

Abstract
The mechanisms by which IL-6 contributes to the pathogenesis of chronic inflammatory diseases and cancer are not fully understood. We previously reported that cyclooxygenase-2 (Cox-2)-dependent PGE2 synthesis regulates macrophage matrix metalloproteinase (MMP)-9 expression, an endopeptidase that participates in diverse pathologic processes. In these studies, we determined whether IL-6 regulates the Cox-2→PGE2→MMP-9 pathway in murine macrophages. IL-6 co-induced Cox-2 and microsomal prostaglandin E synthase-1 (mPGES-1), and inhibited the expression of 15-hydroxyprostaglandin dehydrogenase (15-PGDH), leading to increased levels of PGE2. In addition, IL-6 induced MMP-9 expression, suggesting that the observed proteinase expression was regulated by the synthesis of PGE2. However, inhibition of PGE2 synthesis partially suppressed IL-6–mediated induction of MMP-9. In the canonical model of IL-6-induced signaling, JAK activation triggers STAT and MAPKerk1/2-signaling pathways. Therefore, the ability of structural diverse JAK inhibitors to block IL-6-induced MMP-9 expression was examined. Inhibition of JAK blocked IL-6 induced phosphorylation of STAT3, but failed to block the phosphorylation of MAPKerk1/2, and unexpectedly enhanced MMP-9 expression. In contrast, MEK-1 inhibition blocked IL-6 induced phosphorylation of MAPKerk1/2 and MMP-9 expression without affecting the phosphorylation of STAT3. Thus, IL-6-induced MMP-9 expression is dependent on the activation of MAPKerk1/2 and restrained by a JAK-dependent gene product. Utilizing pharmacologic and genetic approaches, JAK-dependent induction of IL-10 was identified as a potent feedback mechanism controlling IL-6 induced MMP-9 expression. Together, these data reveal that IL-6 induces MMP-9 expression in macrophages via Cox-2-dependent and -independent mechanisms, and identifies a potential mechanism linking IL-6 to the pathogenesis of chronic inflammatory diseases and cancer.
Introduction
IL-6, a pleiotropic cytokine expressed by a variety of immune and non-immune cells, plays an important role in the recruitment and survival of neutrophils and macrophages, regulation of CD4 T cell effector functions, angiogenesis, bone and cartilage metabolism, lipid metabolism, and the expression of acute phase proteins (1,2). Circulating levels of IL-6 are elevated in patients with cancer (3) and several chronic inflammatory diseases including rheumatoid arthritis (4) and cardiovascular disease (5). In addition, adipose tissue is a major source of circulating IL-6, and levels are elevated in obese patients (6). Owing to its multiple roles in the pathogenesis of inflammatory diseases and cancer (1,2), IL-6 has emerged as a major target for therapeutic intervention (7,8).
IL-6-induced biological responses are mediated by the membrane bound IL-6 receptor (IL-6Rα; CD126) (7,9,10). The IL-6/IL-6Rα complex engages transmembrane gp130 (IL-6Rβ; CD130), and the ternary complex dimerizes triggering the binding and phosphorylation of JAK, which then phosphorylates gp130 leading to the activation of STAT and MAPK signaling pathways. Despite the broad biologic activities of IL-6, surprisingly few cell types (e.g., monocyte/macrophages and hepatocytes) express membrane bound IL-6Rα. In contrast, virtually all cells types express gp130, which can bind soluble IL-6/IL-6Rα complexes (i.e. trans-signaling), thereby triggering STAT and MAPK signaling pathways.
Notwithstanding the significant progress in unraveling the many effects of IL-6 on immune and non-immune cells, the mechanisms by which IL-6 contributes to the pathogenesis of chronic inflammatory diseases and cancer is not fully understood. In this regard, evidence derived from mouse models indicates that MMP-9 (type IV collagenase; gelatinase B), a family member of Zn+2-dependent neutral endopeptidases, participates in the pathogenesis of arthritis (11), airway disease (12), cancer (13,14) and cardiovascular diseases (15-17). MMP-9 expression is low or absent in most normal tissues, and markedly elevated during inflammation, wound healing, and neoplasia (18-20). We and others have reported that macrophage MMP-9 expression is stimulated by PGE2 (21-29). Increased Cox-2-dependent synthesis of PGH2 and subsequent isomerization to PGE2 by mPGES-1 (29-34), in combination with reduced catabolism by NAD+-dependent 15-PGDH (35,36), are largely responsible for elevated levels of PGE2 associated with inflammation. Consequently, we determined whether IL-6 regulates the Cox-2→mPGES-1→PGE2→MMP-9 pathway in macrophages.
Results demonstrate that IL-6-induced MMP-9 expression in macrophages via Cox-2-dependent and independent pathways. Because IL-6 can activate both JAK/STAT and MAPK pathways, we explored their roles in regulating MMP-9 expression. Inhibition of MAPKerk1/2 blocked IL-6-mediated induction of MMP-9. In contrast, inhibition of JAK led to a paradoxical increase in MMP-9 expression, which proved to be a consequence of diminished IL-10 levels. To the best of our knowledge, this is the first demonstration that IL-6 induces macrophage expression of MMP-9, which has been directly associated with the pathogenesis of chronic inflammatory diseases and cancer (18-20). In addition, these data suggest that JAK inhibitors have the potential to increase MMP-9 expression in macrophages via the inhibition of IL-10 expression.
Materials and Methods
Macrophages
Thioglycollate-elicited peritoneal macrophages were obtained from Swiss Webster mice by the method of Edelson and Cohn (37) as described previously (38). Mice were injected IP (3 ml/mouse) with 3% Brewer Thioglycollate Medium (DIFCO). Four days later, cells were harvested by lavage with cold Dulbecco's PBS. Peritoneal cells were recovered by centrifugation and resuspended in DMEM supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml) and 4 mM glutamine, and plated into 6-well (2 × 106 cells/well) or 12-well plates (6 × 105 cells/well). Cells were allowed to adhere for 4 h and then washed free of nonadherent cells. The murine macrophage cell line RAW264.7 (39) was obtained from American Type Culture Collection, and maintained as adherent cultures in DMEM-10% FBS. RAW264.7 cells were mechanically harvested and plated into 12-well plates (6 × 105 cells/well). For serum free conditions, macrophages were grown in DMEM supplemented with antibiotics, glutamine and 0.1% BSA (Sigma Aldrich) containing low levels of endotoxin (< 0.1 ng/mg; LE-BSA). Cellgro® DMEM was obtained from Corning and heat inactivated FBS were obtained from Atlanta Biologicals. Antibiotics and glutamine were obtained from Gibco/Life Technologies. Macrophages were incubated with recombinant human IL-6 (Santa Cruz Biotechnology), recombinant mouse IL-10 (BioLegend), rat monoclonal anti-mouse IL-10 or non-immune rat IgG (R&D Systems) celecoxib (LC Laboratories), JAK Inhibitor 1 (Calbiochem/EMD Millipore), Tofacitinib (Selleckchem), or U0126 (Cayman Chemical). All animal studies described in this report have been reviewed and approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee.
Preparation of cell lysates
In experiments designed to monitor levels of phosphorylated and total STAT3 and MAPKerk1/2, cells were harvested in RIPA buffer (Sigma Aldrich) containing 1X protease inhibitor cocktail (Sigma Aldrich) and 1X Halt™ phosphatase inhibitor cocktail (Pierce), briefly sonicated and centrifuged (4,225 × g) for 10 min at 4°C. The supernatants were recovered, normalized for protein and mixed with SDS sample buffer with DTT and boiled for 5 min. Equal amounts of cell lysates were applied to gels based on protein content.
Western blots
Cell lysates were electrophoresed in 8-16% polyacrylamide gels and proteins were transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked in 5% BSA in TBST, washed in TBST and incubated 18 h in blocking buffer containing rabbit anti-mouse phospho-STAT3 (Tyr705) (1:2000; Cell Signaling Technology) or rabbit anti-human phospho-erk1/2 IgG (1:3000; Cell Signaling Technology). Membranes were washed 2X in TBST and incubated 1 h in blocking buffer containing affinity purified goat anti-rabbit IgG conjugated to HRP (1:3000; BioRad Laboratories). The membranes were washed 3X in TBST and bound HRP was visualized utilizing chemiluminescence (Pierce/Thermo Scientific). Membranes were then stripped in 0.1% SDS, 1.0% Tween 20 and 200 mM glycine, pH 2.2 10 min at RT, washed, blocked for 1 h in blocking buffer, and incubated with rabbit anti-mouse STAT3 (1:2000; Cell Signaling Technology) or mouse anti-erk1/2 (KLH-synthetic peptide) (1:3000; Cell Signaling Technology).
Macrophage conditioned media were electrophoresed in gradient gels and proteins were transferred to a PVDF membrane. The membrane was placed in 5% dry defatted milk in TBST for 1 h, washed in TBST and incubated 18 h in 3% blocking buffer containing rabbit anti-mouse MMP-9 IgG (1:2500; Abcam). Membranes were washed 2X in TBST and incubated 1 h in 3% blocking buffer containing goat anti-rabbit IgG conjugated to HRP (1:3000; BioRad Laboratories).
IL-10 knockdowns
RAW264.7 macrophages were transfected with control siRNA (i.e. nonspecific, NS) or IL-10 siRNA (Santa Cruz Biotechnology) utilizing HiPerFect transfection reagent according to the manufacturer's protocol (Qiagen). Cells were aliquoted into a 12-well (3 × 105 cells/well) plate containing siRNA transfection complexes and incubated 30 h. PCR and immunoassay was utilized to determine the extent of IL-10 knockdown.
Real-Time PCR analysis
RNA was prepared using Trizol reagent kits (Invitrogen). RNA (2 μg) was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Invitrogen) and random hexamers. The resulting cDNA diluted 10-fold was used for amplification. For real-time PCR analysis, the reaction volume was 5 μl and contained 1-2 μl of the diluted cDNA, 1X Gene Expression Assay (Life Technologies), 1X PerfeCTa FastMix II, Rox (Quanta) using the StepOne instrument (Applied Biosystems). The following Applied Biosystems Gene Expression Assays were utilized: actin (mm00607939_s1), Cox-2 (mm00478374_m1), MMP-9 (mm00442991_m1), mPGES-1 (mm00452105_m1), IL-6 (mm00446190_m1), 15-PGDH (mm00515121_m1) and IL-10 (mm00439614_m1). The comparative CT (ΔΔCT) method was used to determine the relative target quantity in samples. With the comparative CT method, the StepOne software measures amplification of the target and endogenous control in the samples and in a reference sample. Measurements are normalized using the endogenous control and expressed as relative quantity (RQ).
Determination of PGE2 and IL-10 levels
The concentrations of PGE2 and IL-10 in macrophage conditioned media were determined utilizing the PGE2 EIA kit (monoclonal) (Cayman Chemical) and IL-10 Quantikine Immunoassay (R&D).
Statistics
Relative quantities (RQ) of target gene mRNA and PGE2 or IL-10 levels are expressed as the mean ± SD of 3 independent samples from a single representative experiment. Each experiment was repeated 2-3 times. Where indicated, mean levels of mRNA, PGE2 or IL-10 were compared utilizing t-test (e.g., comparing means of two groups) or single factor analysis of variance (e.g., comparing means of greater than 2 groups). Following the determination of significant differences between the means of a given experiment, subsequent pair-wise comparisons were performed utilizing Newman-Keuls multiple range testing (40).
Results
IL-6 stimulates Cox-2 and mPGES-1 expression, and inhibits 15-PGDH expression in macrophages
We determined whether IL-6 co-induces Cox-2 and mPGES-1 expression in macrophages, leading to increased levels of PGE2. Incubation with LPS, a potent inducer of Cox-2 and mPGES-1 in macrophages, served as a positive control (30,31,41). Levels of Cox-2 and mPGES-1 mRNA were determined utilizing quantitative PCR. Incubation of RAW264.7 macrophages with IL-6 induced Cox-2 and mPGES-1 expression in a dose- and time-dependent manner (Figures 1A and 1B). Since reduced catabolism of prostaglandins contributes to increased levels of PGE2 in inflammatory foci, we also examined the effect of IL-6 on the expression of 15-PGDH, a key enzyme responsible for the catabolism of prostaglandins (35,36). In contrast to Cox-2 and mPGES-1 expression, IL-6 rapidly and potently inhibited 15-PGDH expression in macrophages (Figures 1A and 1B). Consistent with the IL-6-induced reciprocal changes in expression of genes directing the synthesis and degradation of PGE2, levels of PGE2 in media recovered from RAW264.7 cells increased in a dose- and time-dependent manner (Figures 1A and 1B).
Figure 1.

IL-6 induces Cox-2 and mPGES-1 expression, and inhibits 15-PGDH expression in RAW264.7 macrophages. [A] Macrophages were incubated in DMEM-0.1% LE-BSA containing 0 - 100 ng/ml IL-6 or 10 ng/ml LPS. Following 18 h incubation, total RNA and conditioned media were recovered. [B] Macrophages were incubated 0 – 18h in DMEM-0.1% LE-BSA containing 100 ng/ml IL-6 or 10 ng/ml LPS for 18 h. The relative quantity (RQ) of Cox-2, mPGES-1 and 15-PGDH mRNAs were determined by quantitative RT-PCR as described in Materials and Methods. Levels of PGE2 in conditioned media were determined utilizing ELISA. Target gene mRNA and PGE2 levels are expressed as the mean ± SD of 3 independent samples from a representative experiment.
To corroborate and extend these findings to primary macrophages, we examined the effect of IL-6 on the expression of Cox-2, mPGES-1 and 15-PGDH by macrophages recovered from thioglycollate-induced peritonitis (Figure 2). Following 18 h incubation with IL-6, levels of Cox-2 and mPGES-1 mRNA in elicited macrophages were markedly elevated, and levels of 15-PGDH mRNA were reduced. Likewise, levels of PGE2 in media recovered from elicited macrophages treated with IL-6 were increased dramatically.
Figure 2.
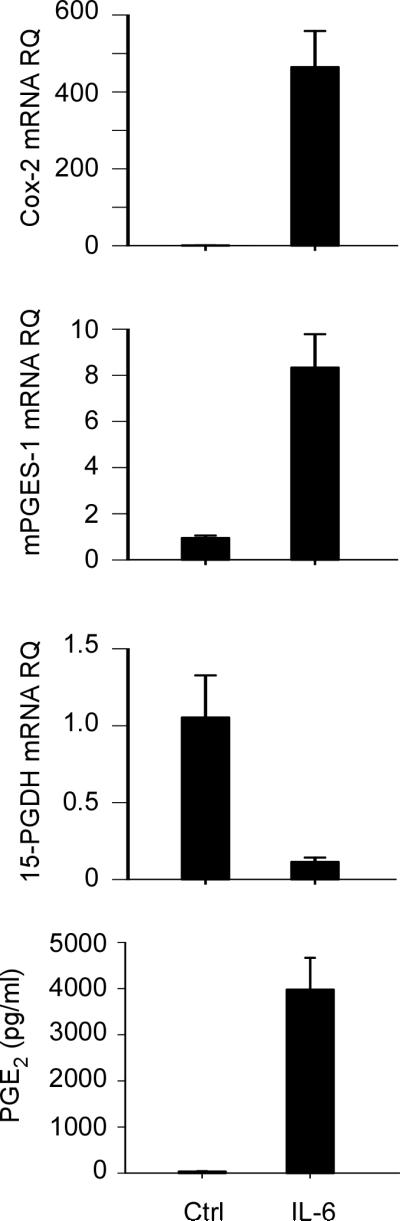
IL-6 induces Cox-2 and mPGES-1, and inhibits 15-PGDH expression in thioglycollate-elicited peritoneal macrophages. Elicited macrophages were incubated in DMEM-0.1% LE-BSA containing 100 ng/ml IL-6 for 18 h and total RNA were recovered. Levels of Cox-2, mPGES-1 and 15-PGDH mRNAs were determined by quantitative RT-PCR, and levels of PGE2 in conditioned media were determined utilizing ELISA. Target gene mRNA and PGE2 levels are expressed as the mean ± SD of 3 independent samples from a representative experiment.
In these studies, murine macrophages were incubated with recombinant human IL-6, which binds and activates the murine IL-6 receptor (42). To confirm that the observed alterations in Cox-2, mPGES-1 and 15-PGDH were due to IL-6 and not a potential contaminant, we pre-incubated human IL-6 with non-immune IgG or neutralizing anti-human IL-6 IgG prior to incubation with macrophages. Anti-IL-6 IgG blocked IL-6-induced alterations in gene expression in murine macrophages (data not shown). Taken together, these data demonstrate that IL-6 stimulates macrophage expression of Cox-2 and mPGES-1, and inhibits the expression of 15-PGDH, leading to increased levels of PGE2.
Cox-2-dependent PGE2 synthesis plays a role in IL-6-mediated induction of MMP-9
Macrophage MMP-9 expression is regulated by Cox-2-dependent synthesis of PGE2 (26-29). Thus we examined whether the observed changes in expression of genes directing the synthesis and degradation of PGE2 in response to IL-6 (Figures 1 and 2), were associated with changes in MMP-9 expression. Treatment of RAW264.7 macrophages with IL-6 resulted in a dose- and time-dependent increase in the levels of MMP-9 mRNA (Figures 3A and 3B). The dose-dependent increases in MMP-9 mRNA were mirrored by an accumulation of MMP-9 in cellular conditioned media (Figure 3A). Treatment with IL-6 similarly stimulated MMP-9 expression in elicited peritoneal macrophages (Figure 3C; p<0.01). IL-6 induced macrophage MMP-9 expression identifies a potential mechanism linking elevated IL-6 levels to the pathogenesis of chronic inflammatory diseases and cancer (18-20).
Figure 3.

IL-6 induces MMP-9 expression in macrophages. [A] RAW264.7 macrophages were incubated 18 h in DMEM-0.1% LE-BSA containing 0 - 100 ng/ml IL-6 or 10 ng/ml LPS. [B] Macrophages were incubated 0 – 18h in DMEM-0.1% LE-BSA containing 100 ng/ml IL-6. [C] Thioglycollate-elicited macrophages were incubated 18 h in DMEM-0.1% LE-BSA containing 100 ng/ml IL-6. Following the indicated incubation, total RNA and conditioned media were recovered. Levels of MMP-9 mRNA and MMP-9 in conditioned media were determined utilizing quantitative RT-PCR and Western blot, respectively. MMP-9 mRNA levels are expressed as the mean ± SD of 3 independent samples from a representative experiment.
In contrast to earlier studies where MMP-1 and MMP-3 induced Cox-2 expression and increased PGE2 synthesis preceded elevated MMP-9 expression (28), IL-6 induced MMP-9 expression parallels the expression of both Cox-2 and mPGES-1 (Figures 1 and 3). These findings raise the possibility that the increased synthesis of PGE2 may not be solely responsible for the induction of MMP-9. However, since macrophage MMP-9 expression is stimulated by PGE2 (26-29), IL-6-induced reciprocal changes in the expression of genes directing the synthesis and degradation of PGE2 may contribute to MMP-9 expression. Therefore, we examined the effect of the selective Cox-2 inhibitor celecoxib on IL-6 induced PGE2 levels and MMP-9 expression in macrophages. The levels of PGE2 in conditioned media recovered from macrophages incubated with IL-6 (18 h) were increased >20-fold, as compared to control macrophages (Figure 4A). Pre-treatment with celecoxib completely inhibited IL-6-induced PGE2 synthesis, but only led to a modest reduction in IL-6-mediated induction of MMP-9 mRNA (Figure 4B; p<0.05) and protein (Figure 4C). These data indicate that IL-6 stimulates MMP-9 expression by both Cox-2-dependent and -independent mechanisms.
Figure 4.

Cox-2-dependent PGE2 synthesis contributes to IL-6-induced MMP-9 expression. [A] RAW264.7 macrophages were pre-incubated in DMEM-0.1% LE-BSA containing DMSO (vehicle control) or 5 μM celecoxib for 30 min, following which 100 ng/ml IL-6 was added. Following 18 h incubation, conditioned media were recovered for determination of [A] PGE2 and [C] MMP-9 levels utilizing ELISA and Western blot, respectively. [B] Total RNA were recovered and levels of MMP-9 mRNA were determined utilizing quantitative RT-PCR. Levels of PGE2 and MMP-9 mRNA are expressed as the mean ± SD of 3 independent samples from a representative experiment.
IL-6-induced MMP-9 expression is MAPKerk1/2-dependent.
In the canonical model of IL-6-induced signaling, binding of IL-6/IL-6Rα to gp130 triggers the activation of the JAK and subsequent phosphorylation of several tyrosines within the cytoplasmic tail of gp130 creating binding sites for STAT and SHP2. The latter is believed to be critical for the activation of MAPKerk1/2 (7,9,10). Thus, inhibition of JAK activity ought to block both STAT and MAPKerk1/2-signaling pathways. Therefore, we examined the effect of two selective inhibitors of JAK on IL-6-induced MMP-9 expression (Figure 5). Unexpectedly, pre-treatment of macrophages with JAK inhibitor 1 (1 – 100 nM) augmented IL-6-mediated induction of MMP-9 mRNA and MMP-9 protein in cellular conditioned media (Figure 5A). Likewise, pre-treatment with Tofacitinib (0.1 – 1 μM), a structurally distinct JAK inhibitor, augmented IL-6-mediated induction of MMP-9 (Figure 5B). In contrast, pre-treatment with the MEK1 inhibitor U0126 (1 - 10 μM) led to a dose-dependent inhibition of IL-6 induced MMP-9 expression (Figure 5C). The failure of JAK inhibitors to block IL-6-induced MMP-9 expression was unexpected. Thus, we monitored their ability to inhibit the phosphorylation of STAT3 and MAPKerk1/2. As anticipated, pre-treatment with JAK Inhibitor 1 (100 nM) or Tofacitinib (1 μM) completely blocked IL-6-induced phosphorylation of STAT3; whereas, IL-6-induced phosphorylation of MAPKerk1/2 was uneffected (Figure 6C). Pre-treatment with U0126 blocked IL-6-induced phosphorylation of MAPKerk1/2 (Figure 6B), but had no effect on IL-6-induced phosphorylation of STAT3 (Figure 6A). Although these data are unexpected based on the established model of IL-6-induced signaling, several investigators have reported that diverse JAK inhibitors block STAT activation without inhibiting MAPKerk1/2 activation (43-47); however, the mechanism(s) underlying JAK-independent activation of MAPKerk1/2 remains undefined at this time.
Figure 5.

Opposing effects of JAK and MEK-1 inhibitors on IL-6-dependent MMP-9 expression. RAW264.7 macrophages were pre-incubated 30 min in DMEM-0.1% LE-BSA containing [A] 0-100 nM JAK Inhibitor 1, [B] 0-1.0 μM Tofacitinib or [C] 0-10 μM U0126, followed by the addition of 100 ng/ml IL-6. Following 18 h incubation, total RNA and conditioned media were recovered. Levels of MMP-9 mRNA and MMP-9 in conditioned media were determined utilizing quantitative RT-PCR and Western blot, respectively. MMP-9 mRNA levels are expressed as the mean ± SD of 3 independent samples from a representative experiment.
Figure 6.

JAK inhibitors block IL-6-induced phosphorylation of STAT3 in macrophages without affecting phosphorylation of MAPKerk1/2. [A] RAW264.7 macrophages were pre-incubated 30 min in DMEM-0.1% LE-BSA containing 100 nM JAK Inhibitor 1, 1.0 μM Tofacitinib or 10 μM U0126, followed by the addition of 100 ng/ml IL-6. Following 90 min incubation, cell lysates were recovered, and levels of phosphorylated and total STAT3 were determined utilizing Western blot. Macrophages were pre-incubated 30 min in DMEM-0.1% LE-BSA containing [B] 0-10 μM U0126, [C] 100 nM JAK Inhibitor 1 or 1.0 μM Tofacitinib followed by the addition of 100 ng/ml IL-6. Following 90 min incubation, cell lysates were recovered, and levels of phosphorylated and total MAPKerk1/2 were determined utilizing Western blot. Data are representative of two separate experiments.
Although IL-6 is one of the most highly induced NF-κB-dependent cytokines (48), NF-κB is not generally reported to mediate IL-6-dependent signaling and gene expression (7,9,10). However, it has been reported that in addition to canonical JAK/STAT signaling, IL-6 induces the accumulation of unphosphorylated STAT3, which forms a complex with unphosphorylated NF-κB that can translocate into the nucleus (49). Therefore, we examined the effect of two cell permeable NF-κB activation inhibitors on IL-6-induced MMP-9 expression. Pre-incubation of RAW264.7 macrophages with several concentrations of NF-κB Activation Inhibitor I (CAS 545380-34-5) or II (CAS 749886-87-1) failed to block or attenuate IL-6-induced MMP-9 expression (data not shown). Taken together, our data demonstrate that IL-6-induced MMP-9 expression in macrophages is dependent on the activation of MAPKerk1/2 and paradoxically enhanced by inhibition of JAK.
Tofacitinib inhibition of IL-6-induced IL-10 enhances MMP-9 expression
The observation that IL-6-induced MMP-9 expression in macrophages was enhanced by JAK inhibition suggested that a JAK-dependent gene product serves to limit IL-6-induced MMP-9 in macrophages. In this regard, the pro-inflammatory properties of LPS are limited, in part, by the JAK-dependent induction of IL-10, a potent anti-inflammatory cytokine (46), which is also reported to inhibit MMP-9 expression (50-53). Therefore, we examined whether IL-6 stimulated IL-10 expression in macrophages. As seen in Figure 7A and B, levels of IL-10 mRNA in macrophages and IL-10 in conditioned media were increased, in a dose-dependent manner. Likewise, IL-6 induced IL-10 expression in a time-dependent manner (Figure 7C and D). When compared to control cells, levels of IL-10 mRNA are increased > 60-fold in as little as 2 h, and >1000 fold at 18 h. Levels of IL-10 in the conditioned media recovered from cells incubated with IL-6 were first elevated at 4 h (124 ± 90 pg/ml; mean ± SD, n=3) and increased to 2031 ± 65 at 18 h. Importantly, IL-6-induced IL-10 expression was inhibited, in a dose-dependent manner, by JAK Inhibitor 1 or Tofacitinib (Figure 7E and F).
Figure 7.

JAK inhibitors block IL-6 induced IL-10 expression in macrophages. [A and B] RAW264.7 macrophages were incubated in DMEM-0.1% LE-BSA containing 0-100 ng/ml IL-6 for 18 h. [C and D] Macrophages were incubated for 18 h in media alone (Ctrl) or 2 - 18 h in media containing 100 ng/ml IL-6. Following the recovery of total RNA and conditioned media, levels of IL-10 mRNA and IL-10 in conditioned media were determined utilizing quantitative RT-PCR and ELISA, respectively. Macrophages were pre-incubated 30 min in DMEM-0.1% LE-BSA containing [E] 0-100 nM JAK Inhibitor 1 or [F] 0-1.0 μM Tofacitinib, following which 100 ng/ml IL-6 was added and cells incubated for 18 h. Following the recovery of total RNA, levels of IL-10 mRNA were determined. IL-10 mRNA levels and IL-10 in conditioned media are expressed as the mean ± SD of 3 independent samples of a representative experiment.
Next, we determined the effect of exogenous IL-10 on IL-6-induced MMP-9 expression. When macrophages were pre-treated (1 h) with as little as 1 ng/ml IL-10, subsequent exposure to IL-6 failed to induced MMP-9 expression (Figure 8A). The IL-10-dependent inhibition of MMP-9 expression was blocked, when macrophages were pre-incubated with neutralizing anti-IL-10 receptor IgG (Figure 8B). These data clearly demonstrate that pre-exposure of macrophages to IL-10 potently inhibits the ability of IL-6 to induce MMP-9 expression. In addition, IL-10 effectively turned off previously up-regulated MMP-9 expression. As seen in Figure 8C, levels of MMP-9 mRNA in macrophages incubated with IL-6 for 18 hours was increased ~3-fold; whereas, levels of MMP-9 mRNA in cells incubated with IL-6 for six hours followed by the addition of IL-10 were similar to control macrophages at 18 hours. Likewise, levels of MMP-9 in conditioned media of cells incubated 6 hours with IL-6 prior to the addition of IL-10 were reduced >50%, as compared macrophages treated with IL-6 alone (Figure 8C). Together, these data demonstrate that exogenously added IL-10 is a potent inhibitor of IL-6 induced MMP-9 expression by macrophages.
Figure 8.

IL-10 potently suppresses IL-6-induced MMP-9 expression in macrophages. [A] RAW264.7 macrophages were pre-incubated 1 h in DMEM-0.1% LE-BSA containing 0-100 ng/ml recombinant mouse IL-10, following which 100 ng/ml IL-6 was added, and incubated 18 h. [B] Macrophages were pre-incubated 1 h in DMEM-0.1% LE-BSA containing non-immune (n) IgG or anti-IL-10 receptor (R), followed by 1h incubation with 10 ng/ml IL-10, and an additional 18 h incubation with 100 ng/ml IL-6. [C] Macrophages were incubated DMEM-0.1% LE-BSA containing 100 ng/ml IL-6 for 6 h, following which 10 ng/ml IL-10 was added and incubated for 18 h. Following the recovery of total RNA and conditioned media, levels of MMP-9 mRNA and MMP-9 in conditioned media were determined utilizing quantitative RT-PCR and Western blot, respectively. Levels of MMP-9 mRNA are expressed as the mean ± SD of 3 independent samples of a representative experiment.
To directly test whether endogenously synthesized IL-10 inhibits IL-6-induced MMP-9 expression, macrophages were treated with IL-6 in the presence of anti-mouse IL-10 IgG or normal IgG. Levels of MMP-9 mRNA in macrophages incubated with IL-6 were increased 5.8 fold over control cells (Figure 9A; P<0.05). In the presence of anti-IL-10, levels of MMP-9 mRNA increased to 14-fold (p<0.01). Likewise, IL-6-induced MMP-9 levels in macrophage conditioned media were increased in the presence of anti-IL-10 IgG (Figure 9A). To corroborate these findings, macrophages were transfected with either nonspecific (NS)-siRNA or IL-10 siRNA, and treated with IL-6 for 18 h. Levels of IL-10 in macrophage conditioned media were significantly (p<0.01) reduced in IL-6 treated cells transfected with IL-10 siRNA (Figure 9B). As predicted, levels of IL-6-induced MMP-9 mRNA were significantly (p<0.01) increased in cells transfected with IL-10 siRNA. Taken together, these data demonstrate that IL-6 induced IL-10 serves to limit MMP-9 expression, and administration of JAK inhibitors, which selectively block IL-10 expression, led to increased MMP-9 expression.
Figure 9.

Inhibition of IL-10 activity or expression enhances IL-6-induced MMP-9 expression in macrophages. [A] RAW264.7 macrophages were incubated in DMEM-0.1% LE-BSA alone (Ctrl) or media supplemented with 100 ng/ml IL-6, IL-6 and 2 ug/ml anti-murine IL-10 IgG, or IL-6 and normal IgG for 18 h. Following the recovery of total RNA, levels of MMP-9 mRNA were determined utilizing quantitative RT-PCR and levels of MMP-9 in conditioned media were determined by Western blot. [B] Macrophages transfected with nonspecific (NS) or IL-10 siRNA received DMEM-0.1% LE-BSA (Ctrl) or media containing 100 ng/ml IL-6, and were incubated 18 h. Total RNA was recovered and levels of MMP-9 mRNA were determined utilizing quantitative RT-PCR, and levels of IL-10 in conditioned media were determined utilizing ELISA. Data are the mean ± SD of 3 independent samples of a representative experiment.
Discussion
In the present study, we have examined the regulatory role of IL-6 on macrophage MMP-9 expression, which participates in diverse physiologic and pathologic processes. Similar to other MMP family members, the degradation of extracellular matrix components is generally thought to be the primary role of MMP-9. However, in addition, MMP-9 modifies the activities of cytokines and chemokines, growth factors, and proteinase inhibitors (18-20). In these studies, we demonstrate that IL-6 rapidly and robustly stimulated an increase in MMP-9 mRNA levels in RAW264.7 macrophages and elicited peritoneal macrophages, which led to an accumulation of MMP-9 in their conditioned media. These data reveal that IL-6, recognized as a critical mediator of inflammation, induces macrophage expression of a metalloproteinase that is directly associated with the pathogenesis of chronic inflammatory diseases and cancer (18-20).
We and others have reported that macrophage MMP-9 expression is stimulated by PGE2 (21-29). PGE2-dependent proteinase expression is dependent on the EP4 prostanoid receptor and subsequent MAPKerk1/2 activation (27,28). Since it is well established that elevated levels of PGE2 associated with inflammation depends on the coupling of Cox-2 and mPGES-1 expression (30-33,41,54-59), as well as reduced catabolism by 15-PGDH (35,36), we examined whether IL-6 regulates the expression of these genes in macrophages. Results demonstrate that IL-6 co-induced Cox-2 and mPGES-1 expression, and inhibited the expression of 15-PGDH. Consistent with these changes in gene expression, levels of PGE2 in macrophage conditioned media were increased. These data suggested a targetable pathway (Cox-2→mPGES-1→PGE2) by which IL-6 induced MMP-9 could be inhibited. However, pre-treatment of macrophages with celecoxib, which completely blocked IL-6-induced PGE2 synthesis, resulted in a partial reduction in MMP-9 mRNA and protein levels.
Insomuch that celecoxib was partially effective, we explored alternative therapeutic approaches to inhibit IL-6 induced MMP-9 expression. In this regard, the IL-6 binding to the IL-6 receptor complex (IL-6Rα/gp130) triggers phosphorylation of JAK, and subsequent activation of STAT and SHP2 (60,61). Phosphorylated STAT dimerizes and translocates to the nucleus to regulate STAT-dependent genes; whereas, the phosphorylation of SHP2 plays an important role in the activation of MAPKerk1/2 and MAPK-dependent gene expression (7,9). Thus, we examined the ability JAK inhibitors to block MMP-9 expression. The inhibition of JAK predictably blocked IL-6 induced activation of STAT3, but failed to block the activation of MAPKerk1/2, and unexpectedly enhanced MMP-9 expression. In contrast, inhibition of MEK-1 blocked IL-6 induced phosphorylation of MAPKerk1/2 and MMP-9 expression without affecting the phosphorylation of STAT3. Thus, IL-6-induced MMP-9 expression is dependent on the activation of MAPKerk1/2 and restrained by a JAK-dependent gene product. Although these data were unexpected based on the established model of IL-6-induced signaling, several investigators have reported that diverse JAK inhibitors, including the highly specific Tofacitinib and Ruxolitinib, effectively block STAT activation without inhibiting MAPKerk1/2 activation (43-47). Although the mechanism(s) underlying JAK-independent activation of MAPKerk1/2 remains undefined at this time, the binding of Src kinase family member HCK to gp130, distal to the SHP2 docking site, reportedly plays a role in IL-6-induced MAPKerk1/2 activation (62).
Utilizing pharmacologic and genetic approaches, we identified IL-10 as the IL-6 induced JAK-dependent suppressor of MMP-9 expression in macrophages. First, the robust induction of IL-10 expression by IL-6 was completely blocked by JAK Inhibitor 1 and Tofacitinib. Second, as little as 1 ng/ml exogenous IL-10 completely blocked IL-6 induced MMP-9 expression. Third, IL-6-induced MMP-9 expression in macrophages was significantly increased, when cells were incubated in the presence of neutralizing anti-IL-10 IgG. Fourth, IL-6-induced MMP-9 expression was significantly increased in macrophages in which IL-10 expression was knocked down via transfection with IL-10 siRNA. Thus, JAK-dependent induction of IL-10 is an important feedback mechanism to control IL-6 induced MMP-9 expression in macrophages. These data are consistent with the recognized role IL-10 plays in limiting the inflammatory response (63). For example, LPS-induced IL-10 inhibits expression of pro-inflammatory cytokines TNFα, IL-6 and IL-12 via activation of the JAK/STAT3 pathway (46). Treatment of cells with the selective JAK inhibitors resulted in enhanced production of LPS-induced inflammatory mediators, which was due to inhibition of IL-10 expression and signaling. Likewise, LPS-induced cytokine production is enhanced by macrophages recovered from IL-10 null mice (46). Taken together, JAK inhibitors have the potential to paradoxically increase expression of inflammatory mediators under defined circumstances by inhibiting IL-10 expression.
In the present studies, we report that IL-6 induced macrophage expression of MMP-9 via Cox-2-dependent and -independent pathways, and required the activation of MAPKerk1/2. Insomuch that MMP-9 plays a causal role in the pathogenesis of chronic inflammatory diseases and cancer, these data reveal a direct mechanism by which elevated IL-6 can contribute to these diseases. Current strategies to block IL-6 induced inflammation utilize anti-IL-6Rα (i.e. Tocilizumab), soluble gp130 and small molecule inhibitors of JAK (i.e. Ruxolitinib and Tofacitinib)(7). As reported here, the use of JAK inhibitors appears to have the unintended consequence of blocking IL-6-induced IL-10 expression resulting in increased MMP-9 expression in macrophages.
Figure 10.

IL-6-mediated induction of MMP-9 is modulated by JAK/STAT-dependent IL-10 expression in macrophages. IL-6, heretofore recognized as a marker of CVD, induces macrophage expression of MMP-9, which is directly associated with the pathogenesis of vascular diseases. IL-6 stimulation of MMP-9 expression was dependent on the activation of MAPKerk1/2, and partially dependent on the synthesis of PGE2, which we have previously shown to bind EP4 and activate MAPKerk1/2. IL-6 also induced IL-10 expression, which potently suppressed MMP-9 expression. Small molecule inhibitors of JAK blocked IL-6-induced IL-10 expression, and augmented MMP-9 expression. Likewise, inhibition of IL-10 expression utilizing siRNA or engagement of the IL-10R utilizing anti-IL-10 led to enhanced MMP-9 expression.
Abbreviations used in this paper
- ApoE
apolipoprotein E
- CVD
cardiovascular disease
- Cox-2
cyclooxygenase-2
- EIA
enzyme immunoassay
- IL-6Rα
IL-6 receptor alpha chain
- IP
intraperitoneal
- LE
low levels of endotoxin
- MMP
matrix metalloproteinase
- mPGES
microsomal prostaglandin synthase
- NS
nonspecific
- PGDH
hydroxyprostaglandin dehydrogenase
- PVDF
polyvinylidene fluoride
- RIPA
radioimmunoprecipitation assay
- RQ
relative quantity
- siRNA
small interfering RNAl
Footnotes
These studies were supported by research grant HL093331 (to DJF) from the National Institutes of Health, and a grant from the New York Crohn's Foundation (to AJD).
Reference List
- 1.Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. (Lond) 2012;122:143–159. doi: 10.1042/CS20110340. [DOI] [PubMed] [Google Scholar]
- 2.Rincon M. Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012;33:571–577. doi: 10.1016/j.it.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 2013;14:e218–e228. doi: 10.1016/S1470-2045(12)70582-X. [DOI] [PubMed] [Google Scholar]
- 4.Schoels MM, van der Heijde D, Breedveld FC, Burmester GR, Dougados M, Emery P, Ferraccioli G, Gabay C, Gibofsky A, Gomez-Reino JJ, Jones G, Kvien TK, Murakami M, Nishimoto N, Smolen JS. Blocking the effects of interleukin-6 in rheumatoid arthritis and other inflammatory rheumatic diseases: systematic literature review and meta-analysis informing a consensus statement. Ann. Rheum. Dis. 2013;72:583–589. doi: 10.1136/annrheumdis-2012-202470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danesh J, Kaptoge S, Mann AG, Sarwar N, Wood A, Angleman SB, Wensley F, Higgins JP, Lennon L, Eiriksdottir G, Rumley A, Whincup PH, Lowe GD, Gudnason V. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS. Med. 2008;5:e78. doi: 10.1371/journal.pmed.0050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eder K, Baffy N, Falus A, Fulop AK. The major inflammatory mediator interleukin-6 and obesity. Inflamm. Res. 2009;58:727–736. doi: 10.1007/s00011-009-0060-4. [DOI] [PubMed] [Google Scholar]
- 7.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waetzig GH, Rose-John S. Hitting a complex target: an update on interleukin-6 trans-signalling. Expert. Opin. Ther. Targets. 2012;16:225–236. doi: 10.1517/14728222.2012.660307. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eulenfeld R, Dittrich A, Khouri C, Muller PJ, Mutze B, Wolf A, Schaper F. Interleukin-6 signalling: more than Jaks and STATs. Eur. J. Cell Biol. 2012;91:486–495. doi: 10.1016/j.ejcb.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J. Immunol. 2002;169:2643–2647. doi: 10.4049/jimmunol.169.5.2643. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez FG, Campbell LG, Liu W, Shipley JM, Itohara S, Patterson GA, Senior RM, Mohanakumar T, Jaramillo A. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am. J. Transplant. 2005;5:671–683. doi: 10.1111/j.1600-6143.2005.00751.x. [DOI] [PubMed] [Google Scholar]
- 13.Chantrain CF, Shimada H, Jodele S, Groshen S, Ye W, Shalinsky DR, Werb Z, Coussens LM, DeClerck YA. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Research. 2004;64:1675–1686. doi: 10.1158/0008-5472.can-03-0160. [DOI] [PubMed] [Google Scholar]
- 14.Masson V, de la Ballina LR, Munaut C, Wielockx B, Jost M, Maillard C, Blacher S, Bajou K, Itoh T, Itohara S, Werb Z, Libert C, Foidart JM, Noel A. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J. 2005;19:234–236. doi: 10.1096/fj.04-2140fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lessner SM, Martinson DE, Galis ZS. Compensatory vascular remodeling during atherosclerotic lesion growth depends on matrix metalloproteinase-9 activity. Arteriosclerosis Thrombosis and Vascular Biology. 2004;24:2123–2129. doi: 10.1161/01.ATV.0000141840.27300.fd. [DOI] [PubMed] [Google Scholar]
- 16.Choi ET, Collins ET, Marine LA, Uberti MG, Uchida H, Leidenfrost JE, Khan MF, Boc KP, Abendschein DR, Parks WC. Matrix metalloproteinase-9 modulation by resident arterial cells is responsible for injury-induced accelerated atherosclerotic plaque development in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1020–1025. doi: 10.1161/01.ATV.0000161275.82687.f6. [DOI] [PubMed] [Google Scholar]
- 17.Ikonomidis JS, Barbour JR, Amani Z, Stroud RE, Herron AR, McClister DM, Jr., Camens SE, Lindsey ML, Mukherjee R, Spinale FG. Effects of deletion of the matrix metalloproteinase 9 gene on development of murine thoracic aortic aneurysms. Circulation. 2005;112:I242–I248. doi: 10.1161/CIRCULATIONAHA.104.526152. [DOI] [PubMed] [Google Scholar]
- 18.Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends in Immunology. 2001;22:571–579. doi: 10.1016/s1471-4906(01)02023-3. [DOI] [PubMed] [Google Scholar]
- 19.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature Reviews Immunology. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 20.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wahl LM, Olsen CE, Sandberg AL, Mergenhagen SE. Prostaglandin regulation of macrophage collagenase production. Proc. Natl. Acad. Sci. U. S. A. 1977;74:4955–4958. doi: 10.1073/pnas.74.11.4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapiro SD, Kobayashi DK, Pentland AP, Welgus HG. Induction of macrophage metalloproteinases by extracellular matrix. Evidence for enzyme- and substrate-specific responses involving prostaglandin-dependent mechanisms. J. Biol. Chem. 1993;268:8170–8175. [PubMed] [Google Scholar]
- 23.Corcoran ML, Kibbey MC, Kleinman HK, Wahl LM. Laminin SIKVAV peptide induction of monocyte/macrophage prostaglandin E2 and matrix metalloproteinases. J. Biol. Chem. 1995;270:10365–10368. doi: 10.1074/jbc.270.18.10365. [DOI] [PubMed] [Google Scholar]
- 24.Shankavaram UT, DeWitt DL, Funk SE, Sage EH, Wahl LM. Regulation of human monocyte matrix metalloproteinases by SPARC. Journal of Cellular Physiology. 1997;173:327–334. doi: 10.1002/(SICI)1097-4652(199712)173:3<327::AID-JCP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 25.Ottino P, Bazan HEP. Corneal stimulation of MMP-1, -9 and uPA by platelet-activating factor is mediated by cyclooxygenase-2 metabolites. Current Eye Research. 2001;23:77–85. doi: 10.1076/ceyr.23.2.77.5471. [DOI] [PubMed] [Google Scholar]
- 26.Khan KMF, Howe LR, Falcone DJ. Extracellular matrix-induced cyclooxygenase-2 regulates macrophage proteinase expression. Journal of Biological Chemistry. 2004;279:22039–22046. doi: 10.1074/jbc.M312735200. [DOI] [PubMed] [Google Scholar]
- 27.Pavlovic S, Du B, Sakamoto K, Khan KMF, Natarajan C, Breyer RM, Dannenberg AJ, Falcone DJ. Targeting prostaglandin E-2 receptors as an alternative strategy to block cyclooxygenase-2-dependent extracellular matrix-induced matrix metalloproteinase-9 expression by macrophages. Journal of Biological Chemistry. 2006;281:3321–3328. doi: 10.1074/jbc.M506846200. [DOI] [PubMed] [Google Scholar]
- 28.Steenport M, Khan KM, Du B, Barnhard SE, Dannenberg AJ, Falcone DJ. Matrix metalloproteinase (MMP)-1 and MMP-3 induce macrophage MMP-9: evidence for the role of TNF-alpha and cyclooxygenase-2. J. Immunol. 2009;183:8119–8127. doi: 10.4049/jimmunol.0901925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan KM, Kothari P, Du B, Dannenberg AJ, Falcone DJ. Matrix metalloproteinase-dependent microsomal prostaglandin E synthase-1 expression in macrophages: role of TNF-alpha and the EP4 prostanoid receptor. J. Immunol. 2012;188:1970–1980. doi: 10.4049/jimmunol.1102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 2002;168:5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 31.Bezugla Y, Kolada A, Kamionka S, Bernard B, Scheibe R, Dieter P. COX-1 and COX-2 contribute differentially to the LPS-induced release of PGE(2) and TxA(2) in liver macrophages. Prostaglandins & Other Lipid Mediators. 2006;79:93–100. doi: 10.1016/j.prostaglandins.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westman M, Korotkova M, af Klint E, Stark A, Audoly LP, Klareskog L, Ulfgren AK, Jakobsson P. Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis and Rheumatism. 2004;50:1774–1780. doi: 10.1002/art.20286. [DOI] [PubMed] [Google Scholar]
- 34.Subbaramaiah K, Yoshimatsu K, Scherl E, Das KM, Glazier KD, Golijanin D, Soslow RA, Tanabe T, Naraba H, Dannenberg AJ. Microsomal prostaglandin E synthase-1 is overexpressed in inflammatory bowel disease. Evidence for involvement of the transcription factor Egr-1. J Biol Chem. 2004;279:12647–12658. doi: 10.1074/jbc.M312972200. [DOI] [PubMed] [Google Scholar]
- 35.Otani T, Yamaguchi K, Scherl E, Du B, Tai HH, Greifer M, Petrovic L, Daikoku T, Dey SK, Subbaramaiah K, Dannenberg AJ. Levels of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase are reduced in inflammatory bowel disease: evidence for involvement of TNF-alpha. Am. J. Physiol Gastrointest. Liver Physiol. 2006;290:G361–G368. doi: 10.1152/ajpgi.00348.2005. [DOI] [PubMed] [Google Scholar]
- 36.Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr. Pharm. Des. 2006;12:955–962. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 37.Edelson PJ, Cohn ZA. Purification and cultivation of monocytes and macrophages. In: Bloom BR, David JR, editors. In vitro methods in cell mediated tumor immunity. Academic Press; New York: 1976. pp. 333–340. [Google Scholar]
- 38.Falcone DJ, Ferenc MJ. Acetyl-LDL stimulates macrophage-dependent plasminogen activation and degradation of extracellular matrix. J. Cellular Physiol. 1988;135:387–396. doi: 10.1002/jcp.1041350305. [DOI] [PubMed] [Google Scholar]
- 39.Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–267. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- 40.Zar JH. Biostatistical Analysis. Second Edition ed. Prentice Hall; Englewood Cliffs, N.J.: 1984. [Google Scholar]
- 41.Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J. Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- 42.Grotzinger J, Kurapkat G, Wollmer A, Kalai M, Rose-John S. The family of the IL-6-type cytokines: specificity and promiscuity of the receptor complexes. Proteins. 1997;27:96–109. [PubMed] [Google Scholar]
- 43.Legendre F, Dudhia J, Pujol JP, Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression. J. Biol. Chem. 2003;278:2903–2912. doi: 10.1074/jbc.M110773200. [DOI] [PubMed] [Google Scholar]
- 44.Wang XQ, Peng YP, Lu JH, Cao BB, Qiu YH. Neuroprotection of interleukin-6 against NMDA attack and its signal transduction by JAK and MAPK. Neurosci. Lett. 2009;450:122–126. doi: 10.1016/j.neulet.2008.11.051. [DOI] [PubMed] [Google Scholar]
- 45.Saito K, Iwashita J, Murata J, Abe T. The tyrosine kinase inhibitor AG490 inhibits growth of cancer cells and activates ERK in LS174T and HT-29 cells. Anticancer Res. 2006;26:1085–1090. [PubMed] [Google Scholar]
- 46.Pattison MJ, MacKenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J. Immunol. 2012;189:2784–2792. doi: 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reiterer G, Yen A. Inhibition of the janus kinase family increases extracellular signal-regulated kinase 1/2 phosphorylation and causes endoreduplication. Cancer Res. 2006;66:9083–9089. doi: 10.1158/0008-5472.CAN-06-0972. [DOI] [PubMed] [Google Scholar]
- 48.Brasier AR. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010;86:211–218. doi: 10.1093/cvr/cvq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J. Clin. Invest. 1995;96:2304–2310. doi: 10.1172/JCI118286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009;104:e9–18. doi: 10.1161/CIRCRESAHA.108.188243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mtairag E, Chollet-Martin S, Oudghiri M, Laquay N, Jacob MP, Michel JB, Feldman LJ. Effects of interleukin-10 on monocyte/endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovascular Research. 2001;49:882–890. doi: 10.1016/s0008-6363(00)00287-x. [DOI] [PubMed] [Google Scholar]
- 53.Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, Nicoletti A. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol. Med. 2003;9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 54.Matsumoto H, Naraba H, Murakami M, Kudo I, Yamaki K, Ueno A, Ohishi S. Concordant induction of prostaglandin E(2) synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E(2) over thromboxane and prostaglandin D-2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochemical and Biophysical Research Communications. 1997;230:110–114. doi: 10.1006/bbrc.1996.5894. [DOI] [PubMed] [Google Scholar]
- 55.Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K, Dannenberg AJ. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 2001;7:3971–3976. [PubMed] [Google Scholar]
- 56.Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin. Cancer Res. 2001;7:2669–2674. [PubMed] [Google Scholar]
- 57.Golijanin D, Tan JY, Kazior A, Cohen EG, Russo P, Dalbagni G, Auborn KJ, Subbaramaiah K, Dannenberg AJ. Cyclooxygenase-2 and microsomal prostaglandin E synthase-1 are overexpressed in squamous cell carcinoma of the penis. Clin. Cancer Res. 2004;10:1024–1031. doi: 10.1158/1078-0432.ccr-1032-3. [DOI] [PubMed] [Google Scholar]
- 58.Boulet L, Ouellet M, Bateman KP, Ethier D, Percival MD, Riendeau D, Mancini JA, Methot N. Deletion of microsomal prostaglandin E-2 (PGE(2)) synthase-1 reduces inducible and basal PGE(2) production and alters the gastric prostanoid profile. Journal of Biological Chemistry. 2004;279:23229–23237. doi: 10.1074/jbc.M400443200. [DOI] [PubMed] [Google Scholar]
- 59.Inada M, Matsumoto C, Uematsu S, Akira S, Miyaura C. Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E-2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. Journal of Immunology. 2006;177:1879–1885. doi: 10.4049/jimmunol.177.3.1879. [DOI] [PubMed] [Google Scholar]
- 60.Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F, Pellegrini S, Yasukawa K, Heinrich P, Stark GR. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 1995;14:1421–1429. doi: 10.1002/j.1460-2075.1995.tb07128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D, Kerr IM, Heinrich PC. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem. J. 1998;335(Pt 3):557–565. doi: 10.1042/bj3350557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schaeffer M, Schneiderbauer M, Weidler S, Tavares R, Warmuth M, de VG, Hallek M. Signaling through a novel domain of gp130 mediates cell proliferation and activation of Hck and Erk kinases. Mol. Cell Biol. 2001;21:8068–8081. doi: 10.1128/MCB.21.23.8068-8081.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev. Immunol. 2012;32:23–63. doi: 10.1615/critrevimmunol.v32.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]