

SUMMARY
GABAa receptors are the primary inhibitory ion channels in the mammalian central nervous system. The A322D mutation in the α1 subunit of GABAa receptors is known to result in its degradation and reduce its cell surface expression, leading to loss of GABAa receptor function in autosomal dominant juvenile myoclonic epilepsy. Here, we show that SAHA, a FDA-approved drug, increases the transcription of the α1(A322D) subunit, enhances its folding and trafficking post-translationally, increases its cell surface level, and restores the GABA-induced maximal current in HEK293 cells expressing α1(A322D)β2γ2 receptors to 10% of that for wild type receptors. To enhance the trafficking efficiency of the α1(A322D) subunit, SAHA increases the BiP protein level and the interaction between the α1(A322D) subunit and calnexin. SAHA is the first reported drug that enhances epilepsy-associated GABAa receptor proteostasis.
INTRODUCTION
The maintenance of protein homeostasis (proteostasis) in each subcellular compartment is necessary for maintaining normal organismal physiology (Balch, et al., 2008; Hartl, et al., 2011). The capacity and throughput of the proteostasis network needs to be adjusted to achieve a delicate balance between protein synthesis, folding, and trafficking vs. degradation while minimizing misfolding and aggregation (Gidalevitz, et al., 2011; Hebert, et al., 2010; Lindquist, 1986; Schroder and Kaufman, 2005; Smith, et al., 2011; Vembar and Brodsky, 2008; Walter and Ron, 2011). Excessive protein misfolding and degradation lead to numerous loss-of-function diseases (Guerriero and Brodsky, 2012; Hebert and Molinari, 2007).
About one third of the eukaryotic proteome, including the membrane proteome, is folded in the endoplasmic reticulum (ER). Ion channel proteins, including GABAa receptors, are co-translationally translocated onto the ER membrane for folding and assembly (Alder and Johnson, 2004; Green and Millar, 1995; Skach, 2009). Cellular folding of ion channels requires the engagement of both the ER and the cytosolic folding machineries because ion channels contain both the ER lumenal and cytosolic components (Braakman and Bulleid, 2011; Bukau, et al., 2006; Deuerling and Bukau, 2004; Frydman, 2001; Hartl and Hayer-Hartl, 2002). The BiP system and the calnexin / calreticulin system are the two major chaperone systems in the ER (Braakman and Bulleid, 2011; Dudek, et al., 2009; Hebert and Molinari, 2012; Hebert, et al., 1995; Helenius and Aebi, 2004; Otero, et al., 2010; Rutkevich and Williams, 2011). BiP (also termed Grp78, Gene name: HSPA5) belongs to the heat shock protein 70 kDa (Hsp70) family. BiP binds to the hydrophobic patches of unfolded proteins, facilitating their folding while preventing aggregation (Dudek, et al., 2009; Flynn, et al., 1991; Otero, et al., 2010; Simons, et al., 1995). However, in association with its co-chaperones, such as ERdj4 or ERdj5, BiP may direct terminally misfolded proteins to the cellular degradation pathway (Dong, et al., 2008; Dudek, et al., 2009; Otero, et al., 2010; Ushioda, et al., 2008). Upon entering the ER, proteins are glycosylated on Asn residues (in the context of an Asn-X-Ser/Thr sequence motif) and attached with the core oligosaccharide Glc3Man9GlcNAc2, where Glc is glucose, Man is mannose, and GlcNAc is N-acetylglucosamine. This N-linked core oligosaccharide serves as a tag for protein maturation (Hebert and Molinari, 2012; Helenius and Aebi, 2004). Calnexin, a membrane bound lectin chaperone, and its soluble orthologue calreticulin assist N-linked glycoprotein folding in the ER with the assistance of protein disulfide isomerases (PDIs) (Helenius, et al., 1997; Rutkevich and Williams, 2011). Although BiP and calnexin were reported to interact with the α1 subunit of GABAa receptors (Connolly, et al., 1996), their role in assisting GABAa receptor folding and trafficking was not explored. Properly folded and assembled ion channels will be exported out of the ER and trafficked to the Golgi, where ion channels undergo additional complex glycosylation events and ultimately reach the plasma membrane in a fully functional state. In the case of misfolded ion channels, the ER quality control machinery recognizes and targets them for the ER-associated degradation (ERAD) pathway, where misfolded ion channels are retrotranslocated into the cytosol and degraded by the proteasome (Vembar and Brodsky, 2008).
GABAa receptors are the major inhibitory neurotransmitter-gated ion channels in the human brain (Macdonald and Olsen, 1994) and belong to the Cys-loop receptor superfamily (Lester, et al., 2004). GABAa receptors are pentameric, sharing common structural characteristics with other Cys-loop receptor members (Dougherty, 2008). The most common GABAa receptor type in the human brain is a heteropentamer, containing two α, two β, and one γ subunits. Each subunit has four transmembrane (TM) helices (TM1-TM4, with the TM2 domain lining the interior of the pore); a large extracellular (or the ER lumenal) N-terminus; and an extracellular (or the ER lumenal) C-terminus. Each heteropentameric GABAa receptor contains two GABA-binding sites. Each GABA binding site lies between one α1 subunit and its adjacent β2 subunit and occupies the N-terminal extracellular domain.
The missense A322D mutation in the TM3 domain of the α1 subunit of GABAa receptors leads to autosomal dominant juvenile myoclonic epilepsy (ADJME), a common form of idiopathic generalized epilepsy representing 5-10% of all epilepsy cases (Cossette, et al., 2002). The A322D mutation in the α1 subunit was reported to result in the misfolding of the α1(A322D) subunit, which led to rapid degradation of the α1(A322D) subunit mainly by the ER-associated degradation pathway (Gallagher, et al., 2007), although the misfolded α1(A322D) subunit might be alternatively targeted to the lysosome for degradation (Bradley, et al., 2008). The consequence is that few α1(A322D) subunits are transported to the plasma membrane, reducing the level of functional heteropentameric GABAa receptors in the cell membrane. As a consequence, the A322D mutation leads to substantially reduced GABA-induced current in electrophysiological experiments. In the case of the few mutant receptors that reach the plasma membrane, they generate GABA-induced currents with different electrophysiological properties compared to WT receptors (Krampfl, et al., 2005; Lachance-Touchette, et al., 2011).
We hypothesized that we could enhance the folding and/or trafficking and thus restore function in the case of epilepsy-associated mutant α1(A322D)β2γ2 GABAa receptors. A number of proteostasis regulators (Balch, et al., 2008) were reported to correct protein misfolding diseases such as cystic fibrosis and lysosomal storage diseases (Hutt, et al., 2010; Mu, et al., 2008). Thus, we tested whether such small molecules could enhance proteostasis of α1(A322D)β2γ2 GABAa receptors. We showed that suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA), potent histone deacetylase (HDAC) inhibitors, increased the total protein level of the α1(A322D) subunit. Our data indicates that SAHA, a FDA-approved drug that crosses the blood-brain barrier (Hockly, et al., 2003), increased the transcription, enhanced the folding and trafficking, and partially corrected function of epilepsy-associated α1(A322D)β2γ2 GABAa receptors, thus representing a potential avenue for the treatment of ADJME that results from GABAa receptor misfolding.
RESULTS
SAHA increases the total protein level of α1(A322D)β2γ2 GABAa receptors.
The A322D mutation in the α1 subunit was reported to decrease both the total and cell surface expression of the α1(A322D) subunit mainly by ERAD (Gallagher, et al., 2007). We tested small molecules that were reported to rescue misfolded proteins in the literature, including celastrol, a potent heat shock response activator; SAHA and TSA, potent HDAC inhibitors; and curcumin and thapsigargin, ER Ca2+ signaling pathway regulators, for their potential to rescue functional GABAa receptor expression. HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors were treated with celastrol (0.5 μM), SAHA (2.5 μM), TSA (0.5 μM), curcumin (1 μM) or thapsigargin (0.5 μM) for 24 h. Western blot analysis showed that both SAHA and TSA, but not other small molecules significantly increased the total protein level of the α1(A322D) subunit (Figure S1A). The different effect between SAHA / TSA and other small molecules might be due to their different regulation of GABAa receptor binding chaperones. Dose-response analysis of SAHA (24 h) showed that SAHA increased the total α1(A322D) subunit significantly at as low as 0.5 μM, and the saturation effect of SAHA was achieved at 2.5 μM (Figure S1B). Time-course study showed that SAHA’s effect on increasing total α1(A322D) subunit was achieved as early as 4 h and maximized at 24 h; longer SAHA treatment didn’t further increase total α1(A322D) subunit, which might be due to the instability of SAHA in cell culture media (Figure S1C). SAHA treatment (2.5 μM, 24 h) or TSA treatment (0.5 μM, 24 h) increased the total protein levels of α1, β2 and γ2 subunits in HEK293 cells expressing either WT or α1(A322D)β2γ2 GABAa receptors (Figure 1A, see Figure 1B for quantification). The total protein level of the α1(A322D) subunit is 25% of that of the WT α1 subunit because the A322D mutation resulted in extensive ERAD of the α1(A322D) subunit (Figure 1B). In HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors, SAHA treatment (2.5 μM, 24 h) increased the total protein level of the α1(A322D) subunit by 5.0-fold, the β2 subunit by 2.0-fold, and the γ2 subunit by 1.4-fold, whereas TSA treatment (0.5 μM, 24 h) increased the total protein level of the α1(A322D) subunit by 3.0-fold, the β2 subunit by 2.7-fold, and the γ2 subunit by 1.9-fold (Figure 1B). An increase in WT α1, β2, or γ2 subunits afforded by SAHA treatment might be due to inefficient folding and trafficking or an increase in the synthesis of WT subunits. However, a more dramatic increase of the total protein was observed in HEK293 cells expressing α1(A322D) subunits than that expressing WT α1 subunits, possibly because SAHA’s effect is more dramatic on misfolding-prone α1(A322D) subunits. An MTT cell toxicity assay showed that SAHA treatment (2.5 μM, 24 h) of the HEK293 cells expressing either WT or α1(A322D)β2γ2 GABAa receptors did not substantially reduce the cell viability (Figure S1D), consistent with the fact that SAHA is well-tolerated in the clinical trial (Kelly, et al., 2003). Quantitative RT-PCR experiment showed that SAHA treatment (2.5 μM, 24 h) increased the mRNA level of the α1(A322D) subunit by 3.8-fold in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors (Figure S1E), indicating that SAHA treatment increased the protein synthesis load of the α1(A322D) subunit, partially contributing to the 5.0-fold increase of the protein level of the α1(A322D) subunit.
Figure 1.
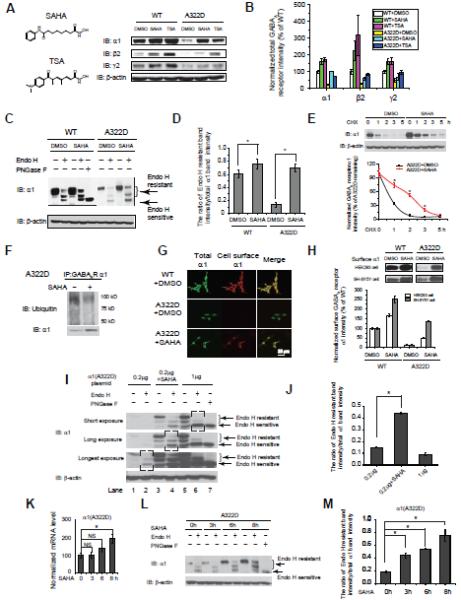
SAHA enhances the folding and trafficking of the α1(A322D) subunit in cells stably expressing epilepsy-associated α1(A322D)β2γ2 GABAa receptors.
(A and B) SAHA (2.5 μM, 24 h) or TSA (0.5 μM, 24 h) increases the total protein level of α1, β2, and γ2 subunits in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAa receptors (n = 3) (A). The chemical structures of SAHA and TSA are shown on left of (A). Quantification is shown in (B). IB: immunoblotting.
(C and D) SAHA (2.5 μM, 24 h) increases the endo H-resistant post-ER glycoform of the α1 subunit in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAa receptors (n = 3) (C). PNGase F treatment serves as a control for unglycosylated α1 subunit. Quantification of the ratio of endo H-resistant / total α1subunit bands is shown in (D).
(E) SAHA (2.5 μM, 24 h) decreases the degradation rate of the α1(A322D) subunit in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors using cycloheximide (CHX) pulse-chase analysis.
(F) SAHA (2.5 μM, 24 h) decreases ubiquitinated α1(A322D) subunit in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors.
(G) SAHA (2.5 μM, 24 h) increases the α1(A322D) subunit surface expression in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors using indirect immunofluorescence analysis (n = 3).
(H) SAHA (2.5 μM, 24 h) increases the surface protein expression of the α1 subunit in HEK293 cells and SH-SY5Y cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAa receptors according to surface biotinylation analysis (n = 3).
(I and J) An increased protein synthesis load of the misfolding-prone α1(A322D) subunit does not lead to increased trafficking efficiency of the α1(A322D) subunit. HEK293 cells were transiently transfected with 0.2 μg or 1.0 μg of α1(A322D) plasmids for 48 h or transfected with 0.2 μg of α1(A322D) plasmids for 48 h and treated with SAHA (2.5 μM, 24 h) together with same amount of β2 and γ2 subunit plasmids before being lysed for endo H digestion analysis (I). Quantification is shown in (J). Because of the large dynamic range of blot intensity, for clear visualization, we put the same blot under three different exposure times to quantify the trafficking efficiency of the α1(A322D) subunit under three different conditions. We used the three black-boxed regions in Figure 1I to achieve accurate quantification with appropriate exposure.
(K) SAHA (2.5 μM) increases the mRNA level of the α1(A322D) subunit significantly after 8 h treatment, but not before 6 h treatment using quantitative RT-PCR analysis in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors. NS: not significant.
(L and M) SAHA (2.5 μM) increases the ratio of endo H resistant / total α1(A322D) subunit significantly as early as 3 h treatment (L). Quantification is shown in (M).
Each data point in (B) (D) (E) (H) (J) (K) and (M) is reported as mean ± SEM. * p < 0.05 See also Figure S1.
SAHA enhances the folding and trafficking, and reduces the ERAD of the α1(A322D) subunit post-translationally.
To determine whether the increased total α1(A322D) subunit protein after SAHA treatment folded properly in the ER, we treated the cell lysates with endoglycosidase H (endo H) enzyme and then analyzed them using Western blot. The endo H enzyme selectively cleaves after asparaginyl-N-acetyl-D-glucosamine in the N-linked glycans incorporated on the α1 subunit in the ER, but it is not possible to remove this oligosaccharide chain after the high-mannose form is enzymatically remodeled in the Golgi. Therefore, endo H resistant α1 subunit bands represent properly folded, post-ER α1 subunit glycoforms, which traffic at least to the Golgi compartment. The Peptide-N-Glycosidase F (PNGase F) enzyme cleaves between the innermost GlcNAc and asparagine residues from N-linked glycoproteins, serving as a control for unglycosylated α1 subunits (Figure 1C, lane 5). After endo H digestion, subunits with a MW equal to unglycosylated α1 subunits were considered endo H-sensitive, whereas those with higher MW were considered endo H-resistant (Figure 1C, lanes 2, 4, 7, and 9). There are two endo H-resistant bands because the α1 subunit has two glycosylation sites (Asn38 and Asn138) in the ER. This is consistent with a report of the endo H-resistant β2 subunits (Lo, et al., 2010). The upper two endo H-resistant α1(A322D) subunit bands are 23% of those of the WT α1 subunit (Figure 1C, cf. lane 7 to lane 2). SAHA treatment (2.5 μM, 24 h) in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors clearly increased the upper two endo H-resistant α1(A322D) subunit bands (Figure 1C, cf. lane 9 to lane 7), indicating that SAHA treatment increased properly folded, post-ER glycoforms of the α1(A322D) subunit. The ratio of endo H resistant / total α1(A322D) serves as a measure of trafficking efficiency of the α1(A322D) subunit. SAHA treatment (2.5 μM, 24 h) significantly increased the ratio of endo H resistant / total α1(A322D) (Figure 1C, cf. lane 9 to lane 7, see Figure 1D for quantification), indicating that SAHA treatment increased trafficking efficiency of the α1(A322D) subunit.
Furthermore, we used cycloheximide (CHX)-chase experiments to quantify whether SAHA influenced the degradation rate of the α1(A322D) subunit. The α1(A322D) subunit has a half-life of 40 min when fitted to a single exponential function (Figure 1E), consistent with the reported [35S]methionine-chase result (Bradley, et al., 2008; Gallagher, et al., 2007). SAHA treatment (2.5 μM, 24 h) increased the half-life of the α1(A322D) subunit from 40 min to 125 min (Figure 1E), indicating that SAHA reduced the degradation of the α1(A322D) subunit. To determine whether SAHA influenced the proteasome degradation of the α1(A322D) subunit, HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors were immunoprecipitated using anti-α1 antibody and blotted for ubiquitin. SAHA treatment (2.5 μM, 24 h) clearly decreased the intensity of ubiquitinated α1(A322D) subunit (Figure 1F), indicating that SAHA reduced the ERAD of the α1(A322D) subunit. To determine whether SAHA influenced the lysosomal degradation of the α1(A322D) subunit, we used a lysosomal protease inhibitor leupeptin with or without SAHA in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors. Leupeptin treatment (25 μM, 24 h) increased the total protein level of the α1(A322D) subunit (Figure S1F), consistent with a literature report that the lysosome is involved in the degradation of the α1(A322D) subunit (Bradley, et al., 2008). Co-application of SAHA and leupeptin produced slightly synergistic effect to increase the total protein level of the α1(A322D) subunit (Figure S1F), indicating that SAHA did not influence the lysosomal degradation of the α1(A322D) subunit. To determine whether SAHA influenced the endocytosis of the α1(A322D) subunit, we used a dynamin 1 inhibitor dynole 34-2 with or without SAHA in HEK293 cells stably expressing α1(A322D)β2γ2 GABAA receptors. Dynole 34-2 treatment (2.5 μM, 24 h) increased the surface α1(A322D) subunit (Figure S1G), consistent with a literature report that the α1(A322D) subunit undergoes dynamin-1 dependent endocytosis on the plasma membrane (Bradley, et al., 2008). Co-application of SAHA and dynole 34-2 produced additive effect to increase the surface α1(A322D) subunit (Figure S1G), indicating that SAHA did not influence the dynamin-1 dependent endocytosis of the α1(A322D) subunit.
To determine whether properly folded α1(A322D) subunits after SAHA treatment were successfully trafficked to the plasma membrane, we performed an indirect immunofluorescence microscope experiment. The application of the anti-α1 antibody without a prior membrane permeabilization step enables us to visualize the cell surface α1 subunit. In HEK293 cells expressing WT GABAa receptors, as expected, a substantial amount of α1 subunit staining was observed for both total α1 subunit (Figure 1G, top row, green) and cell surface α1 subunit (Figure 1G, top row, red). The merge between the total α1 subunit and cell surface α1 subunit is colored yellow (Figure 1G, third column). The A322D mutation led to very weak total α1 subunit staining (Figure 1G, second row, green) and essentially no cell surface α1 subunit staining (Figure 1G, second row) because of extensive ERAD. SAHA treatment (2.5 μM, 24 h) resulted in a dramatic increase in both total α1(A322D) subunit staining (Figure 1G, third row, green) and cell surface α1(A322D) subunit staining (Figure 1G, third row, red), indicating that SAHA treatment enhanced the trafficking of the α1(A322D) subunit to the cell surface.
We further confirmed and quantified the increased cell surface α1 subunit level using cell surface biotinylation and Western blot analysis. SAHA treatment (2.5 μM, 24 h) increased the surface α1(A322D) subunit level 5.0-fold (to 49% of the surface WT α1 subunit level) in HEK293 cells and 11.5-fold (to 137% of the surface WT α1 subunit level) in SH-SY5Y cells expressing α1(A322D)β2γ2 GABAa receptors (Figure 1H). SAHA treatment (2.5 μM, 24 h) also modestly increased the surface WT α1 subunit in HEK293 cells or SH-SY5Y cells stably expressing WT α1β2γ2 GABAa receptors (Figure 1H). This might be due to an increase of the synthesis or an enhanced trafficking of WT α1 subunit.
To show that an increased trafficking efficiency of the misfolding-prone α1(A322D) subunit was due to SAHA’s effect post-translationally and not from an increased protein synthesis load, we carried out the following experiments. HEK293 cells were transiently transfected with 0.2 μg or 1.0 μg of α1(A322D) plasmids for 48 h, or transfected with 0.2 μg of α1(A322D) plasmids for 48 h and treated with SAHA for 24 h before being lysed for endo H digestion analysis. Using these two plasmid amounts was led by a previous report, showing that the total α1(A322D) subunit protein expression level increased linearly with the transfected α1(A322D) plasmids from 0 – 2 μg (Ding, et al., 2010). We observed that HEK293 cells transfected with 1.0 μg of α1(A322D) plasmids led to more total α1(A322D) protein load than HEK293 cells transfected with 0.2 μg of α1(A322D) plasmids in the presence or absence of SAHA (Figure 1I, cf. lane 5 to lane 3 and lane 1). However, the ratio of endo H resistant / total α1(A322D) in HEK293 cells did not increase due to increased amount of transfected plasmids (i.e., 0.2 μg to 1.0 μg) (Figure 1I, cf. lane 6 to lane 2, see Figure 1J for quantification), indicating that increasing protein synthesis load of the α1(A322D) subunit did not increase trafficking efficiency of the α1(A322D) subunit. This was consistent with a previous report, showing that increasing protein synthesis load of α1(A322D) subunit did not increase its cell surface expression (Ding, et al., 2010). Furthermore, an increased trafficking efficiency of the α1(A322D) subunit afforded by SAHA treatment in HEK293 cells transfected with 0.2 μg of α1(A322D) plasmids was in sharp contrast to the un-increased trafficking efficiency in HEK293 cells transfected with 1.0 μg of α1(A322D) plasmids (Figure 1I, cf. lane 4 to lane 6, see Figure 1J for quantification), indicating that SAHA’s effect on enhancing the trafficking efficiency of the α1(A322D) subunit was indeed a post-translational effect instead of due to increased protein synthesis load of the α1(A322D) subunit.
A time-course study showed that SAHA (2.5 μM) increased the mRNA level of the α1(A322D) subunit significantly after 8 h treatment, but not before 6 h treatment using quantitative RT-PCR analysis in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figure 1K). However, SAHA (2.5 μM) increased the ratio of endo H resistant / total α1(A322D) subunit significantly as early as 3 h treatment (Figure 1L, see Figure 1M for quantification), indicating that an increased trafficking efficiency of the α1(A322D) afforded by SAHA occurred before an increased transcriptional level of the α1(A322D). Similar case was reported: SAHA did not increase the transcription of ΔF508 CFTR before 8 h treatment while SAHA increased mature ΔF508 CFTR protein level as early as after 4 h treatment (Hutt, et al., 2010). Therefore, our result is consistent with SAHA's effect on increasing the trafficking efficiency of the misfolding-prone α1(A322D) subunit post-translationally.
SAHA significantly increases the peak amplitude of GABA-induced chloride currents in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors.
The A322D mutation in the α1 subunit leads to significantly lowered peak amplitude of GABA-induced currents in whole-cell patch-clamp electrophysiological experiments (Krampfl, et al., 2005; Lachance-Touchette, et al., 2011). In our whole-cell patch-clamp experimental setup, dose-response analysis showed that the EC50 value (the concentration of a drug that yields 50% of its maximal current) of GABA was 12.7 ± 4.5 μM (Figure S2) in HEK293 cells expressing WT α1β2γ2 GABAa receptors, consistent with the reported EC50 value of 11.2 ± 0.6 μM (Krampfl, et al., 2005). Without drug treatment, the peak current was 1.0 ± 0.4 pA in response to GABA (3 mM) in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors, and 138 ± 12 pA in response to GABA (1 mM) in HEK293 cells expressing WT α1β2γ2 GABAa receptors (Figure 2A). The activation and desensitization kinetics are different between WT and α1(A322D)β2γ2 GABAa receptors, consistent with the literature report (Krampfl, et al., 2005; Lachance-Touchette, et al., 2011). SAHA (2.5 μM, 24 h) treatment significantly increased the GABA-induced peak current to 14 ± 3 pA in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figure 2A), amounting to 10% of the GABA-induced peak current in HEK293 cells expressing WT GABAa receptors. Quantification of the GABA-induced currents is shown in Figure 2B. These data indicate that SAHA partially restored the function of epilepsy-associated α1(A322D)β2γ2 GABAa receptors. Acute application of SAHA (2.5 μM, 1 min) in the external perfusion recording media to HEK293 cells expressing GABAa receptors during the whole-cell patch-clamp experiment did not significantly change the GABA-induced peak current (data not shown), indicating that SAHA did not act as an agonist/antagonist for GABAa receptors and its function required transcriptional and/or post-translational events.
Figure 2.
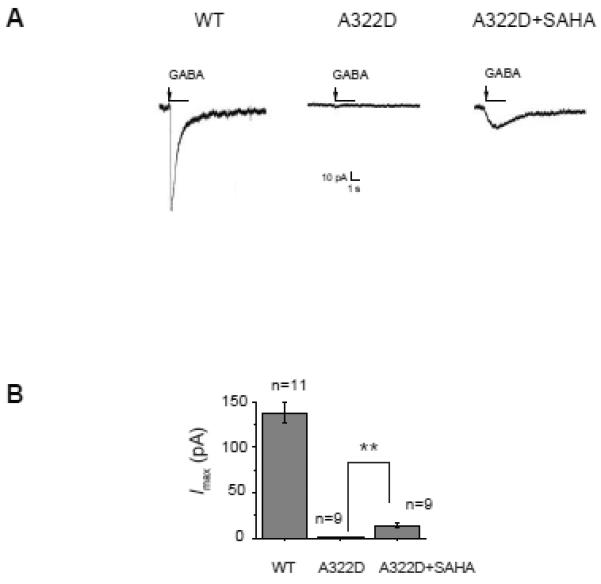
SAHA (2.5 μM, 24 h) increases the peak amplitude of GABA-induced chloride currents in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors. Representative whole-cell patch clamping recording traces in HEK293 cells expressing WT or α1(A322D)β2γ2 GABAa receptors are shown in (A), and quantification of the peak currents is shown in (B). Each point is reported as mean ± SEM. ** p < 0.01 See also Figure S2.
SAHA increases BiP level in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors without an apparent activation of the IRE1 arm of the unfolded protein response.
We hypothesized that SAHA partially corrects the folding deficiency of the α1(A322D) subunit by regulating the expression and/or activity of molecular chaperones in the ER and/or the cytoplasm because the α1(A322D) subunit has both ER lumen and cytoplasmic domains. We tested whether SAHA treatment altered the protein level of major molecular chaperones in the cytoplasm (Hsp70 and Hsp90) and in the ER (calnexin, calreticulin, Grp94 and BiP) and proteins that are involved in the ERAD pathway (Bag2 and Derlin 1). Only the protein level of BiP was significantly upregulated by SAHA treatment (2.5 μM, 24 h): BiP protein level was increased by 2.7-fold in HEK293 cells expressing WT GABAa receptors and 2.1-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figures 3A and 3B). Furthermore, the mRNA level of BiP was increased by 5.6-fold in HEK293 cells expressing WT GABAa receptors and 7.6-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors after SAHA treatment (2.5 μM, 24 h) using quantitative RT-PCR analysis (Figure 3C), indicating that SAHA increased the BiP level mainly through transcriptional regulation.
Figure 3.
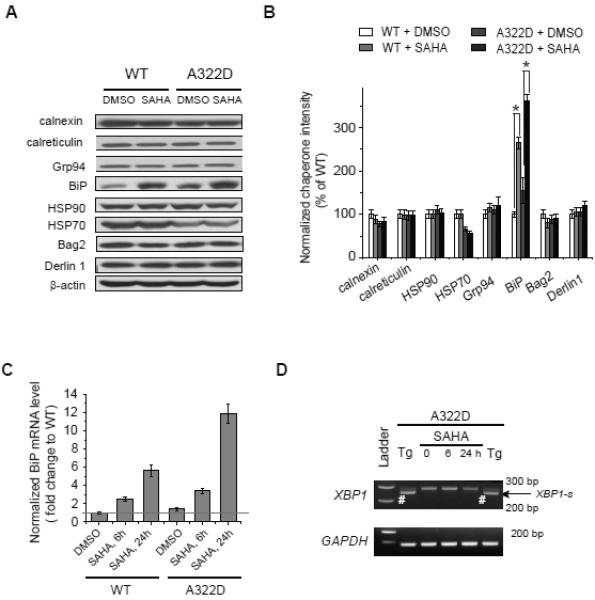
SAHA increases the protein and mRNA level of BiP without an apparent induction of the XBP1 splicing.
(A and B) SAHA (2.5 μM, 24 h) only increases the protein level of BiP among tested major ER and cytosolic chaperones and ERAD factors in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAa receptors (n = 3) (A). Quantification is shown in (B).
(C) AHA (2.5 μM) increases the mRNA level of BiP using quantitative RT-PCR analysis.
(D) SAHA (2.5 μM) does not induce the splicing of XBP1 using RT-PCR analysis. Thapsigargin (Tg) is used as a positive control to induce XBP1 splicing. GAPDH is used as a loading control. Each data point in (B) and (C) is reported as mean ± SEM. * p < 0.05 See also Figure S3 and Table S1.
HSPA5 (Gene name of BiP) is an important target gene in the activation of the unfolded protein response (UPR), which is a well-established ER stress responsive pathway (Schroder and Kaufman, 2005; Walter and Ron, 2011). The ER responds to the accumulation of unfolded proteins by activating up to three integrated intracellular signaling pathways (IRE1, ATF6 and PERK), collectively referred to as the UPR. The UPR regulates the expression of numerous genes comprising the cellular proteostasis pathways (Adachi, et al., 2008; Lee, et al., 2003; Okada, et al., 2002). Because HSPA5 is a target of the IRE1 arm of the UPR, we tested whether SAHA treatment increased BiP mRNA level by activating the IRE1 arm. IRE1 responds to stress by oligomerization, resulting in trans-autophosphorylation that activates its endonuclease function, which precisely splices the mRNA that encodes the transcription factor XBP1 (Schroder and Kaufman, 2005; Walter and Ron, 2011). We used RT-PCR to detect the spliced form of Xbp-1 in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors after an incubation with SAHA (2.5 μM). SAHA treatment did not led to the splicing of XBP1, indicating no activation of the IRE1 arm of the UPR (Figure 3D). Used as a positive control, thapsigargin (Tg, 0.5 μM, 6 h) resulted in XBP1 splicing (Figure 3D, lanes 2 and 6, bottom band, white pound). Therefore, SAHA used another pathway to regulate the mRNA level of BiP (see below, and Figure 6C). Tg (0.5 μM) and tunicamycin (Tm, 10 μg/ mL), potent UPR inducers, increased the protein level of BiP, but not calnexin (Figures S3A and S3B), indicating that in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors, activation of the IRE1 arm did not increase calnexin protein level.
Figure 6.
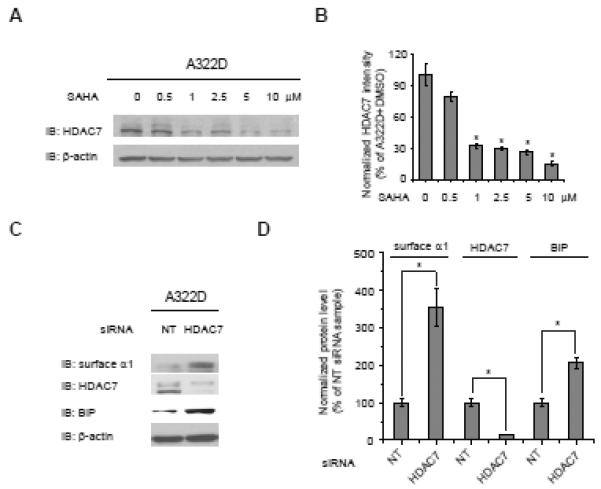
Effect of SAHA on HDAC7 in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors.
(A and B) SAHA treatment (24 h) decreases the HDAC7 protein level in a dose-dependent manner (A). Quantification is shown in (B).
(C and D) HDAC7 knockdown increases the protein level of cell surface α1(A322D) subunit and BiP. HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors were transfected with siRNA against HDAC7 or non-targeting (NT) control for 48 h before protein analysis (C). Quantification is shown in (D).
Each data point in (B) and (D) is reported as mean ± SEM. * p < 0.05
SAHA enhances the interaction between BiP and the α1 subunit in the ER, and BiP promotes GABAa receptor folding and trafficking.
BiP, an abundant ER resident Hsp70 family chaperone, binds to hydrophobic patches of unfolded proteins and facilitates their folding while preventing aggregation (Rudiger, et al., 1997). Because BiP binds GABAa receptors (Connolly, et al., 1996), which have large ER lumenal components, we continued to test whether SAHA enhanced the interaction between BiP and the α1 subunit in the ER. HEK293 cells that express WT or α1(A322D)β2γ2 GABAa receptors were treated with DMSO vehicle control or SAHA (2.5 μM, 24 h), and the ER fractions were enriched. Clearly, SAHA treatment (2.5 μM, 24 h) significantly increased the ratio of immunoprecipitated BiP / α1 by 21-fold in HEK293 cells expressing WT α1β2γ2 GABAa receptors and 2.9-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figure 4A, cf. lane 2 to lane 1, and lane 4 to lane 3, see Figure 4B for quantification), indicating that SAHA treatment enhanced the interaction between α1 subunits and BiP in the ER. We also performed a reverse immunoprecipitation experiment, confirming that SAHA treatment enhanced the interaction between the α1(A322D) subunit and BiP (Figure 4C). However, whether an enhanced interaction between BiP and the α1(A322D) subunit promotes the maturation of the α1(A322D) subunit merits further study.
Figure 4.
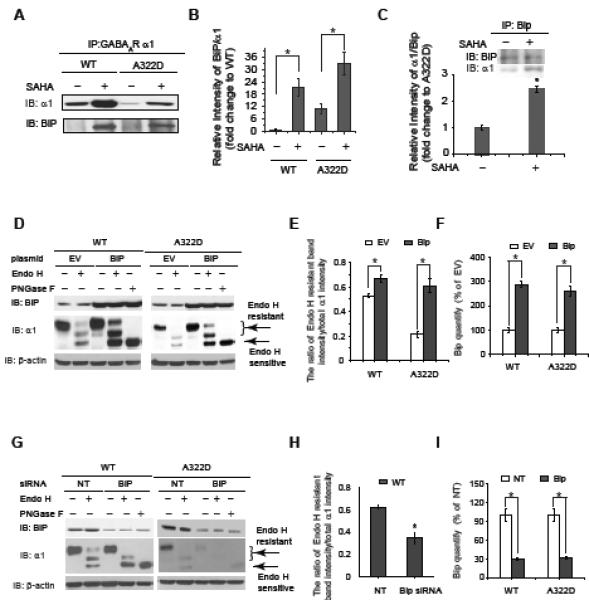
SAHA enhances the interaction between BiP and the α1 subunit, and BiP promotes the α1 subunit folding in the ER.
(A and B) SAHA treatment (2.5 μM, 24 h) enhances the interaction between the α1 subunit and BiP in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAa receptors. Quantification of the relative intensity of BiP/α1 is shown in (B).
(C) Reverse immunoprecipitation confirms that SAHA treatment enhances the interaction between the α1(A322D) subunit and BiP.
(D, E and F) BiP overexpression increases endo H-resistant post-ER glycoform of the α1 subunit (n = 3) (D). PNGase F treatment serves as a control for unglycosylated α1 subunit. Quantification of the ratio of endo H-resistant / total α1 subunit bands is shown in (E), and quantification of BiP overexpression is shown in (F). EV: empty vector.
(G, H and I) BiP knockdown decreases endo H-resistant post-ER glycoform of the α1 subunit (n = 3) (G). Quantification of the ratio of endo H-resistant / total α1 subunit bands is shown in (H), and quantification of BiP knockdown is shown in (I). NT: non-targeting.
Each data point in (B), (C), (E), (F), (H) and (I) is reported as mean ± SEM. * p < 0.05
Because BiP can either promote its client protein folding or target its client protein for degradation, we tested how overexpression or knockdown of BiP influenced the protein homeostasis of the α1 subunit. A 2.8-fold overexpression of BiP significantly increased the ratio of endo H-resistant α1 / total α1 subunit by 1.3-fold in HEK293 cells expressing WT α1β2γ2 GABAa receptors (Figure 4D, cf. lane 4 to lane 2), and a 2.6-fold overexpression of BiP significantly increased the ratio of endo H-resistant α1 / total α1 subunit by 3.0-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figure 4D, cf. lane 9 to lane 7) (see Figures 4E and 4F for quantification), indicating that BiP overexpression increased the trafficking efficiency of the α1 subunit. Knockdown of BiP (69% knockdown efficiency) clearly decreased the intensity of endo H-resistant α1(A322D) subunit (Figure 4G, cf. lane 9 to lane 7), and knockdown of BiP (71% knockdown efficiency) significantly decreased the ratio of endo H resistant α1 / total α1 subunit (Figure 4G, cf. lane 4 to lane 2, see Figures 4H and 4I for quantification), indicating that BiP knockdown decreased the trafficking efficiency of the α1 subunit. Therefore, BiP promotes the folding and maturation of the α1 subunit. Because SAHA increases the BiP expression (Figure 3A) and BiP overexpression enhances the maturation of the α1(A322D) subunit (Figure 4D), SAHA enhances the maturation of the α1(A322D) subunit by increasing the BiP level.
SAHA enhances the interaction between calnexin and the α1 subunit, and calnexin promotes GABAa receptor folding and trafficking in a glycan-dependent way.
Because the α1 subunit is a glycoprotein and calnexin (CNX) and calreticulin are major lectin chaperones in the ER that assist glycosylated protein folding (Helenius, et al., 1997), we tested whether the folding of the α1 subunit was facilitated by calnexin and/or calreticulin in addition to BiP in the ER. Although SAHA did not increase the protein level of calnexin or calreticulin (Figure 3A), we hypothesized that SAHA increased the interaction between the α1 subunit and calnexin and/or calreticulin to enhance the folding of the α1 subunit. Using enriched ER fractions for immunoprecipitation, SAHA treatment (2.5 μM, 24 h) significantly increased the ratio of immunoprecipitated calnexin / α1 by 3.3-fold in HEK293 cells expressing WT α1β2γ2 GABAa receptors and 1.5-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors (Figure 5A, cf. lane 2 to lane 1, and lane 4 to lane 3, see Figure 5B for quantification), indicating that SAHA treatment enhanced the interaction between the α1 subunit and calnexin. We also performed a reverse immunoprecipitation experiment, confirming that SAHA treatment enhanced the interaction between the α1(A322D) subunit and calnexin (Figure 5C). However, an interaction was not detected between the α1(A322D) subunit and endogenous calreticulin or overexpressed HA-tagged calreticulin (Figure S4).
Figure 5.
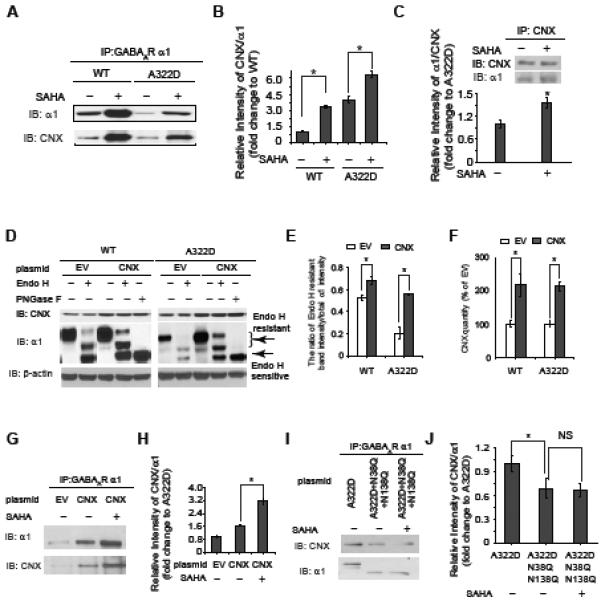
SAHA enhances the interaction between calnexin and the α1 subunit, and calnexin (CNX) promotes the α1 subunit folding in the ER.
(A and B) SAHA treatment (2.5 μM, 24 h) enhances the interaction between the α1 subunit and calnexin in HEK293 cells stably expressing WT α1β2γ2 or α1(A322D)β2γ2 GABAa receptors. Quantification of the relative intensity of calnexin/ α1 is shown in (B).
(C) Reverse immunoprecipitation confirms that SAHA treatment (2.5 μM, 24 h) enhances the interaction between the α1(A322D) subunit and calnexin.
(D, E and F) CNX overexpression increases endo H-resistant post-ER glycoform of the α1 subunit (n = 3) (D). PNGase F treatment serves as a control for unglycosylated α1 subunit. Quantification of the ratio of endo H-resistant / total α1 subunit bands is shown in (E), and quantification of CNX overexpression is shown in (F). EV: empty vector.
(G and H) SAHA treatment (2.5 μM, 24 h) enhances the interaction between the α1(A322D) subunit and overexpressed calnexin in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (G). Quantification of the blots is shown in (H).
(I and J) Calnexin interacts with the α1(A322D) subunit in a glycan-dependent way. The N38Q/N138Q mutations in the α1(A322D) subunit disrupt the interaction between the N-glycans in the α1(A322D) subunit and calnexin (I). SAHA treatment does not change the interaction between the N38Q/N138Q mutant α1(A322D) subunit and calnexin (I). Quantification of the blots is shown in (J). NS: not significant.
Each data point in (B), (C), (E), (F), (H) and (J) is reported as mean ± SEM. * p < 0.05 See also Figure S4.
Overexpression of calnexin significantly increased the ratio of endo H-resistant / total α1 subunit by 1.3-fold in HEK293 cells expressing WT α1β2γ2 GABAa receptors (Figure 5D, cf. lane 4 to lane 2) and 2.7-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figure 5D, cf. lane 9 to lane 7) (see Figures 5E and 5F for quantification), indicating that calnexin overexpression increased the trafficking efficiency of the α1 subunit. Therefore, calnexin promotes the folding and trafficking of the α1 subunit. To explore how the interaction between calnexin and the α1(A322D) subunit influences the α1(A322D) subunit folding, HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors were transfected with calnexin or calreticulin for 48 h and treated with SAHA (2.5 μM, 24 h). SAHA treatment significantly increased the ratio of immunoprecipitated calnexin / α1(A322D) (Figure 5G, cf. lane 3 to lane 2, see Figure 5H for quantification), whereas no interaction was detected between the α1(A322D) subunit and overexpressed HA-tagged calreticulin after SAHA treatment (Figure S4, lane 3). This is consistent with SAHA’s role in promoting the maturation of the α1(A322D) subunit by enhancing the interaction between the α1(A322D) subunit and calnexin (also see below, Figure 5I).
Furthermore, we asked whether calnexin used a glycan-dependent pathway to promote the α1(A322D) subunit folding in the ER. Upon entering the ER, at two glycosylation sites (Asn38 and Asn138), the α1 subunit is attached with the core oligosaccharide Glc3Man9GlcNAc2. The outmost two glucoses are removed by glucosidase I and II, which generates the monoglucosylated oligosaccharide Glc1Man9GlcNAc2, a substrate for calnexin. The N38Q/N138Q double mutations in α1(A322D) would prevent a glycan-dependent binding of calnexin to the N38Q/N138Q mutant α1(A322D) subunit. The N38Q/N138Q mutant α1(A322D) subunit had a lower molecular weight than the α1(A322D) subunit (Figure 5I, cf. lane 2 to lane 1), presumably due to loss of glycans. The N38Q/N138Q mutations significantly decreased the ratio of immunoprecipitated calnexin / α1(A322D) in HEK293 cells (Figure 5I, cf. lane 2 to lane 1, see Figure 5J for quantification), indicating that calnexin partially binds to N-glycans of the α1(A322D) subunit. SAHA treatment did not significantly change the ratio of immunoprecipitated calnexin / N38Q/N138Q mutant α1(A322D) subunit (Figure 5I, cf. lane 3 to lane 2, see Figure 5J for quantification), indicating that a glycan-binding activity of calnexin is important in regulating SAHA’s function to promote α1(A322D) subunit folding.
SAHA decreases HDAC7 protein level, and HDAC7 knockdown increases protein level of cell surface α1(A322D) subunit and BiP.
SAHA was reported to selectively suppress HDAC7 expression (Dokmanovic, et al., 2007), and HDAC7 plays a central role in regulating SAHA’s function of rescuing mutant CFTR (Hutt, et al., 2010). However, how HDAC7 influences protein folding in the ER was not well understood. Therefore, we continued to test the role of HDAC7 in restoring GABAa receptor function, possibly by regulating BiP level. SAHA decreased HDAC7 protein level in a dose-dependent manner from 0.5 – 10 μM in HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors (Figures 6A and 6B). HDAC7 knockdown (87% knockdown efficiency) significantly increased the protein level of cell surface α1(A322D) subunit (Figures 6C and 6D) and total α1(A322D) subunit (data not shown), indicating that HDAC7 regulated the protein synthetic load of the α1(A322D) subunit. Knockdown of HDAC7 significantly increased BiP protein level in HEK293 cells expressing α1(A322D)β2γ2 GABAa receptors (Figures 6C and 6D), indicating that HDAC7 could enhance the surface expression of the α1(A322D) subunit partially by upregulating BiP expression. This also implies that SAHA treatment increased the BiP level by suppressing HDAC7 without an apparent induction of the IRE1 arm of the UPR (Figure 3D).
DISCUSSION
Here, we reported that SAHA enhances epilepsy-associated α1(A322D)β2γ2 GABAa receptor proteostasis. The plasma concentration of SAHA in the context of Phase I clinical trials exceeded 2.5 μM (Kelly, et al., 2003). Thus, our finding that at 2.5 μM, SAHA effectively partially restored epilepsy-associated α1(A322D)β2γ2 GABAa receptor proteostasis (Figures 1 and 2) may be clinical significant. As SAHA at this concentration only restores a GABA-induced maximal current in HEK293 cells expressing α1(A322D)β2γ2 receptors to 10% of that in HEK293 cells expressing wild type receptors, it needs to be evaluated whether this will have a significant in vivo effect. Seizures are threshold events. A small change in GABAa receptor functions, for example by the application of a GABAa receptor antagonist called bicuculline, can change the seizure threshold (Hentschke, et al., 2006). The conversion of an essentially non-functional α1(A322D)β2γ2 receptor to a receptor with 10% wild type receptor function may change a seizure threshold to a clinically beneficial level. SAHA did not rescue trafficking-deficient mutant hERG channel folding and trafficking (unpublished data), indicating that SAHA has certain selectivity in enhancing GABAa receptor folding. As SAHA crosses the blood-brain barrier (Hockly, et al., 2003), is well tolerated in our cell culture study and in clinical trials, and only 8% of genes are significantly changed after SAHA treatment in numerous cell lines by DNA microarray analysis (Glaser, et al., 2003), this makes it a promising test candidate for the amelioration of idiopathic epilepsy resulting from GABAa receptor misfolding.
GABAa receptors are heteropentameric receptors. The missense A322D mutation is in the TM3 domain of the α1 subunit, close to the cytosolic part. The introduction of a negative charge in TM3 leads to a collapse of the TM3 helix from the ER membrane, which triggers the misfolding and degradation of the whole α1(A322D) subunit. SAHA treatment increased the population of properly folded α1(A322D) subunits, which will assemble efficiently with β2 and γ2 subunits. Fully assembled GABAa receptors will be able to traffic to the plasma membrane for their function. The A322D mutation in the α1 subunit has a dominant negative effect, leading to reduced cell surface expression of WT α1 subunit as well as WT α3 subunits by associating with those WT subunits in the ER and trapping them in the ER for degradation (Ding, et al., 2010). Therefore, simple introduction of WT α1 subunit into patients with the A322D mutation might not prevent the seizure symptoms. It might be necessary to rescue the misfolding behavior of the α1(A322D) subunit in the ER per se to restore its function.
Although SAHA was reported to enhance proteostasis of mutant CFTR (Hutt, et al., 2010), lysosomal glucocerebrosidase (Lu, et al., 2011), and α1-antitrypsin (Bouchecareilh, et al., 2012), the proposed mechanism was very different. In restoration of the function of ΔF508 CFTR, SAHA was shown to mainly use the HDAC7 pathway without activation of the UPR or heat shock response; SAHA did not increase the BiP protein level, and the interaction between CFTR and calnexin was not explored in CFBE40o- cells stably expressing ΔF508 CFTR (Hutt, et al., 2010). In restoration of the function of L444P glucocerebrosidase (GCase), SAHA was shown to decrease the interaction between L444P GCase and Hsp90, but not Hsp70 or TCP1; however, the role of SAHA’s effect on ER chaperones was not explored although the folding of L444P GCase occurs in the ER (Lu, et al., 2011). In restoration of the function of Z-variant of α1-antitrypsin (Z-α1AT), SAHA was shown to act by reducing the interaction between Z-α1AT and calnexin, and calnexin silencing restored Z-α1AT maturation; SAHA did not increase the BiP protein level in HCT116 cells stably expressing Z-α1AT (Bouchecareilh, et al., 2012). Here, we showed that SAHA enhanced GABAa receptor folding and trafficking by increasing the BiP protein level and enhancing the interaction between the α1(A322D) subunit and calnexin. An increased expression of BiP by SAHA was not reported in cells expressing other misfolded proteins; SAHA increased BiP level by inhibiting HDAC7 (Figure 6C). A decreased interaction between calnexin and the α1(A322D) subunit was shown to reduce the maturation of the α1(A322D) subunit (Figure 5I), and calnexin overexpression promoted the α1(A322D) subunit maturation (Figure 5D). This is in contrast to the Z-α1AT case, indicating that the role of calnexin in assisting protein folding might depend on its specific client protein. It was reported that calnexin has a potential acetylation site at Lys137 (Choudhary, et al., 2009); it will be intriguing to explore how SAHA increased the interaction between α1(A322D) subunit and calnexin, which might be by inhibiting HDACs in the ER. The critical involvement of ER chaperones to assist the folding of the α1 subunit might partially be due to the topology of the α1 subunit: the α1 subunit has a large 224-residue ER lumen N-terminus. This is in contrast to the topology of CFTR, which has relatively short ER lumen components.
In a biological system, the protein quality control machinery is finely tuned to meet the demand of protein synthesis, folding, trafficking and degradation (Balch, et al., 2008). Our results showed that an increased protein load did not lead to an increased protein trafficking efficiency for misfolding-prone α1(A322D) subunits (Figure 1I). Similar case was reported: increasing protein synthesis of R752W hERG channels using the 4-PBA small molecule did not restore their maturation (Ficker, et al., 2000). Simply overloading newly synthesized misfolding-prone proteins in the ER itself is not sufficient to enhance their trafficking efficiency or maturation. Therefore, to rescue a misfolding-prone α1(A322D) subunit, it is important to rescue the misfolding behavior of the α1(A322D) subunit in the ER per se.
SAHA increased the transcriptional level and thus newly synthesized protein load of the α1(A322D) subunit (Figure S1E). Furthermore, SAHA enhanced the folding and trafficking of the α1(A322D) subunit post-translationally by promoting BiP and calnexin-assisted folding in the ER (Figures 1, 4 and 5). Therefore, SAHA increased both the transcription and the trafficking efficiency of the α1(A322D) subunit, leading to greater rescue of functional α1(A322D)β2γ2 GABAa receptors on the plasma membrane (Figure 2) than either effect alone.
SIGNIFICANCE
We reported that SAHA, a FDA-approved drug that crosses the blood-brain barrier, enhances the function of epilepsy-associated α1(A322D)β2γ2 GABAa receptors. The A322D mutation in the α1 subunit resulted in extensive ERAD of the α1(A322D) subunit and loss of function of GABAa receptors. SAHA enhanced the folding and trafficking of the α1(A322D) subunit post-translationally by promoting BiP and calnexin-assisted folding in the ER in addition to its role in increasing the transcription of the α1(A322D) subunit. This significantly restored partial function of α1(A322D)β2γ2 GABAa receptors on the plasma membrane, which suggests SAHA’s promise to ameliorate ADJME resulting from GABAa receptor misfolding. We also reported that both BiP and calnexin, two abundant ER chaperones, are sufficient to promote the α1(A322D) subunit folding in the ER. Therefore, we might be able to target BiP or calnexin to enhance GABAa receptor proteostasis, which might be extended to treat a number of protein misfolding diseases.
EXPERIMENTAL PROCEDURES
Cycloheximide-chase assay
HEK293 cells stably expressing α1(A322D)β2γ2 GABAa receptors were seeded at 2.5 × 105 cells per well in 6-well plates and incubated at 37°C overnight. Cells were then treated with SAHA for 24 h prior to cycloheximide-chase. To stop protein translation, cells were treated with 100 μg/mL cycloheximide (Ameresco). Cells were then chased for the indicated time, harvested, and lysed.
Confocal Immunofluorescence
The labeling of cell surface and total α1 subunits and confocal immunofluorescence microscopy analysis were performed according to published procedure (Eshaq, et al., 2010). See Extended Experimental Procedures for detail.
Biotinylation of Cell Surface Proteins
Cells were plated in 10-cm dishes for surface biotinylation experiments according to published procedure (Lachance-Touchette, et al., 2011). See Extended Experimental Procedures for detail.
Whole-Cell Patch Clamp Electrophysiology Recording
Whole-cell currents were recorded 48 h post transfection using HEK293 cells using published procedure (Eshaq, et al., 2010). See Extended Experimental Procedures for detail.
Quantitative RT-PCR
The relative expression levels of target genes were analyzed using quantitative RT-PCR according to published procedure (Mu, et al., 2008). See Extended Experimental Procedures for detail.
RT-PCR Analysis of Xbp-1 Splicing
After reverse transcription, PCR reactions were performed using cDNA, Taq DNA polymerase, and primers against XBP1 and GAPDH (listed in Table S1). PCR products were separated on a 2.5% agarose gel. Unspliced Xbp-1 yielded a 289 bp amplicon, and spliced Xbp-1 yielded a 263 bp amplicon.
Western blot band intensity quantification
We used NIH image J software to quantify the Western blot band intensity, following the step-by- step instructions from http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j. This quantification method has been widely used (Lafon, et al., 2012).
Statistical analysis
All data are presented as mean ± SEM, and any statistical significance was calculated using two-tailed Student’s t-Test.
Supplementary Material
HIGHLIGHTS.
SAHA enhances α1(A322D)β2γ2 GABAa receptor folding, trafficking and function
SAHA increases the trafficking efficiency of α1(A322D) subunit post-translationally
BiP promotes GABAa receptor folding in the ER
• Calnexin promotes GABAa receptor folding in the ER in a glycan-dependent way
ACKNOLEDGEMENTS
We thank the following for their generosity in providing us with plasmids: Professor Tohru Mizushima (Kumamoto University) for pcDNA3.1-BiP and pCR(calreticulin)-HA and Professor Michael Brenner (Harvard Medical School) for Apr-M8-CNX. This work was supported by the Research Start-up Fund from Case Western Reserve University School of Medicine, Epilepsy Foundation of America (225243), and the Clinical Translational Science Collaborative of Cleveland CTSA (UL1RR024989) from the National Center for Research Resources and the National Center for Advancing Translational Sciences of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supporting Information includes Extended Experimental Procedures, four figures, and one Table.
REFERENCES
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 Is a Transcription Factor Specializing in the Regulation of Quality Control Proteins in the Endoplasmic Reticulum. Cell Struct. Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- Alder NN, Johnson AE. Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J. Biol. Chem. 2004;279:22787–22790. doi: 10.1074/jbc.R400002200. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Bouchecareilh M, Hutt DM, Szajner P, Flotte TR, Balch WE. Histone Deacetylase Inhibitor (HDACi) Suberoylanilide Hydroxamic Acid (SAHA)-mediated Correction of alpha 1-Antitrypsin Deficiency. J. Biol. Chem. 2012;287:38265–38278. doi: 10.1074/jbc.M112.404707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ. Protein Folding and Modification in the Mammalian Endoplasmic Reticulum. Annu. Rev. Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Bradley CA, Taghibiglou C, Collingridge GL, Wang YT. Mechanisms involved in the reduction of GABA(A) receptor alpha 1-subunit expression caused by the epilepsy mutation A322D in the trafficking-competent receptor. J. Biol. Chem. 2008;283:22043–22050. doi: 10.1074/jbc.M801708200. [DOI] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J. Biol. Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- Cossette P, Liu LD, Brisebois K, Dong HH, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Bukau B. Chaperone-assisted folding of newly synthesized proteins in the cytosol. Crit. Rev. Biochem. Molec. Biol. 2004;39:261–277. doi: 10.1080/10409230490892496. [DOI] [PubMed] [Google Scholar]
- Ding L, Feng H-J, Macdonald RL, Botzolakis EJ, Hu N, Gallagher MJ. GABA(A) Receptor alpha 1 Subunit Mutation A322D Associated with Autosomal Dominant Juvenile Myoclonic Epilepsy Reduces the Expression and Alters the Composition of Wild Type GABA(A) Receptors. J. Biol. Chem. 2010;285:26390–26405. doi: 10.1074/jbc.M110.142299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokmanovic M, Perez G, Xu W, Ngo L, Clarke C, Parmigiani RB, Marks PA. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol. Cancer Ther. 2007;6:2525–2534. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell. 2008;19:2620–2630. doi: 10.1091/mbc.E07-07-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty DA. Cys-loop neuroreceptors: Structure to the rescue? Chem. Rev. 2008;108:1642–1653. doi: 10.1021/cr078207z. [DOI] [PubMed] [Google Scholar]
- Dudek J, Benedix J, Cappel S, Greiner M, Jalal C, Mueller L, Zimmermann R. Functions and pathologies of BiP and its interaction partners. Cell. Mol. Life Sci. 2009;66:1556–1569. doi: 10.1007/s00018-009-8745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaq RS, Stahl LD, Stone R, II, Smith SS, Robinson LC, Leidenheimer NJ. GABA acts as a ligand chaperone in the early secretory pathway to promote cell surface expression of GABA(A) receptors. Brain Res. 2010;1346:1–13. doi: 10.1016/j.brainres.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Thomas D, Viswanathan PC, Dennis AT, Priori SG, Napolitano C, Memmi M, Wible BA, Kaufman ES, Iyengar S, et al. Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am. J. Physiol.-Heart C. 2000;279:H1748–H1756. doi: 10.1152/ajpheart.2000.279.4.H1748. [DOI] [PubMed] [Google Scholar]
- Flynn GC, Pohl J, Flocco MT, Rothman JE. Peptide-binding specificity of the molecular chaperone BiP. Nature. 1991;353:726–730. doi: 10.1038/353726a0. [DOI] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: The role of molecular chaperones. Annu. Rev. Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Ding L, Maheshwari A, Macdonald RL. The GABA(A) receptor alpha 1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc. Natl. Acad. Sci. USA. 2007;104:12999–13004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T, Prahlad V, Morimoto RI. The Stress of Protein Misfolding: From Single Cells to Multicellular Organisms. Cold Spring Harb. Perspect. Biol. 2011;3:a009704. doi: 10.1101/cshperspect.a009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003;2:151–163. [PubMed] [Google Scholar]
- Green WN, Millar NS. Ion-Channel Assembly. Trends Neurosci. 1995;18:280–287. [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Protein folding - Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Bernasconi R, Molinari M. ERAD substrates: Which way out? Semin. Cell Dev. Biol. 2010;21:526–532. doi: 10.1016/j.semcdb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci. 2012;37:404–410. doi: 10.1016/j.tibs.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Simons JF, Peterson JR, Helenius A. Calnexin, calreticulin, and Bip/Kar2p in protein folding. Cold Spring Harb. Symp. Quant. Biol. 1995;60:405–415. doi: 10.1101/sqb.1995.060.01.045. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Helenius A, Trombetta ES, Hebert DN, Simons JF. Calnexin, calreticulin and the folding of glycoproteins. Trends Cell Biol. 1997;7:193–200. doi: 10.1016/S0962-8924(97)01032-5. [DOI] [PubMed] [Google Scholar]
- Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, Jentsch TJ, Gal A, Hubner CA. Mice with a targeted disruption of the Cl- /HCO3- exchanger AE3 display a reduced seizure threshold. Mol. Cell. Biol. 2006;26:182–191. doi: 10.1128/MCB.26.1.182-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou XB, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PAS, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt DM, Herman D, Rodrigues APC, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WK, O'Connor O, Richon VM, Curley T, Richardson S, Thapi D, Chiao JH, Rifkind RA, Marks PA, Scher HI. Phase I clinical trial of an oral histone deacetylase inhibitor: Suberoylanilide hydroxamic acid (SARA) Clin. Cancer Res. 2003;9:6245S–6246S. [PubMed] [Google Scholar]
- Krampfl K, Maljevic S, Cossette P, Ziegler E, Rouleau GA, Lerche H, Bufler J. Molecular analysis of the A322D mutation in the GABA(A) receptor alpha(1)-subunit causing juvenile myoclonic epilepsy. Eur. J. Neurosci. 2005;22:10–20. doi: 10.1111/j.1460-9568.2005.04168.x. [DOI] [PubMed] [Google Scholar]
- Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, Lortie A, Carmant L, Bedford F, Bowie D, et al. Novel alpha 1 and gamma 2 GABA(A) receptor subunit mutations in families with idiopathic generalized epilepsy. Eur. J. Neurosci. 2011;34:237–249. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- Lafon A, Petty E, Pillus L. Functional antagonism between Sas3 and Gcn5 acetyltransferases and ISWI chromatin remodelers. PLoS Genet. 2012;8:e1002994. doi: 10.1371/journal.pgen.1002994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Cys-loop receptors: new twists and turns. Trends Neurosci. 2004;27:329–336. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu. Rev. Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Lo W.-y., Lagrange AH, Hernandez CC, Harrison R, Dell A, Haslam SM, Sheehan JH, Macdonald RL. Glycosylation of beta 2 Subunits Regulates GABA(A) Receptor Biogenesis and Channel Gating. J. Biol. Chem. 2010;285:31348–31361. doi: 10.1074/jbc.M110.151449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Yang C, Chen M, Ye DY, Lonser RR, Brady RO, Zhuang Z. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc. Natl. Acad. Sci. USA. 2011;108:21200–21205. doi: 10.1073/pnas.1119181109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABA(A) receptor channels. Annu. Rev. Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- Mu TW, Ong DST, Wang YJ, Balch WE, Yates JR, Segatori L, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002;366:585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudiger S, Germeroth L, SchneiderMergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkevich LA, Williams DB. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr. Opin. Cell Biol. 2011;23:157–166. doi: 10.1016/j.ceb.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Simons JF, Ferronovick S, Rose MD, Helenius A. Bip/Kar2p serves as a molecular chaperone during carboxypeptidase-Y folding in yeast. J. Cell Biol. 1995;130:41–49. doi: 10.1083/jcb.130.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skach WR. Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 2009;16:606–612. doi: 10.1038/nsmb.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.