in the Appendix.
To determine the location of the crossover point that formed the duplication, the hybrid junction rrn loci were amplified from independently isolated duplications and their sequences were compared to those of the two parental rrn sequences. Figure 6 describes the exchange points of 26 independent rrnE/B duplications and 7 rrnH/B duplications isolated from recA+ and recA mutant strains. For each duplication, the exchange fell within one of three regions of extended sequence identity. Most crossover events occurred within region III, the largest window of perfect identity (2390 bp for rrnE/B or 2490 bp for rrnH/B), while fewer events occurred within the other two smaller windows, region I (756 bp for rrnE/B and rrnH/B) or region II (497 bp for rrnE/B and 413 bp for rrnH/B). Duplications trapped in rec+ and recA backgrounds showed a similar distribution of exchange points.
The results in Figure 6 suggest either that rrn-mediated duplications form by the same pathway in rec+ and recA strains or that homologous recombination and single-strand annealing pathways lead to the same distribution of exchange points. The results do not exclude the possibility that topoisomerase catalyzes exchanges within regions of shared rrn sequence identity. In discussing a model for rrn duplication, we favor the idea that DNA breaks often occur within the transcribed rrn region and produce gaps or breaks that can initiate duplications by two alternative pathways.
Duplication rates are strongly reduced in strains deficient for Holliday structure resolution
Standard pathways for homologous recombination (RecBC and RecFOR) involve early events that process breaks and gaps leading to RecA-catalyzed strand invasion. This produces Holliday junctions that are moved on recombining duplexes by RuvAB (Heller and Marians 2005; reviewed by West 2003). The later events that stabilize and ultimately resolve these Holliday structures are achieved by two alternative enzymes: (1) the RuvABC resolvasome and (2) the RecG branch-migration enzyme (Benson et al. 1991; Wardrope and Leach 2009). If one resolution pathway is inactivated by mutation, the other is still available. Cells lacking both RuvC and RecG are severely deficient in recombination, presumably due to their inability to stabilize and resolve Holliday junctions. As seen in Figure 7, a recG mutation alone had little effect on duplication rate. The RecG enzyme is associated with the RecBCD pathway, which below we argue does not contribute to duplication. Strains with a single ruvAB or ruvC mutation caused a significant drop in duplication rate. The largest decrease was seen in the ruvC, recG double mutant, which showed a 30-fold drop in duplication rate compared to that of wild type. This suggests that rrn-mediated duplications form by a recombination process that involves Holliday structures and requires their resolution by either RuvC or RecG. Of the residual duplications that formed at a reduced rate in the ruvC recG mutant, three junctions were sequenced and showed rrnE/B exchanges within the rrn sequence (see Figure 6), suggesting formation by single-strand annealing. The importance of Holliday structure resolution to duplication formation suggests that homologous recombination is normally involved in duplication formation despite the failure of recA, -B, or -F mutations to reduce the duplication formation rate. In addition to blocking recombination, the ruvC recG mutations may cause toxic accumulation of Holliday structures as seen previously in Neisseria (Sechman et al. 2006) and yeast (Fabre et al. 2002). This possibility will be discussed later.
Figure 7.

Duplication rate is reduced in strains defective in Holliday structure resolution. The duplication rate is defined as the frequency of duplications attained after 33 generations of growth. Bars in red indicate strains that showed a significant reduction in duplication rate. The bar at the far right indicates the rate in a strain lacking both rrnE and rrnH, both of which lie on the clockwise side of the assayed purHD operon.
Defects in early homologous recombination steps restore normal duplication rates to ruvC recG mutants
It was surprising that single recA, -B, and -F mutations and even a recB, recF double mutation, which block early steps in recombination, failed to affect a process that requires the late step of Holliday structure resolution. We expected a single recB or recF mutation to cause a less severe drop than a recA or a recB, recF double mutant. To pursue this, these early mutations were added to a ruvC, recG strain, defective for duplication. As seen in Figure 8, each of the single mutations restored a normal duplication rate. Thus, duplications of purHD formed at normal rates in strains lacking all of these major players in homologous recombination: RecA, RecB, RecF, RuvC, and RecG.
Figure 8.

The duplication defect of ruvC recG is corrected by mutations in recA, -B, or -F. Duplication rates are the frequency of purHD duplications achieved by growing a cell lacking a duplication for 33 generations. White bars indicate the frequency in recA+ strains carrying the indicated mutations. Red bars indicate the frequency in the same strains with an added recA, recB, or recF mutation.
To explain the normal duplication rate in the suppressed ruvC recG mutants, we suggest that blockage of early recombination steps activates an alternative pathway that can form duplications by single-strand annealing and does not require RuvC or RecG. That is, duplications may form by homologous recombination in wild-type strains (despite the phenotypes seen in Figure 5). However, blockage of early recombination steps allows gaps and breaks to accumulate and provides ends that can form a duplication by single-strand annealing without need for strand invasion or Holliday structure resolution. When Holliday resolution is blocked, recombination cannot be completed, but the annealing pathway opened by early mutations supports duplication by an alternative annealing route for which Holliday resolution is irrelevant. A curious aspect of this suppression is that individual recB, recF, and recA mutations restored duplication. One might have expected that recA or the recB, recF mutation combination would be more effective than the recB or recF mutations individually. An explanation is suggested below.
Duplication by single-strand annealing raises a mechanistic problem. While it is easy to visualize use of an annealing pathway to form a deletion using resected ends at a chromosome break, formation of a duplication from one break would seem to require separation of strands for the entire duplicated region. Duplication by annealing is easier to imagine with two breaks, one near each participating rrn locus in different sister chromosomes. Because of frequent DNA breakage in rrn loci, delays in their repair caused by a recA (or recF or recB) mutation could allow damage to accumulate and assure coexistence of breaks in different rrn loci.
We suggest that, in the simple ruvC recG mutant, the early recombination functions (RecFORA and RecBCDA) continue to consume gaps and breaks that might otherwise provide ends for single-strand annealing. These early pathways try repeatedly but unsuccessfully to convert initial substrates into resolvable Holliday structures. Ultimately, these attempts may lead to loss of rrn sequences from the processed ends and thereby abort attempts at duplication. When RecA or the proteins that load RecA onto single strands (RecB or RecF) are eliminated, gaps and breaks remain unrepaired and can provide single-strand ends for use in annealing. These strands will not be coated with RecA protein and thus may be particularly active in annealing. In addition, these early blocks cause unrepaired gaps and breaks to accumulate, increasing the probability of non-allelic rrn loci in different sister chromosomes having simultaneous gaps or breaks. These simultaneous lesions may be essential to duplication formation by annealing as described below. The early blocks in recombination could also reduce any toxic effects of accumulated Holliday structures (see below).
The model described above is diagrammed in Figure 9 as it applies to duplication initiated by gaps. The gap may be generated by digesting one end of a nick or could be formed in one step following cutting of a hairpin structure in the nontranscribed strand of the rrn sequence (Figure 9, left). The RecFORA pathway starts repair of this gap using a sequence from a different rrn locus and generates Holliday structures that must a resolved by RuvC and RecG to complete recombinational duplication. When a recF mutation blocks early steps in gap repair, the gaps persist and accumulate such that two participating rrn loci can have single-strand ends with rrn sequences that can anneal (Figure 9, right). The annealing pathway activated by loss of gap repair (the recF mutation) replaces the lost recombinational duplication pathway and explains why recF mutants showed no duplication defect (Figure 5). This activated pathway also suppresses the defect seen in a ruvC recG double mutant, since the new pathway does not involve Holliday structures. This model does not explain why single recF or recB mutations did not show some partial loss of duplication ability or partial suppression of ruvC, recG. To approach this, we looked for reasons that one kind of damage (perhaps double-strand breaks) might be prevented from making duplications.
Figure 9.

Formation of duplications at gaps by RecFORA recombination and by annealing in recF mutants. The model assumes that duplications are normally made primarily by the RecFORA pathway and are initiated by nicks within the rrn sequence. Strand exchange leads to Holliday structures that can be resolved to produce a duplication (left). In the absence of RecFORA, unrepaired nicks and gaps accumulate in both rrn cistrons and can produce single strands that are uncoated by RecA and available for formation of duplications by single-strand annealing (right). Activation of this pathway by recF mutation renders the RuvC and RecG enzymes dispensable and allows suppression of a ruvC, recG double mutation.
All seven rrn loci are devoid of Chi sites
One aspect of the results in Figure 8 does not fit with the model as described above. It seemed reasonable that blockage of both RecFORA and RecBCDA pathways might be required to fully suppress the RuvC RecG defect. In fact, duplication rates were fully restored to the ruvC recG mutant by either a recB or a recF mutation individually. This could indicate that RecB and RecF act together to generate the Holliday structure. However, an alternative explanation was suggested by examining the sequences of rrn loci.
Chi sites are required for RecBCD-mediated homologous recombination. Repair of a double-strand break by RecBCD involves degrading double-stranded DNA until a Chi sequence is reached (reviewed by Kreuzer 2005 and by Myers and Stahl 1994). At this point, only the 5′ end is further resected, leaving a 3′ overhang ending at the point Chi was encountered. A break within or immediately origin-distal to the rrn sequence could generate an exchange between non-allelic rrn sequences only if Chi sites were present within the rrn locus. Figure 10 shows the positions of all Chi sequences in the 75-kb regions surrounding each the seven rrn loci of S. enterica. It is apparent that Chi sites in the useful orientation are absent from all seven rrn loci and from all of their immediate origin-proximal 5-kb regions. Our calculations suggest that, given the frequency of Chi within the seven pictured regions (each 75 kb), the random chance of all rrn loci lacking Chi is ∼0.5% and the probability of having no Chi sites in either the rrn or the origin-proximal 5-kb regions is 3 × 10−5.
Figure 10.

Distribution of Chi sites in and near rrn loci. The seven rrn sequences (5.5 kb) are aligned with each other, leaving 35 kb of sequences on either side of rrn that are completely different for each locus. In the diagram, large green dots indicate Chi sequences oriented to activate a RecBCD enzyme moving toward the replication origin (right to left). Smaller red bars indicate Chi sequences in the opposite orientation.
This Chi distribution suggests that, whenever RecBCD acts on a break within rrn sequences, it digests rrn material and produces a 3′ end that can initiate an exchange only outside of rrn. In the absence of Chi sequences, RecBCD can still repair DNA breaks occurring within the rrn region but does so by exchanges outside of the rrn sequence. With no Chi sites within rrn loci, double-strand breaks cannot initiate RecBCD-mediated duplications or deletions, which require a recombination exchange between the rrn sequences of two different rrn loci. Data in Figure 6 (above) show that all of the duplications analyzed did arise by exchanges within the rrn sequence. This suggests that RecBCD activity at double-strand breaks could not be responsible for duplications arising in wild-type strains. This explains why a single recB mutation did not reduce the normal duplication rate—RecBCD does not contribute to duplication. Any duplications made by recombination must reflect the activity of RecFORA at gaps. However, the ability of a recB mutation to fully suppress the ruvC recG mutations must be explained.
The processing of double-strand breaks within rrn is diagrammed in Figure 11. In a normal strain (top left) the break can be repaired by RecBCD, but the process removes the rrn sequence adjacent to the break and leads to an exchange point outside of rrn, allowing repair of the break, but no contribution to duplication. In the absence of RecBCD, breaks accumulate and different rrn loci can provide ends for single-strand annealing. In a simple recB mutant, this additional annealing makes only a small contribution to duplication rate above the background of recombinational duplication by gap repair. However, when late steps in recombination are blocked in a ruvC recG double mutant, the recB mutation makes a large contribution to duplication rate by allowing double-strand breaks to provide ends that initiate annealing. That is, the recB mutation opens a new annealing pathway that uses double-strand breaks within rrn as initiating structures. Such structures could not contribute in any RecBCD-proficient situation (wild type, recF, or recA) and make a small contribution in any background that already has an active pathway for duplication gap repair by either recombination or annealing.
Figure 11.

Repair of rrn double-strand breaks and effect of a recB mutation on duplication formation. Breaks within an rrn locus are subject to repair by RecBCDA, which can restore a replication fork. However, the absence of Chi sequences within rrn loci prevents RecBCD from making exchanges between paralogous rrn loci (left). In the absence of RecBCDA, breaks are not resected and can provide ends with rrn sequence. The resulting single-strand rrn sequences are free of RecA protein and can engage in single-strand annealing with non-allelic rrn sequences.
Thus, there may be three ways of making an rrn duplication. 1) Gaps can initiate recombinational duplication (using RecFORA) in wild type. 2) Gaps can initiate duplication by annealing in recF or recA cells. 3) Double-strand breaks can initiate annealing only in recB mutants. By allowing use of a new initiating structure, a recB mutation might be expected to increase the duplication rate over that seen either in wild type or in recA or recF mutants. This makes a prediction that is tested below with mixed results.
Duplication rate is stimulated slightly by recB mutation
In the process of duplication suggested above, purHD duplications arise in rec+ strains predominantly by homologous recombination (RecFOR acting at gaps) and form exclusively by single-strand annealing in recA or recF mutants. Double-strand breaks within rrn cannot initiate duplications by recombination (using RecBCDA), but do initiate annealing in recB mutants because rrn sequences are left at the unresected break. The increased annealing allowed by a recB mutation under some circumstances should increase the rate of duplication formation. In a strain that has functional RecFORA, the effect of the recB mutation would likely be small, since it would enhance an already high rate of duplications due to RecFORA acting at gaps. Similarly, the enhancement might be small in a recF mutant because gaps provide a good source of duplication by annealing. The magnitude of duplication enhancement provided by a recB mutation would depend on the relative frequency of gaps and breaks. We have looked for evidence of this enhancement.
The recB mutation shows no significant enhancement in Figure 5, in wild type (above a background of gap-stimulated recombination), or in a recF strain (above a background of gap-initiated annealing). The increase in rate caused by recB in a recF, recA background does not appear significant (Figure 12). However, a slight effect was seen in a recA mutant background (Figure 12). This difference is on the border of significance, given the variability of these measurements, but the duplication rate seen in the recA, recB double mutant is the highest that we have measured in any strain. This rate may reflect annealing initiated by both gaps and breaks.
Figure 12.

Duplication rates are stimulated by recB mutations in a recA mutant strain. Addition of a recB mutation causes a small increase in the purHD duplication rate in strains lacking recA. In recA mutants, duplications may form entirely by single-strand annealing. Absence of RecB may leave undigested rrn sequences at double-strand breaks, enhancing the likelihood of duplication by annealing.
Effects of mismatch repair on duplication formation
The seven rrn loci of Salmonella differ at many points in their sequence (see Figure 6 and Appendix). Since duplications form by exchanges between different rrn loci, it seemed possible that recombination or even annealing might be subject to rejection of duplexes by the methyl-directed mismatch repair system (MMR) (Petit et al. 1991; Friedberg et al. 2005). This idea is supported by the observation (Figure 13) that a mutS mutation stimulated duplication in the recA+ background. No effect of mutS was seen in the recA mutant background, so we suggest that rrn heteroduplexes formed by single-strand annealing are not subject to MMR rejection. This result differs from earlier findings using shorter and more closely positioned repeats, for which MMR played a big role in annealing (Bzymek and Lovett 2001).
Figure 13.

Effects of various mutations on duplication rates with and without RecA. Duplication rates were measured in recA+ (green bars) and recA (yellow bars) strains carrying additional mutations in genes involved in mismatch repair, recombination, SOS response, and, possibly, in single-strand annealing.
Testing the role of other recombination/repair functions in duplication
If duplications form efficiently by annealing in recA mutant strains, then mutations that inhibit annealing might decrease duplication rate in a recA mutant strain and might show no defect in recA+ strains, where duplications form primarily by homologous recombination. A series of mutations defective in various recombination and DNA repair functions were tested for their effect on duplication rates in recA+ and recA− strains (see Figure 13). While these effects are too small be interpreted, several seem interesting.
Most notable is the effect of recO mutations, which remove a central part of the gap repair mechanism (RecFORA). One might expect this mutation to behave as did the recF mutation described above. In a recA+ background, the recO mutation caused a significant drop in duplication rate, suggesting that it might eliminate the gap-initiated recombination that normally forms a duplication in a wild-type cell. The recF mutation (Figure 5) did not cause this drop, arguably because it activated the alternative annealing pathway. This might suggest that recO defects are less able to prevent loading of RecA on single strands and thus less able to activate annealing. In keeping with this idea, the recO mutation had no effect on duplication in a recA mutant, where no RecA protein is available to be loaded.
The sbcCD mutation caused a small reduction in duplication in an otherwise rec+ strain but not in a recA mutant. SbcCD degrades hairpin structures in single-strand DNA (Eykelenboom et al. 2008; Darmon et al. 2010) and might be expected to contribute to creation of gaps in ribosomal DNA (rDNA). Large gaps could be particularly important for recombination, which requires RecA loading, while smaller gaps are sufficient for annealing in a recA mutant where the only ends are needed and RecA loading is not an issue.
The sbcB and xonA mutations (dominant and null alleles of the gene for a 3′ exonuclease ExoI) may cause a small decrease in both pathways. ExoI and the RecJ single-strand 5′ exonuclease could be imagined to contribute by extending single-strand nicks to gaps, but recJ mutations showed little effect on duplication rate. The ligB gene encodes a nonessential homolog of the standard DNA ligase (ligA) (Sriskanda and Shuman 2001) and made no significant contribution to duplication formation.
There was also very little effect of lexANull and lexAInd mutations, which cause the LexA-repressed SOS DNA repair system to be either constitutively expressed or uninducible, respectively. The same result applied to both recA+ and recA strains. This suggests that none of the 40 enzymes controlled by the SOS response is limiting for rrn-mediated duplication by either recombination or annealing (Friedberg et al. 2005).
ruvC recG mutation combination does not affect duplication rate of the distant pyrD locus
The dependencies described above apply to duplications of the purHD locus, which is flanked by rrn loci. In contrast, the pyrD gene is located on the opposite side of the chromosome (see Figure 2) and is not closely flanked by rrn or any other major direct-order sequence repeats. The pyrD duplication rate is the lowest of all Salmonella loci tested at 4 × 10−6/cell/generation, 750-fold lower than the rate for rrn-mediated duplication of purHD (Reams et al. 2010). The pyrD duplication rate is unaffected by a recA mutation (like that of purHD locus). Unlike purHD, the pyrD locus duplicates at a normal rate in ruvC, recG mutants (see Figure 14).
Figure 14.

Effect of recombination defects on pyrD duplications. The pyrD locus is located far from the purHD site (see Figure 2) and is not flanked by direct-order rrn loci or by any other major sequence repeat.
In understanding the different effects of Holliday resolution in these two regions, it should be noted that these loci duplicate at very different rates (10−6 for pyrD and 10−3 for purHD). In addition, the pyrD locus is not flanked by extensive sequence repeats while purHD is closely flanked by the rrnB and rrnE loci. These differences suggest that pyrD normally duplicates by pathways that are independent of RecA and involve no Holliday structures (or RuvC, RecG activity). These duplications must use the very short available homologies, which has been verified by sequencing junctions of pyrD duplications (E. Kofoid, unpublished results). It seems likely that pyrD duplicates (at a low rate) by nonrecombination pathways that use single-strand annealing or the palindrome-processing pathway described previously for tandem inversion duplications (TIDs) (Kugelberg et al. 2006) and discussed below. While the duplication rate of pyrD is very low, this behavior seems likely to be representative of most areas of the chromosome, which have no flanking extensive sequence repeats and no mechanisms to stimulate formation of gaps or breaks. A purHD from which the repeated rrnB and rrnH loci were deleted behaved much like pyrD. Without flanking rrn repeats, the purHD duplication rate falls 100-fold (see Table 2) and is no longer affected by ruvC, recG mutations (A. Reams, unpublished results).
Discussion
Three unexpected results are described here. First, formation rates of duplications arising between extensive repeated rrn sequences are unaffected by the absence of enzymes essential for homologous recombination (RecA, RecB, and RecF). Second, while strains lacking these early recombination functions form duplications at normal rates, mutants (ruvC recG) blocked for late recombination steps (Holliday structure resolution) show a 30-fold decrease in duplication rates. Third, the reduced duplication rates seen in the absence of RuvC and RecG are corrected by single blocks in earlier steps of homologous recombination (RecA, RecB, or RecF). These observations may reflect shifts in channeling a rich source of gaps and breaks between two alternative pathways of duplication formation–recombination and single-strand annealing.
In many assay systems, duplications mediated by large direct repeats depend heavily on RecA (Romero and Palacios 1997). However, these assays detect duplications by positive selections that demand multiple gene copies and may favor cells with higher amplifications. Since the amplification of a duplication to higher copy numbers is heavily RecA dependent (Kugelberg et al. 2006, 2010; Poteete 2009; Reams et al. 2010), we suspect that the frequently reported RecA dependency of duplication reported in other systems may reflect a need for higher amplification, which can depend on RecA even when the initial duplication event is RecA independent. The assay used here traps duplications without favoring higher copy numbers. In addition, this assay is not affected by the lethal effects of recombination mutations tested. That is, reduced viability of certain mutant strains (e.g., recA, ruvC) does not lower the measured duplication rate since the trapping assay detects the ratio of duplication to nonduplication cells in the viable population.
A different but not mutually exclusive explanation for the RecA independence of rrn-mediated duplications is that the repeated rrn sequences may be subject to frequent formation of gaps and breaks. A precedent for the of role of introduced breaks may be the low recombination dependence of duplications between IS3 elements, which depend on the availability of transposase to make nicks in the two recombining sequences (Reams et al. 2012). Nicks and or breaks in both recombining sequences can allow accumulation of the multiple simultaneous breaks needed for duplication by single-strand annealing and allow duplications to form without recombination. When these lesions are not processed by recombinational repair enzymes, they may be free to initiate an alternative annealing pathway for duplication formation.
The 5.5-kb rrn repeats are heavily transcribed and rich in palindromic sequences. These genes are GC-rich and GC-skewed such that the nontranscribed strand is G-rich and may be subject to formation of R-loops with hairpin structures. In Salmonella, several rrn sequences have extended palindromic intervening sequences (IVS in Figure 6) that extend natural palindromes that are inherent in the ribosomal RNA (rRNA) sequences. These extensions must be cut out of rRNA prior to ribosome assembly (Mattatall and Sanderson 1996, 1998). The distribution of these palindromic sequences among various rrn loci is described in the Appendix. Degradation of hairpin structures in the nontranscribed strand of rDNA would generate gaps in one step. The heavy transcription of rrn regions may temporarily expose single stands, making them available for pairing and susceptible to nucleases. The heavy transcription of rrn loci is likely to require activity of topoisomerases and gyrases, which, we speculate, may cause a significant rate of DNA breakage. These enzymes are able to catalyze illegitimate recombination (Ikeda et al. 1982) and make double-strand breaks in the presence of inhibitors or following collisions with replication forks (Gellert et al. 1977; Pohlhaus and Kreuzer 2005). While the frequent breakage of rrn sequences is speculative, it should be remembered that rrn duplications form at a very high frequency (10−3/cell/generation) and end in specific short regions (5.5 kb), suggesting a need for frequent internal initiating structures.
A model for formation of rrn duplications
We propose that the results described above are consistent with the idea that rrn loci are subject to frequent nicks and breaks that can be processed in alternative ways and form duplications by the several pathways described below.
In wild-type strains, duplications are made primarily by homologous recombination between sister strands using the RecFORA pathway (see Figure 9, left). Single-strand gaps within rrn sequences are loaded with RecA protein and invade a non-allelic rrn sequence of a sister chromosome, thereby generating Holliday structures that can be resolved to leave a duplication.
Elimination of the RecFORA pathway prevents recombinational formation of duplications, but allows unrepaired gaps to accumulate in both participating rrn loci. These gaps provide ends that can participate in an alternative single-strand annealing pathway (see Figure 9, right). The absence of RecA or failure to load RecA onto the gaps leaves single-strand ends free to engage in single-strand annealing. Evidence that RecA inhibits annealing was previously reported in E. coli (Bzymek and Lovett 2001), and the yeast RecA homolog Rad51 also inhibits single-strand annealing (McDonald and Rothstein 1994; Ivanov et al. 1996; Stark et al. 2004).
Double-strand breaks within rrn loci cannot initiate duplications because Chi sites are absent from all rrn loci (see Figure 10). Repair of rrn breaks by RecBCD is expected to occur by exchanges outside of rrn (Figure 11, left). Thus, a recB mutant shows no loss of duplication rate.
In a ruvC recG mutant, recombination is blocked by inability to resolve Holliday structures. (see Figure 9, left). In this mutant, however, the RecFORA enzymes continue to convert gaps into unresolvable Holliday structures. By processing gaps that might have led to annealing, the active RecFORA functions block the alternative annealing route to duplication. Repeated attempts to establish Holliday structures may ultimately lead to loss of rrn sequences from ends and thus prevent rrn rearrangements. The few rrn duplications formed in a ruvC recG mutant form by exchanges within rrn like those thought to form by annealing. The possibility of toxic Holliday structures is discussed below.
When recA or recF mutations are added to a ruvC recG mutant, gaps are allowed to accumulate and open the compensating annealing pathway, just as was seen in strains with only a recA or recF mutation. Thus, in the absence of these early recombination steps, single-strand annealing is stimulated and RuvC and RecG functions become irrelevant since there is no way to make a Holliday structure.
Addition of a recB mutation to a ruvC recG double mutant prevents digestion of rrn sequence from double-strand breaks within rrn and allows double-strand ends to initiate duplication by annealing. Thus, double-strand breaks within rrn, which do not normally contribute to recombinational duplication (due to lack of Chi), can contribute to duplication by annealing in the absence of RecBCD degradation.
A remarkable aspect of the reported results is that a similar rate of duplication was seen regardless of the pathway used. If the activated annealing pathway makes up for a loss in recombination events and leaves the duplication rate largely unchanged, the two pathways must function at the same rate. Similarly, when a recB mutation is added to a ruvC, recG mutant, the final rate is very similar to that seen in wild type, in recA, or in recF. We suggest that the similarity in rates reflects dependence on a limiting resource, the gaps and breaks that initiate the exchange, regardless of the pathway used. That is, gaps form at a limiting rate and are essentially all ultimately repaired by either one pathway or the other. The similarity of rates when double-strand annealing is allowed by a recB mutation may suggest that the rate of forming breaks within rrn is equivalent or a bit lower to than that of forming gaps. It should be kept in mind that the exchange events measured here arise at a very high rate—10−3/cell/division.
Requirements for activation of annealing pathways
This model suggests that normal recombination functions not only contribute to duplication by homologous recombination, but also minimize the likelihood of single-strand annealing by processing structures such as nicks or gaps and coating single strands with RecA protein. This inhibition may minimize rearrangements between long direct repeats that are prone to frequent damage, such as rrn sequences. The model also suggests that annealing can be initiated either by single-strand nicks and gaps or by double-strand breaks. In the case of rrn loci, formation of these lesions may be stimulated by the high transcription rate and abundance of included palindromic sequences. A previous report on E. coli has shown that deletions arising by single-strand annealing are strongly enhanced by intervening palindromes (Bzymek and Lovett 2001).
The ability of recombination pathways to block rearrangement by annealing may also involve coating single strands with RecA protein. Coated single strands (i.e., RecA filaments) are impaired for annealing. Evidence for the stimulation of single-strand annealing by the absence of a functional RecA has been previously reported in E. coli (Bzymek and Lovett 2001). The activation of the annealing pathway may require elimination of RecA or the proteins that load RecA onto a single strand (i.e., RecFOR or RecBCD). This fits with a recF mutant, which can accumulate unrepaired nicks and gaps as outlined above, and also fails to load RecA on single-strand regions. Similarly, a recB mutant fails to remove rrn sequence from ends at a break and also fails to load RecA onto single-strand ends.
While the genes involved in homologous recombination have been studied extensively in E. coli and Salmonella, relatively very little is known about the annealing process. Several proteins have been associated with single-strand annealing in bacteria, including SbcCD (Bzymek and Lovett 2001; Goldfless et al. 2006). Figure 11 shows the effects of removing these functions from a recA mutant strain, in which duplications are believed to arise exclusively by annealing (Figure 13). Of the enzymes tested, only recQ and recJ mutations appeared to reduce duplication by annealing, but their effect was small. No effect was seen for SbcCD, although this nuclease might have been expected to cut palindromic structures in rrn genes and contribute to initiation by both pathways (Connelly et al. 1998). Duplication formation in a wild-type strain, presumed to involve RecA, was stimulated by a mismatch-repair defect (mutS), suggesting rejection of mismatched heteroduplexes, whereas the RecA-independent annealing pathway was unaffected. These results are consistent with the idea that rrn-mediated duplications arise by two alternative pathways, each dependent on different sets of genetic components.
Lethal effects of accumulated Holliday structures
The ruvC recG mutation combination is known to severely reduce recombination rates and lowers cell viability to 20% in an overnight culture of E. coli (Lloyd 1991). Recently, lethality has been attributed to the toxic effects of a failure to resolve Holliday structures in both yeast (Fabre et al. 2002) and Neisseria (Sechman et al. 2006). The general loss of viability in E. coli ruvC recG mutants is thus likely to reflect toxic Holliday structures formed by sister-strand exchanges following occasional spontaneous damage. This general loss of viability is not likely be responsible for the reduced duplication rate described here (Figure 7 and Figure 8) because the measured duplication rate is based on the relative frequency of cells with and without a duplication, which are subject to the same loss of viability. However, it is possible that the 30-fold drop in duplication rate is associated with lethal effects of Holliday structures formed in the course of recombination between rrn loci. That is, ruvC and recG mutations block recombination between rrn sequences and cause accumulation of a lethal intermediate. Such lethality is specific to the production of rrn duplications, but is peripheral to the general model presented here as long as it requires rrn-rrn recombination and duplication would be prevented with or without the lethal effect. The recA, recB, and recF mutations that suppress ruvC, recG not only allow initiating structures to accumulate and activate an annealing pathway, but also prevent formation of toxic Holliday structures. In Neisseria, pilin variation is achieved by recombination between an expression site and silent copies of the pilin gene (Sechman et al. 2006). These exchanges are lethal in strains unable to resolve Holliday structures, and lethality requires both RecA and a mechanism to stimulate the exchange (Cahoon and Seifert 2011). Thus lethality seen in pilin variation and possibly in rrn recombination may reflect high exchange rates in specific restricted sequences.
Similarity to previously reported results for bacterial conjugation
A pattern of genetic dependencies similar to those reported here was seen previously for plasmid conjugation in E. coli (Benson et al. 1991). The conjugative F′lac+ plasmid is transferred between cells as a single strand, whose recombination with the chromosome is low. Acquisition of the plasmid is reduced in ruv mutants and this defect is corrected by an added recA mutation. These results suggest that plasmid circularization normally occurs by recombination requiring Holliday structure resolution, but removal of recA activates an alternative RecA-independent pathway that may involve annealing.
Comparing these results to previous studies of rDNA recombination in yeast
In budding yeast, rrn loci are present as a tandem array of >100 identical copies of a 9-kb repeat, in which copy number is subject to frequent change (Linskens and Huberman 1988). Duplication of an rDNA sequence has been attributed to gene conversion rather than unequal recombination because addition of a copy is seldom accompanied by a corresponding deletion. That is, the exchange that generates a duplication is usually nonreciprocal (Gangloff et al. 1996). While standard conversion events would be expected to require canonical recombination functions, the rate of rDNA copy-number increase is not reduced by lack of Rad51, Rad52, or Rad59 (Houseley and Tollervey 2011). Although the process of yeast rDNA copy-number control is complex (Fierro 1999; Pâques and Haber 1999), it may resemble the process described here in that copy-number changes are caused by homologous recombination, but can also arise by alternative annealing pathways that are activated when recombination is prevented. In addition, the copy-number changes in yeast rDNA copies may be more similar to the second step of amplification (e.g., increasing from a tandem duplication to three tandem copies). In bacteria, multiple, closely positioned substantial repeats interact to cause copy-number changes by a process that is highly RecA dependent. In contrast, the de novo bacterial rrn duplications described here involve interactions between two separate imperfect repeats that are highly transcribed and have the potential to form secondary structures.
Alternatives to duplication by the single-strand-annealing pathway
Single-strand annealing is proposed as the RecA-independent alternative pathway for duplication formation. Annealing avoids the need for strand invasion, but requires simultaneous breaks or nicks to generate single strands from different rrn sequences. A completely different RecA-independent duplication pathway has been proposed to explain the origin of short duplications that amplify during growth under selection in the Cairns system (Cairns and Foster 1991; Roth et al. 2006). In this pathway, a quasi-palindromic sequence initiates repair replication from a snap-back structure that eventually switches template strands, perhaps stimulated by a second palindrome, to make three copies of the intervening region (Kugelberg et al. 2010). The resulting triplication has direct-order copies flanking a central inverse order copy—a symmetrical tandem inversion duplication (sTID). The two internal quasi-palindromic sTID junctions are prone to deletions because of their propensity to form secondary hairpin structures. Such structures, singly or together, can be deleted to leave a TID with asymmetric junctions or a simple direct repeat duplication. The first step of this pathway has been clearly demonstrated by Leach and coworkers (Darmon et al. 2010), and palindromes have been shown to stimulate deletion formation in several systems (Sinden et al. 1991; Bzymek and Lovett 2001).
Palindrome processing by the TID pathway is attractive for rrn duplications because palindromes within the locus could be used for both initiation and strand switching. However, the large duplicated regions, such as rrnB-rrnH (790 kb), would necessitate repair synthesis of extensive chromosomal regions. Observed TID amplifications are generally shorter (10–30 kb) (Araya et al. 2010; Kugelberg et al. 2010). Palindrome processing also does not easily explain the reduced duplication rate seen in a ruvC recG double mutant. While the TID mechanism remains a formally possible way to form rrn duplications, we suggest that this pathway is more likely to apply to the RecA-independent duplications that arise at low rates in regions without long direct-order flanking repeats.
Acknowledgments
We thank Sophie Maisnier-Patin, John Paul Aboubechara, Semarhy Quinones-Soto, Douglas Huseby, Wolf Heyer and Steven Kowalczykowski for helpful suggestions. This work was supported by National Institutes of Health grant GM27068.
Appendix: Sequence Differences Between rrn Loci in Salmonella enterica
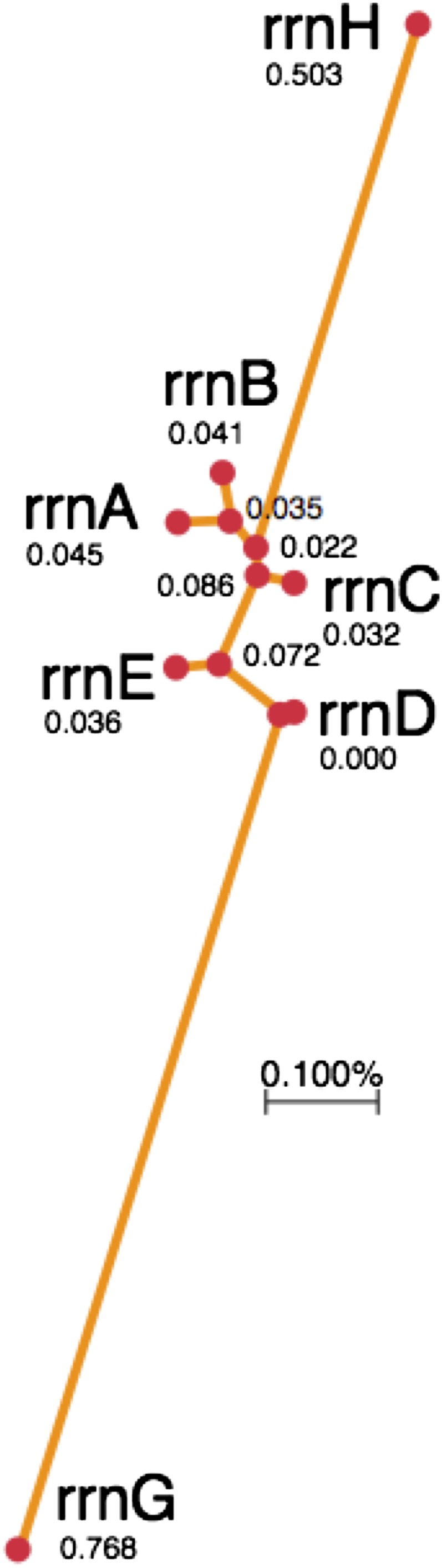
The figure below describes the sequence variation between the seven rrn loci of S. enterica. The central cluster of most similar rrn genes (rrnA,- B,-C, -D, -E) contains those located nearest the replication origin of the Salmonella chromosome (see Figure 2). All of the loci in this cluster have an intervening sequence in helix 25 of their 23S RNA gene that is removed from the messenger RNA prior to ribosome assembly (Mattatall and Sanderson 1996, 1998). The most dissimilar loci (rrnH and rrnG) show ∼1.5% sequence difference. The rrnG locus has an intervening sequence only in helix 45 of its 23S RNA gene. The rrnH gene has an intervening sequence in both helix 25 and helix 45.
Footnotes
Communicating editor: S. Sandler
Literature Cited
- Anderson P., Roth J., 1981. Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons. Proc. Natl. Acad. Sci. USA 78: 3113–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya C. L., Payen C., Dunham M. J., Fields S., 2010. Whole-genome sequencing of a laboratory-evolved yeast strain. BMC Genomics 11: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson F., Collier S., Lloyd R. G., 1991. Evidence of abortive recombination in ruv mutants of Escherichia coli K12. Mol. Gen. Genet. 225: 266–272. [DOI] [PubMed] [Google Scholar]
- Bzymek M., Lovett S. T., 2001. Evidence for two mechanisms of palindrome-stimulated deletion in Escherichia coli: single-strand annealing and replication slipped mispairing. Genetics 158: 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon L. A., Seifert H. S., 2011. Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Mol. Microbiol. 81: 1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon L. A., Seifert H. S., 2013. Transcription of a cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog. 9: e1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J., Foster P. L., 1991. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly J. C., Kirkham L. A., Leach D. R., 1998. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. USA 95: 7969–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court D. L., Sawitzke J. A., Thomason L. C., 2002. Genetic engineering using homologous recombination. Annu. Rev. Genet. 36: 361–388. [DOI] [PubMed] [Google Scholar]
- Darmon E., Eykelenboom J. K., Lincker F., Jones L. H., White M., et al. , 2010. E. coli SbcCD and RecA control chromosomal rearrangement induced by an interrupted palindrome. Mol. Cell 39: 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis P. P., Ehrenberg M., Bremer H., 2004. Control of rRNA synthesis in Escherichia coli: a systems biology approach. Microbiol Mol Biol Rev 68: 639–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eykelenboom J. K., Blackwood J. K., Okely E., Leach D. R., 2008. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol. Cell 29: 644–651. [DOI] [PubMed] [Google Scholar]
- Fabre F., Chan A., Heyer W. D., Gangloff S., 2002. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. USA 99: 16887–16892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro F, M. J., 1999. Molecular mechanisms of chromosomal rearrangement in fungi. Crit. Rev. Microbiol. 25: 1–17. [DOI] [PubMed] [Google Scholar]
- Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz A., 2005. DNA Repair and Mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- Gangloff S., Zou H., Rothstein R., 1996. Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO J. 15: 1715–1725. [PMC free article] [PubMed] [Google Scholar]
- Gellert M., Mizuuchi K., O’Dea M. H., Itoh T., Tomizawa J. I., 1977. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl. Acad. Sci. USA 74: 4772–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfless S. J., Morag A. S., Belisle K. A., Sutera V. A., Jr, Lovett S. T., 2006. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol. Cell 21: 595–604. [DOI] [PubMed] [Google Scholar]
- Heller R. C., Marians K. J., 2005. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell 17: 733–743. [DOI] [PubMed] [Google Scholar]
- Helm R. A., Maloy S., 2001. Rapid approach to determine rrn arrangement in Salmonella serovars. Appl. Environ. Microbiol. 67: 3295–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm R. A., Lee A. G., Christman H. D., Maloy S., 2003. Genomic rearrangements at rrn operons in Salmonella. Genetics 165: 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J., Tollervey D., 2011. Repeat expansion in the budding yeast ribosomal DNA can occur independently of the canonical homologous recombination machinery. Nucleic Acids Res. 39: 8778–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H., Aoki K., Naito A., 1982. Illegitimate recombination mediated in vitro by DNA gyrase of Escherichia coli: structure of recombinant DNA molecules. Proc. Natl. Acad. Sci. USA 79: 3724–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov E. L., Sugawara N., Fishman-Lobell J., Haber J. E., 1996. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer K. N., 2005. Interplay between DNA replication and recombination in prokaryotes. Annu. Rev. Microbiol. 59: 43–67. [DOI] [PubMed] [Google Scholar]
- Kugelberg E., Kofoid E., Reams A. B., Andersson D. I., Roth J. R., 2006. Multiple pathways of selected gene amplification during adaptive mutation. Proc. Natl. Acad. Sci. USA 103: 17319–17324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelberg E., Kofoid E., Andersson D. I., Lu Y., Mellor J., et al. , 2010. The tandem inversion duplication in Salmonella enterica: selection drives unstable precursors to final mutation types. Genetics 185: 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linskens M. H., Huberman J. A., 1988. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 8: 4927–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. L., Sanderson K. E., 1998. Homologous recombination between rrn operons rearranges the chromosome in host-specialized species of Salmonella. FEMS Microbiol. Lett. 164: 275–281. [DOI] [PubMed] [Google Scholar]
- Lloyd R. G., 1991. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J. Bacteriol. 173: 5414–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattatall N. R., Sanderson K. E., 1996. Salmonella typhimurium LT2 possesses three distinct 23S rRNA intervening sequences. J. Bacteriol. 178: 2272–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattatall N. R., Sanderson K. E., 1998. RNase III deficient Salmonella typhimurium LT2 contains intervening sequences (IVSs) in its 23S rRNA. FEMS Microbiol. Lett. 159: 179–185. [DOI] [PubMed] [Google Scholar]
- McDonald J. P., Rothstein R., 1994. Unrepaired heteroduplex DNA in Saccharomyces cerevisiae is decreased in RAD1 RAD52-independent recombination. Genetics 137: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers R. S., Stahl F. W., 1994. Chi and the RecBC D enzyme of Escherichia coli. Annu. Rev. Genet. 28: 49–70. [DOI] [PubMed] [Google Scholar]
- Pâques F., Haber J. E., 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63: 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit M. A., Dimpfl J., Radman M., Echols H., 1991. Control of large chromosomal duplications in Escherichia coli by the mismatch repair system. Genetics 129: 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlhaus J. R., Kreuzer K. N., 2005. Norfloxacin-induced DNA gyrase cleavage complexes block Escherichia coli replication forks, causing double-stranded breaks in vivo. Mol. Microbiol. 56: 1416–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteete A. R., 2009. Expansion of a chromosomal repeat in Escherichia coli: roles of replication, repair, and recombination functions. BMC Mol. Biol. 10: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reams A. B., Neidle E. L., 2003. Genome plasticity in Acinetobacter: new degradative capabilities acquired by the spontaneous amplification of large chromosomal segments. Mol. Microbiol. 47: 1291–1304. [DOI] [PubMed] [Google Scholar]
- Reams A. B., Neidle E. L., 2004. Gene amplification involves site-specific short homology-independent illegitimate recombination in Acinetobacter sp. strain ADP1. J. Mol. Biol. 338: 643–656. [DOI] [PubMed] [Google Scholar]
- Reams A. B., Kofoid E., Savageau M., Roth J. R., 2010. Duplication frequency in a population of Salmonella enterica rapidly approaches steady state with or without recombination. Genetics 184: 1077–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reams A. B., Kofoid E., Kugelberg E., Roth J. R., 2012. Multiple pathways of duplication formation with and without recombination (RecA) in Salmonella enterica. Genetics 192: 397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D., Palacios R., 1997. Gene amplification and genomic plasticity in prokaryotes. Annu. Rev. Genet. 31: 91–111. [DOI] [PubMed] [Google Scholar]
- Roth J. R., Benson N., Galitski T., Haack K., Lawrence J. G., et al. , 1996. Rearrangement of the bacterial chromosome: formation and applications, pp. 2256–2276 in Escherichia coli and Salmonella, edited by Neidhardt F. C. ASM Press, Washington, DC. [Google Scholar]
- Roth J. R., Kugelberg E., Reams A. B., Kofoid E., Andersson D. I., 2006. Origin of mutations under selection: the adaptive mutation controversy. Annu. Rev. Microbiol. 60: 477–501. [DOI] [PubMed] [Google Scholar]
- Sechman E. V., Kline K. A., Seifert H. S., 2006. Loss of both Holliday junction processing pathways is synthetically lethal in the presence of gonococcal pilin antigenic variation. Mol. Microbiol. 61: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden R. R., Zheng G. X., Brankamp R. G., Allen K. N., 1991. On the deletion of inverted repeated DNA in Escherichia coli: effects of length, thermal stability, and cruciform formation in vivo. Genetics 129: 991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriskanda V., Shuman S., 2001. A second NAD(+)-dependent DNA ligase (LigB) in Escherichia coli. Nucleic Acids Res. 29: 4930–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark J. M., Pierce A. J., Oh J., Pastink A., Jasin M., 2004. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 24: 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardrope L., E. O., and D. Leach, 2009. Resolution of joint molecules by RuvABC and RecG following cleavage of the Escherichia coli chromosome by EcoKI. PLoS ONE 4: e6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S. C., 2003. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 4: 435–445. [DOI] [PubMed] [Google Scholar]