

Abstract
Immunologic derangements in rheumatoid arthritis (RA) patients likely contribute to premature atherosclerotic cardiovascular disease (CVD). Traditional CVD risk factors do not reliably identify at-risk RA patients, probably because disease-associated mechanisms are not taken into account. The purpose of this study was to determine whether plasma from subjects with RA exhibits atheroma-promoting properties leading to disruption of cholesterol homeostasis in human monocytes/macrophages. Twenty-one healthy controls (HC) and 22 RA patients were enrolled in an IRB approved study at Winthrop University Hospital. Naïve THP-1 macrophages were exposed to plasma from each HC and RA patient. Following incubation, RNA and protein were isolated. QRT-PCR and Western blotting techniques were then used to measure expression of proteins responsible for cholesterol efflux (ATP binding cassette transporter (ABC)A1, ABCG1, 27-hydroxylase) and cholesterol uptake (CD36, ScR-A1, lectin oxidized low density lipoprotein receptor (LOX)-1, CXCL16). To confirm the pro-atherogenic effects of RA plasma on macrophages, foam cell formation was quantified. Results showed that RA plasma downregulates cholesterol efflux proteins and upregulates scavenger receptors CD36, LOX1 and CXCL16. These pro-atherogenic changes in gene expression in the presence of RA plasma are associated with augmented lipid accumulation and foam cell formation by THP-1 macrophages. RA plasma induces macrophage cholesterol overload. Demonstration of disrupted cholesterol homeostasis mediated by RA plasma provides further evidence of the involvement of the immune system in atherogenesis. Our data suggest that chronic exposure to RA plasma adversely affects the capacity of monocytes/macrophages in the arterial wall to metabolize cholesterol and maintain lipid homeostasis, thereby contributing to the development of premature atherosclerosis.
Keywords: Rheumatoid arthritis, atherosclerosis, cholesterol transport, macrophage
Introduction
The risk of cardiovascular disease (CVD) in patients with rheumatoid arthritis (RA) is very high and, at present, the elevation in risk attributed to RA is considered comparable to that seen with type 2 diabetes.1,2 The pathogenesis of early CVD in RA is complex and can be explained only partially by traditional CVD risk factors, such as hypertension, dyslipidemia, and smoking.3,4 RA-associated factors that contribute to CVD are upregulation of cytokines and adhesion molecules and increased serum oxidized lipids (such as oxidized low-density lipoprotein (LDL)).5–7 To date, traditional CVD risk assessments in RA patients do not provide insight into abnormalities in cholesterol management at the cellular level. Previous work by our group has demonstrated that plasma from patients with systemic lupus erythematosus (SLE) impairs cholesterol metabolism in cultured human macrophages.8,9 Here, we explore the nature of plasma from individuals with RA: inflammatory markers and pro-atherogenic effects on cholesterol transport in THP-1 human macrophages, a well-accepted model for atherosclerosis.10
Cholesterol balance in macrophages depends on three major pathways: cholesterol uptake by designated receptors, intracellular catabolism, and efflux of cholesterol to extracellular acceptors. Lipid uptake into the cells is mostly accomplished by the scavenger receptors CD36, scavenger receptor (SR)-A1, LOX-1 and CXCL16.11–13 Intracellular cholesterol processing is represented by the cholesterol P450 27-hydroxylase, an enzyme that converts cholesterol to oxysterols that are polar and thus more readily extracted from cells than cholesterol.14 Cholesterol efflux from cells of the arterial wall to extracellular acceptors involves the ATP binding cassette transporters (ABC) A1 and G1.15,16 Overall, expression of the proteins mentioned above comprises a cholesterol transport gene profile which reflects cellular influx, processing and elimination of cholesterol and its derivatives.
We demonstrate here that RA plasma disrupts the balance between efflux and influx in THP-1 human macrophages, resulting in lipid accumulation as manifested by increased foam cell transformation upon cholesterol-loading.
Materials and methods
Subject inclusion and exclusion criteria
Human subject studies were performed under protocol #08310 approved by the Institutional Review Board of Winthrop University Hospital. Written informed consent was obtained from all enrolled subjects of the study: 21 healthy controls (HC) and 22 RA patients between age 40 and 60 years, all females. RA patients fulfilled the 2010 revised criteria of the American College of Rheumatology for classification of RA.17 Patients with previous documentation of a diagnosis of a connective tissue disorder other than RA were excluded.
Information on duration of disease, lipid profile, C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), rheumatoid factor (RF) and cyclic citrullinated peptide antibody (CCP) was collected. Plasma samples from patients and controls were tested for levels of interleukin (IL)-6, IL-10, interferon (INF)-γ, and tumor necrosis factor (TNF)-α by enzyme-linked immunosorbent assay (ELISA) kit (Thermoscentific, Rockford, IL).
Cell culture and experimental conditions
THP-1 monocytes were grown at 37°C in a 5% CO2 atmosphere in RPMI 1640 as described.8 To facilitate differentiation into macrophages, THP-1 monocytes were treated with 100 nM phorbol 12-myristate 13-acetate (PMA) for 24 h at 37°C. Upon PMA removal, the macrophages were cultured for another 24 h prior to treatment. THP-1 macrophages were incubated in 12-well plates for 18 h in RPMI medium containing each of the following: 10% fetal calf serum; 10% pooled normal human plasma (Innovative Research, Novi, MI); 10% plasma from each HC; 10% plasma from each RA patient. No fetal calf serum was added to cells incubated in human plasma.
RNA isolation and gene expression analysis by QRT-PCR
RNA was isolated using 1 ml Trizol reagent per 106 cells and dissolved in nuclease-free water. The quantity of total RNA from each condition was measured by absorption at 260 and 280 wavelengths by ultraviolet spectrophotometry (Hitachi U2010 spectrophotometer).
ABCA1, ABCG1, 27-hydroxylase, LOX-1, ScR-A1 and CD36 mRNA were quantified by real-time PCR as described with specific primers.8,18 To detect CXCL16 the following primers were used: F 5′-ACTACACGACGTTCCAGCTCC-3′; R 5′-CTTTGTCCGAGGACAGTGATC-3′. Each reaction was done in triplicate for each plasma sample. The CT value for each gene was normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and the relative expression level was calculated as the mean value of the untreated THP-1 as 1.0. Non-template controls were included for each primer pair to check for significant levels of any contaminants. A melting-curve analysis was performed to assess the specificity of the amplified PCR products.
All results presented below use as a baseline (set at 100%) mRNA and protein levels in THP-1 macrophages exposed to 10% commercial pooled plasma. Plasma from each individual subject was evaluated in triplicate and the mean of these three independent experiments was used for analysis. The data represent the results from 22 RA and 21 HC subjects±SEM.
Protein extraction and Western blot analysis
THP-1 macrophages were incubated in 12-well plates for 24 h under the conditions described above. Total cell lysates were prepared as described previously.8,9 Protein samples (20 μg/lane) were fractionated on 8% SDS-PAGE, and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membrane was blocked for 1 h in blocking solution (5% nonfat dry milk (Bio-Rad) in 1X Tris-buffered saline/1% Tween 20 (TTBS)) and then immersed in different dilutions of primary antibody for protein detection. To detect and quantify expression of 27-hydroxylase, a 1 : 300 dilution of primary antibody in blocking solution was used overnight at 4°C. 1 : 500 dilutions of primary antibody were used for detection of ABCA1, ABCG1, CD36, SR-A1, LOX-1 and CXCL16. Rabbit anti-human ABCA1 (sc-20794) (Santa Cruz, CA) and ABCG1 (ab-36969) (Abcam Inc., Cambrige, MA) were used for detection of ABCA1 and ABCG1, respectively. Rabbit anti-human LOX-1 (ab60178), CD36 (ab64014), SR-A1 (ab36625), and CXCL16 (ab101404) were purchased from Abcam Inc. (Cambrige, MA). As a loading control, on the same transferred membrane, β-actin was detected using mouse anti-human β-actin antibody (ab8227, Abcam Inc., Cambrige, MA).
The immunoreactive proteins were detected using Pierce ECL Western Blot substrate system, and film development in SRX-101 A (Konica Minolta Holdings, Inc., Tokyo, Japan). Stripping and reprobing of the membranes were performed according to the manufacturer’s protocol (Thermoscientific, Rockford, IL). Band intensities for Western blot protein samples were quantified using Kodak Digital Science 1D, version 2.0.3, after imaging with Kodak Digital Science Electrophoresis Documentation and Analysis System 120.
Foam cell formation and cholesterol influx analysis
Differentiated THP-1 macrophages were cholesterol-loaded with 50 μg/ml oxidized LDL (oxLDL) (Intracel, Frederick, MD) for 24 h. For the foam cell differentiation cells were incubated for another 18 h with 50 μg/ml oxidized LDL in the presence of 10% plasma from each RA or 10% plasma from each HC subject. Next, cells were stained with 0.2% Oil Red O (Sigma) and visualized via light microscopy (Axiovert 25; Carl Zeiss, Gottingen, Germany) as described.9 The number of foam cells formed in each condition was calculated manually in triplicate for each plasma sample and presented as percentage of total cells.
For the cholesterol influx assay, oxLDL-loaded macrophages were exposed to 10% plasma from each RA or 10% plasma from each HC subject for another 3 h in the presence of 5 μg/ml 1,1′-dioctadecyl-3,3,3′,3′-tetramethylin docarbocyaninet (Dil)-oxLDL (Intracel, Frederick, MD). After incubation, accumulation of Dil-oxLDL in cells was determined by the fluorescent intensity of accumulated DiloxLDL with a Nikon A1 microscopy unit with 40× magnification and photographed with DS-Ri1 digital camera. Fluorescent intensity was quantified from at least three random fields (1024×1024 pixels) per slide, from three slides per experimental condition and graphed.
Data analysis
Statistical analysis was performed using Graphpad Prism, version 5.01 (GraphPad Software, San Diego, CA). All data were analyzed by one-way analysis of variance, and pairwise multiple comparisons were made between control and treatment conditions using Bonferroni correction. Probability values less than 0.05 were regarded as significant.
Results
Clinical characteristics and immunological status of RA patients
RA patients were all females with a mean age of 50±5 years. Eighty-two percent of RA patients were Caucasian, 14% Hispanic, and 5% African American (Table 1). Control subjects were 21 healthy volunteers (HC), 100% females, 50±7 years old. Seventy-one percent of HC were Caucasian, 24% were Hispanic, and 5% were Asian.
Table 1.
Demographic and clinical characteristics of rheumatoid arthritis group
| Age (years): mean ± SD | 49.9 ± 5.47 |
|---|---|
| Ethnicity (%) | |
| African American | 4.5 |
| Asian | 0 |
| Caucasian | 81.8 |
| Hispanic | 13.6 |
| Gender: % female:male | 100:0 |
| Disease duration (months): mean±SD | 85.0±9.7 |
| Age of onset (years): mean±SD | 41.68±10.17 |
| Serum cholesterol>200 mg/dl (%) | 52.9 |
| LDL cholesterol>130mg/dl (%) | 30.8 |
| HDL cholesterol<35 mg/dl (%) | 20.8 |
| Anti-CCP>50 U/ml (%) | 42.0 |
| RF>14U/ml (%) | 76.9 |
| CRP>3 mg/L (%) | 31.0 |
| ESR>20 mm/h (%) | 27.3 |
| HAQ score (%) | |
| Mild (0.25–1.5) | 43.75 |
| Moderate (1.6–5.0) | 31.25 |
| Severe (>5) | 25 |
| DMARDs (%) | 90.9 |
| NSAID (%) | 40.9 |
| Biologic (%) | 63.6 |
| Prednisone (%) | 27.3 |
| MTX (%) | 36.3 |
The mean duration of disease for RA patients was 7 years: 22% of all enrolled patients were diagnosed with RA less than 24 months prior to enrollment. Seventy-seven had an elevated RF, 42% of the patients had an elevated anti-CCP level (Table 1).
Mean levels of pro-inflammatory cytokines were significantly higher in the plasma from RA patients as compared to the HC group (Table 2): IL-6 (4.66±0.438 pg/ml vs. 1.71±0.045 pg/ml, P < 0.01), TNF-α (4.343±0.40 vs. 1.297±0.221 ng/ml ng/ml, P < 0.001) and IFN-γ (171.2±125.5 pg/ml vs. 9.54±0.7 pg/ml, P < 0.001). The anti-inflammatory cytokine IL-10 was also elevated in our RA subjects (94.74±16.32 pg/ml vs. 12±4.0 pg/ml, P < 0.001).
Table 2.
Plasma cytokine levels in healthy volunteers and RA patients
| Cytokine | Concentration in HC plasma (pg/ml) |
Concentration in RA plasma (pg/ml) |
|---|---|---|
| TNF-α | 1.297±0.221 | 4.434±0.40*** |
| IL-6 | 1.71±0.045 | 4.66±0.438** |
| IFN-γ | 9.54±0.7 | 340.5±36.1*** |
| IL-10 | 12±4.0 | 94.74±16.32*** |
P<0.01;
P<0.001.
RA patient plasma, but not plasma from healthy volunteers, increases scavenger receptors CD36, LOX-1 and CXCL16 while not altering SR-A1
The presence of RA plasma markedly augmented cholesterol influx in THP-1 macrophages, significantly affecting expression of scavenger receptors CD36 and LOX-1 (Figure 1). Incubation of macrophages with 10% RA plasma increased CD36 mRNA by 134.45±38.56% (P < 0.001) and LOX-1 by 68.9±12.45% (P < 0.001) above cells exposed to 10% HC plasma. Protein expression of CD36 and LOX-1 was upregulated to 175.45±22.76% (P < 0.01) and 169.85±43.16% (P < 0.01), respectively. The mRNA level of CXCL16 in RA plasma-incubated THP-1 macrophages was significantly higher 124.45±12.56% (P < 0.05) than the corresponding mRNA level in macrophages exposed to HC plasma. However, CXCL16 protein expression did not exhibit statistically significant upregulation. Mean levels of SR-A1 in THP-1 macrophages exposed to RA plasma were not significantly different from those exposed to HC plasma (Figure 1). In all cases, HC plasma was nearly indistinguishable from commercial plasma.
Figure 1.
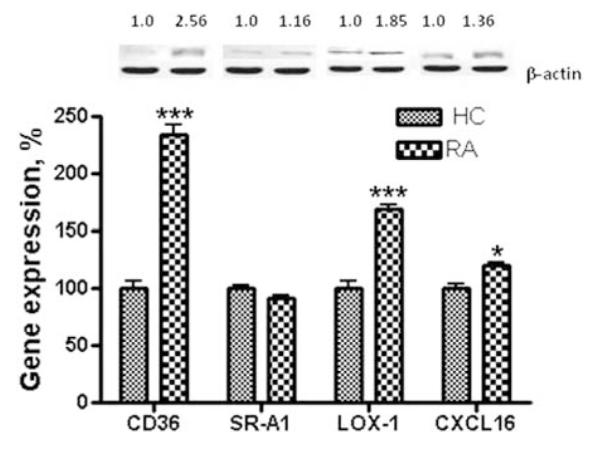
Effect of RA plasma on the expression of scavenger receptors in human THP-1 macrophages. THP-1 macrophages were incubated in the presence of 10% plasma from each of 22 RA or each of 21 HC subjects for 18 h and 24 h, as indicated in methods. Following incubation, total RNA isolated from cells of each condition was reverse transcribed and amplified by QRT-PCR with GAPDH message as an internal standard. Gene expression levels were graphed as relative mRNA expression with mean of HC set at 100%. The data represent the mean for 22 RA or 21 HC±SEM of three independent experiments. Protein expression is presented as relative comparison between protein abundance of the sample and loading control (β-actin). *P < 0.05; ***P < 0.001 versus THP-1 macrophages exposed to HC plasma
RA patient plasma, but not plasma from healthy volunteers, downregulates expression of the cholesterol efflux proteins ABCA1, ABCG1 and 27 - hydroxylase
We observed significant downregulation of cholesterol efflux proteins in THP-1 human macrophages upon exposure to 10% plasma from RA patients (Figure 2). Thus, the mean mRNA and protein levels of ABCA1 were decreased to 55.29±10.23% (P < 0.001) and 55.05±8.3% (P < 0.01), respectively compared to levels in cells treated with HC plasma. The mean values for ABCG1 mRNA and protein were 73.65±9.67% (P < 0.05) and 67.88.6±29.34% (NS), respectively upon exposure to RA plasma versus HC plasma. The average expression of 27-hydroxylase in THP-1 macrophages in the presence of RA plasma was diminished to 26.5±8.56% (P < 0.01) for mRNA and to 40.12±10.23% (P < 0.001) for protein (Figure 2).
Figure 2.
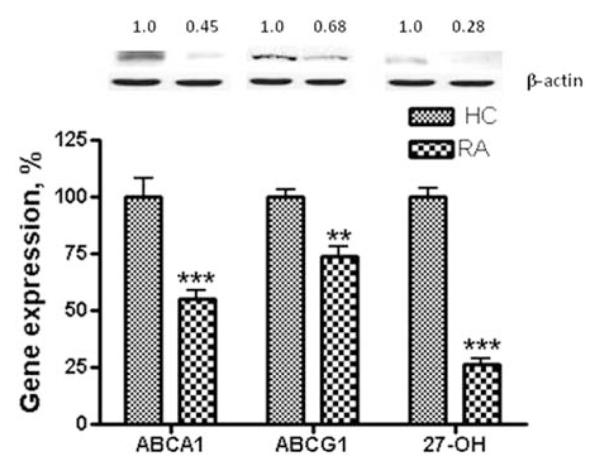
Effect of RA plasma on the expression of cholesterol efflux proteins in human THP-1 macrophages. THP-1 macrophages were incubated in the presence of 10% plasma from each of 22 RA or each of 21 HC subjects for 18 h and 24 h, as indicated in Materials and methods section. Following incubation, total RNA isolated from cells of each condition was reverse transcribed and amplified by QRT-PCR with GAPDH message as an internal standard. Gene expression levels were graphed as relative mRNA expression with mean of HC set at 100%. The data represent the mean for 22 RA or 21 HC±SEM of three independent experiments. Protein expression is presented as relative comparison between protein abundance of the sample and loading control (β-actin). 27-OH=27-hydroxylase. **P < 0.01; ***P < 0.001 versus THP-1 macrophages exposed to HC plasma
RA patient plasma, but not plasma from healthy volunteers, promotes foam cell transformation in THP-1 macrophages
As additional evidence for the pro-atherogenic properties of RA plasma, we observed a significant increase in uptake of pro-atherogenic lipids (oxLDL) and formation of foam cells by THP-1 macrophages in the presence of plasma (Figure 3). Foam cell formation, assessed by Oil-red-O staining, was enhanced by 64.26±12.3% % (P < 0.05) in THP-1 macrophages exposed to each RA plasma in triplicate compared to those exposed to 21 individual HC plasmas, each in trip-licate and by 85.36±9.4% % (P < 0.001) compared to macrophages incubated with medium alone (Figure 3(a)).
Figure 3.

Foam cell formation (FCF) in THP-1 macrophages exposed to plasma from RA patients and HC. Cultured THP-1 macrophages were exposed to RA or HC plasma (3 h for oxLDL uptake assay and 24 h for FCF analysis) as indicated in methods. (a) FCF was calculated as a percentage of oil-red-O stained cells. All results are expressed as mean for 22 RA or 21 HC±SEM of three independent experiments. *P < 0.05 versus % FCF in THP-1 macrophages exposed to HC plasma. (b) Accumulation of Dil-oxLDL in cells was determined by fluorescent intensity (FU) of accumulated Dil-oxLDL. Fluorescent intensity was quantified and graphed. ##P < 0.01 versus FU of THP-1 macrophages exposed to HC plasma
The uptake of (Dil)-oxLDL by THP-1 macrophages doubled in the presence of 10% RA plasma as compared to cells incubated in the presence of 10% HC plasma (Figure 3(b)).
Discussion
Numerous epidemiologic and clinical studies indicate that RA is an independent risk factor for CVD.1–3 Traditional CVD risk assessments in RA patients do not reflect abnormalities in cholesterol handling in cells that participate in atherosclerosis within the artery.19 This study demonstrates that RA plasma disrupts cholesterol transport gene expression in human macrophages: downregulating the cholesterol efflux proteins ABCA1, ABCG1 and 27-hydroxylase while upregulating the scavenger receptors CD36, LOX1 and CXCL16. Our data may help to elucidate the underlying process by which long-term RA leads to CVD. RA plasma compromises the capacity of macrophages in the arterial wall to metabolize cholesterol and maintain lipid homeostasis, enhancing foam cell formation and accelerating atherosclerosis.
Multiple factors within RA plasma may be responsible for its observed atheroma-promoting properties.20 Pro-inflammatory cytokines are key mediators of inflammation in the bones and joints of RA patients that may amplify atherosclerosis progression by eliciting dyslipidemia, endothelial dysfunction and oxidative stress.7,21 Elevated levels of CRP or an increased ESR may independently predict increased disability, progression of joint disease and CVD.6 IFN-γ is known to be predominantly proatherogenic and has been found by our group to suppress 27-hydroxylase.22 The present study extends our previous finding that plasma from patients with SLE augments CD36, attenuates 27-hydroxylase and increases foam cell transformation in THP-1 macrophages.8,9 Further exploration is needed to determine the specific mediators involved in disruption of cholesterol transport in both SLE and RA. Prospective longitudinal studies should reveal whether the cholesterol metabolic profile offers predictive value in identifying RA patients with high CVD risk.
ACKNOWLEDGEMENTS
This work was supported by an Innovative Research Grant from the Arthritis Foundation National Center and by the Elizabeth Daniell Research Fund.
Footnotes
Author contributions: All authors participated in the design, interpretation of the studies and analysis of the data. EB, KB, LB, GR, and SEC made substantial contributions to acquisition of data. IV, SM, MJL, and ABR conducted the experiments. IV, ABR, and SEC wrote the manuscript. All authors read and approved the final manuscript.
REFERENCES
- 1.Charles-Schoeman C. Cardiovascular disease and rheumatoid arthritis: an update. Curr Rheumatol Rep. 2012;14:455–462. doi: 10.1007/s11926-012-0271-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stamatelopoulos KS, Kitas GD, Papamichael CM, Chryssohoou E, Kyrkou K, Georgiopoulos G, Protogerou A, Panoulas VF, Sandoo A, Tentolouris N, Mavrikakis M, Sfikakis PP. Atherosclerosis in rheumatoid arthritis versus diabetes: a comparative study. Arterioscler Thromb Vasc Biol. 2009;29:1702–1708. doi: 10.1161/ATVBAHA.109.190108. [DOI] [PubMed] [Google Scholar]
- 3.Crowson CS, Matteson EL, Roger VL, Therneau TM, Gabriel SE. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am J Cardiol. 2012;110:420–424. doi: 10.1016/j.amjcard.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524–1529. doi: 10.1136/annrheumdis-2011-200726. [DOI] [PubMed] [Google Scholar]
- 5.Sandoo A, Kitas GD, Carroll D, Veldhuijzen van Zanten JJ. The role of inflammation and cardiovascular disease risk on microvascular and macrovascular endothelial function in patients with rheumatoid arthritis: a cross-sectional and longitudinal study. Arthritis Res Ther. 2012;14:R117. doi: 10.1186/ar3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. 2009;6:410–417. doi: 10.1038/nrcardio.2009.57. [DOI] [PubMed] [Google Scholar]
- 7.Libby P. Role of inflammation in atherosclerosis associated with rheumatoid arthritis. Am J Med. 2008;121(10 Suppl 1):S21–31. doi: 10.1016/j.amjmed.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Reiss AB, Anwar K, Merrill JT, Chan ES, Awadallah NW, Cronstein BN, Michael Belmont H, Belilos E, Rosenblum G, Belostocki K, Bonetti L, Hasneen K, Carsons SE. Plasma from systemic lupus patients compromises cholesterol homeostasis: a potential mechanism linking autoimmunity to atherosclerotic cardiovascular disease. Rheumatol Int. 2010;30:591–598. doi: 10.1007/s00296-009-1020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiss AB, Wan DW, Anwar K, Merrill JT, Wirkowski PA, Shah N, Cronstein BN, Chan ES, Carsons SE. Enhanced CD36 scavenger receptor expression in THP-1 human monocytes in the presence of lupus plasma: linking autoimmunity and atherosclerosis. Exp Biol Med (Maywood) 2009;234:354–360. doi: 10.3181/0806-BC-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis. 2012;221:2–11. doi: 10.1016/j.atherosclerosis.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Reiss AB, Glass AD. CD36 and ScR-A: scavenger receptors that mediate uptake of oxidized low-density lipoprotein and foam cell formation. In: Reiss AB, Carsons S, Cronstein BN, editors. Proteins involved in the pathogenesis of atherosclerosis. ■Research Signpost; 2006. pp. 1–12. [Google Scholar]
- 12.Pirillo A, Reduzzi A, Ferri N, Kuhn H, Corsini A, Catapano AL. Upregulation of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) by 15-lipoxygenase-modified LDL in endothelial cells. Atherosclerosis. 2011;214:331–337. doi: 10.1016/j.atherosclerosis.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Lehrke M, Millington SC, Lefterova M, Cumaranatunge RG, Szapary P, Wilensky R, Rader DJ, Lazar MA, Reilly MP. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J Am Coll Cardiol. 2007;49:442–449. doi: 10.1016/j.jacc.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 14.Reiss AB, Awadallah NW, Cronstein BN. Cytochrome P450 cholesterol 27-hydroxylase: an anti-atherogenic enzyme. Recent Res Devel in Lipids Res. 2000;4:39–50. [Google Scholar]
- 15.Voloshyna I, Reiss AB. The ABC transporters in lipid flux and atherosclerosis. Prog Lipid Res. 2011;50:213–224. doi: 10.1016/j.plipres.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, III, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Ménard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovský J, Wolfe F, Hawker G. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–2581. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 18.Voloshyna I, Hai O, Littlefield MJ, Carsons SE, Reiss AB. Resveratrol mediates anti-atherogenic effects on cholesterol flux in human macrophages and endothelium via PPAR-γ and adenosine. Eur J Pharmacol. 2013;698:299–309. doi: 10.1016/j.ejphar.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 19.Popa CD, Arts E, Fransen J, van Riel PL. Atherogenic index and high-density lipoprotein cholesterol as cardiovascular risk determinants in rheumatoid arthritis: the impact of therapy with biologicals. Mediators Inflamm. 2012;2012:785946. doi: 10.1155/2012/785946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahlenberg JM, Kaplan MJ. Mechanisms of premature atherosclerosis in rheumatoid arthritis and lupus. Annu Rev Med. 2013;64:249–263. doi: 10.1146/annurev-med-060911-090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirjafari H, Al-Husain A, Bruce IN. Cardiovascular risk factors in inflammatory arthritis. Curr Opin Lipidol. 2011;22:296–301. doi: 10.1097/MOL.0b013e3283488c50. [DOI] [PubMed] [Google Scholar]
- 22.Reiss AB, Patel CA, Rahman MM, Chan ES, Hasneen K, Montesinos MC, Trachman JD, Cronstein BN. Interferon-gamma impedes reverse cholesterol transport and promotes foam cell transformation in THP-1 human monocytes/macrophages. Med Sci Monit. 2004;10:BR420–425. [PubMed] [Google Scholar]