

Abstract
Mitochondrial oxidative phosphorylation is the major source of energy in cardiac muscle. In the streptozotocin-induced diabetic (STZ-DM) mice, myocardial oxidative phosphorylation was perturbated and oxidative phosphorylation complex V (ATP synthase) activity was significantly reduced. To determine the independent effects of hyperglycemia and insulin deficiency on the changes of myocardial complex V, we used phlorizin (Ph) to normalize blood glucose in the diabetic mice. Ph treatment did not improve myocardial complex V activity in the STZ-DM mice, whereas insulin treatment normalized myocardial complex V activity in the diabetic mice. Therefore, the reduction of complex V activity was caused by insulin deficiency and not by hyperglycemia in STZ-DM myocardium. Acute insulin stimulation induced phosphorylation of Akt and translocation of Akt to mitochondria in myocardium. Translocation of phospho-Akt to mitochondria was enhanced in the STZ-DM mice and was blunted in the diet-induced diabetic mice. In parallel, insulin activation of complex V was enhanced in the STZ-DM myocardium and suppressed in the diet-induced diabetic myocardium. In vivo inhibition of Akt blocked insulin stimulation of phospho-Akt translocation and blunted activation of complex V. Insulin-activated Akt translocation to mitochondria in cardiac muscle is a novel paradigm that may have important implications on myocardial bioenergetics.
Keywords: Akt translocation, Mitochondria, Diabetes, Cardiac muscle, Oxidative phosphorylation, Insulin
1. Introduction
Mitochondria are the most abundant organelles in cardiac muscle, responsible for producing the majority of myocardial energy through oxidative phosphorylation. In addition, mitochondria play key roles in the regulation of oxidative stress and apoptosis signaling [1,2]. The proteins involved in the propagation of oxidative phosphorylation (complex I through V) are located in the inner membrane of mitochondria, and the energy produced from electron transport chain help pump protons out of the inner membrane to maintain an electrochemical gradient across mitochondria membranes [1]. Maintenance of adequate electrochemical gradient prevents mitochondria membrane depolarization and is essential to allow ATP production and prevent buildup of oxidative stress and induction of apoptosis [3].
Diabetic patients have a reduced myocardial phosphocreatine/ATP ratio, indicating impaired high energy phosphate metabolism and energy deficit [4,5]. Myocardial respiration through oxidative phosphorylation is reduced in the myocardium of rodent Type 2 diabetes models [6–9]. Understanding how oxidative phosphorylation is dysregulated in the diabetic myocardium will help identify potential targets that can be used toward developing new strategies to modulate mitochondrial function and improve myocardial protection in diabetic patients.
Since diabetic cardiomyopathy has been observed in Type 1 and 2 diabetic patients, it is likely caused by metabolic perturbations that are common in both Type 1 and 2 diabetes. Hyperglycemia has traditionally been tagged as a key factor contributing to the development of cardiac metabolic dysregulation in diabetes [10,11]. However, a causal relationship between hyperglycemia and myocardial mitochondria dysfunction has not been established. In this study, we have characterized the changes of oxidative phosphorylation complex activities in a murine model of insulin-deficient diabetes, and investigated whether correcting hyperglycemia alone (without normalizing insulin levels) can improve mitochondrial oxidative phosphorylation.
Insulin receptor signaling likely plays a key role in the regulation of myocardial oxidative phosphorylation because insulin receptor KO mice showed decreased oxidative phosphorylation and exacerbated ventricular dysfunction [12]. Although insulin receptor signaling is highly complex and interacts with many signaling molecules, the phosphatidylinositol 3-kinase (PI3K)–Akt/protein kinase B (PKB) pathway is responsible for most of the metabolic actions of insulin and represents an important pathway of insulin signaling network [13,14]. Akt/PKB is a serine/threonine kinase directly downstream from PI3K and mediates most of the metabolic actions of insulin [15]. In the second part of this study, we explored whether insulin receptor signaling can reach mitochondria through translocation of Akt in cardiac muscle and whether insulin can acutely modulate oxidative phosphorylation complex V (ATP synthase) activity through activation of PI3K–Akt pathway.
2. Research design and methods
2.1. Materials
Streptozotocin, NADH, antimycin A, sucrose, fructose, lauryl maltoside, potassium cyanide, 3-dimethoxy-5-methyl-6-n-decyl-1,4-benzoquinone (DB), cytochrome c, rotenone, 2,6-dichlorophenolindophenol (DCPIP), decylubiquinone, sodium dithionite, carbonyl cyanide m-chlorophenylhydrazone, phosphoenolpyruvate (PEP), pyruvate kinase/lactate dehydrogenase, adenosine triphosphate (ATP), oligomycin, 5,5′-dithiobis(2-nitrobenzoic acid), acetyl CoA, oxalacetic acid and Phlorizin were purchased from Sigma-Aldrich (St. Louis, MO). Bovine serum albumine (BSA) was purchased from Fisher Scientific (Fairlawn, NJ). Anti-Akt, anti-phospho serine 473-Akt, and anti-insulin receptor beta subunit antibodies were purchased from Cell-Signaling Technology (Danvers, MA, USA). Anti-porin, anti-complex Vα, Vβ, Vd, and inhibitory factor antibodies and complex V immunocapture kit were purchased from MitoScience (Eugene, Oregon, USA). Recombinant human insulin was from Novo Nordisk (Princeton, NJ), and long-acting insulin glargine was from Sanofi-Aventis (Bridgewater, NJ). Power SYBR mix was obtained from Applied Biosystems (Foster City, CA). Other chemicals were purchased from Sigma or Fisher Scientific.
2.2. Experimental animals
C57BL/6 mice and specialized murine diet were purchased from Harlan Co. (Indianapolis, IN). Streptozotocin (STZ)-induced diabetes was obtained by injecting STZ (160 mg/kg body weight, i.p.) into C57BL/6 mice. Blood glucose levels were monitored by tail-vein sampling. The diabetic mice were harvested at indicated intervals after the onset of diabetes (random glucose >200 mg/dL). When indicated, the diabetic mice were treated with insulin glargine (up to 2 U per day in two divided doses, accordingly to plasma glucose levels) or phlorizin (500 mg/Kg body weight per day) to normalize blood glucose as we previously reported [16]. High fat/high fructose (HFF)-induced diabetes was obtained by feeding the mice with a high fat (42% fat) chow and 60% fructose drinking water for 6 weeks. For acute insulin effects, insulin (1 U per kg body weight) was injected into the inferior vena cava under anesthesia. The animal experimental protocol was approved by the Institutional Animal Care and Use Committee at University of California, Irvine.
2.3. Cardiomyocytes culture
Primary cultures of neonatal cardiomyocytes were prepared from Sprague–Dawley rats according to a protocol we previously described [17]. Cardiomyocytes were plated in 100-mm Petri dishes and incubated at 37 °C, 5% CO2. When indicated, after overnight serum deprivation, cardiomyocytes were incubated with insulin at indicated time intervals.
2.4. Mitochondria preparation
Mice were anesthetized with ketamine/xylazine as we previously described [18]. Myocardium was collected, frozen in liquid nitrogen, pulverized, and stored at −70 °C until further use. The pulverized myocardium were homogenized in a mitochondria isolation buffer (225 mM mannitol, 75 mM sucrose,10 mM MOPS [pH 7.2],1 mM EGTA, 0.5% BSA, 3 μg/ml aprotinin, 3 μg/ml leupeptin, 2 mM phenylmethyl–sulfonyl fluoride (PMSF), 20 mM NaF,10 mM NaPP, and 2 mM Na3VO4) on ice with Dounce homogenizer. For neonatal cardiomyocytes, the cells were scraped from the plates and homogenized in the same mitochondria isolation buffer with Dounce homogenizer. The homogenized mixture was centrifuged at 1000 g, 4 °C for 15 min to remove cell debris, and the supernatants were span down at 16,100 g, 4 °C for 15 min to obtain mitochondria pellet. The pellets were resuspended in mitochondria isolation buffer and stored at −70 °C until use. To remove non-mitochondria proteins, the mitochondria preps were digested with 50 μg/ml proteinase K for 30 min on ice [19].
2.5. Oxidative phosphorylation complex activity assays
Mitochondria membranes were ruptured by freeze–thaw cycles, and OXPHO complex activities were measured as previously described [20]. In brief, equal amounts of mitochondria proteins were assayed as outlined below.
2.5.1. Complex I+III
1 ml reaction buffer consisted of 10 mM Tris–HCl [pH 8.0], 1 mg/ml BSA, 8 μM oxidized cytochrome c, and 40.8 μM KCN was added to the cuvette, and incubated at 37 °C for 3 min with mitochondria proteins. The reaction was started by adding 0.8 mM NADH. Cytochrome c reduction at 550–540 nm was recorded for 3 min. 4 μM rotenone was added and absorbance at 550–540 nm was recorded for 3 min to quantify the rotenone-sensitive activity.
2.5.2. Complex II
1 ml reaction buffer consisted of 10 mM KH2PO4 [pH7.8], EDTA 2 mM, 1 mg/ml BSA, 80 μM DCPIP, 240 μM KCN, 4 μM rotenone and 200 μM ATP was placed in cuvette and equal amount of mitochondria proteins was added to each reaction sample. The reaction was started by adding 80 μM decylubiquinone. The activities were measured by changes of absorbance at 600 nm for 3 min.
2.5.3. Complex IV
Equal amounts of mitochondria proteins were mixed with 1 ml reaction buffer containing 10 mM KH2PO4 [pH 6.5], 1 mg/ml BSA, 0.25 M sucrose and placed in the cuvette. The reaction was started by adding 10 μM reduced cytochrome c. The activity was recorded by the absorbance at 550–540 nm for 3 min.
2.5.4. Complex V
Fresh mitochondria proteins were added to 800 μl pre-warmed distilled water and 200 μl pre-warmed reaction buffer containing 50 mM Tris–HCl [pH8.0], 1 mM NADH, 5 mg/ml BSA, 20 mM MgCl2, 50 mM KCl, 2.5 mM ATP, 15 μM carbonyl cyanide m-chlorophenylhydrazone, 10 mM phosphoenol pyruvate, 5 μM antimycin and 4 U of lactate dehydrogenase and pyruvate kinase at 37 °C. The activity was measured by the absorbance at 340 nm for 3 min. 12 μM oligomycin was added to the reaction mixture to determine the oligomycin-sensitive complex V activity.
2.6. Mitochondria abundance
Mitochondria content was determined by the ratio of mitochondria DNA to nuclear DNA in the myocardium. Pulverized myocardium was incubated with 50 mM Tris–HCl [pH 7.4], 100 mM EDTA, 400 mM NaCl, 0.5% SDS and 50 mg/ml proteinase K overnight at 55 °C. After precipitation with 1.25 M NaCl, DNA was extracted with ethanol. Quantitative real-time PCR was used to determine the copy number of ND5 and β-actin DNA. The following primer sets were used for qPCR. Mitochondria ND-5: forward 5′-TGGATGATGGTACGGACGAA-3′, reverse 5′-TGCGGTTATAGAGGATTGCTT GT-3′. Nuclear β-actin: forward 5′-TGTTCCCTTCCACAGGGTGT-3′, reverse 5′-TCCCAGTTGGTAACAATGCCA-3′. PCR was performed with ABI 7900 real-time thermocycler coupled with SYBR Green: Stage 1, 50 °C for 2 min, stage 2, 95 °C for 10 min, and stage 3, 40 cycles of 95 °C for 15 s then 60 °C for 60 s. Each sample was analyzed in triplicates, and the ND5 and β-actin copy numbers were determined by the Comparative Threshold Cycle method (ABI User Bulletin #2). Standard curves were obtained by serial dilutions of known ND5 and β-actin cDNA fragments corresponding to the primer sets.
2.7. Western blots
Equal amounts of proteins from each sample were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane, and incubated with a blocking buffer (3% BSA in 20 mM Tris–HCl [pH7.5], 137 mM NaCl, and 0.1% Tween 20) for 1 h at room temperature. The membranes were incubated sequentially with primary antibodies for 2 h at room temperature, washed three times with TBS-T (20 mM Tris–HCl [pH7.5], 137 mM NaCl, and 0.1% Tween 20), and incubated with respective horseradish peroxidase-conjugated secondary antibodies (1:5000 to 1:20,000 dilution in TBS-T) for 1 h at room temperature. The membranes were three times with TBS-T, then incubated with West Pico Chemiluminescent Substrate to visualize the proteins (Thermo Scientific, Pittsburgh, PA).
2.8. Statistical analysis
The data were presented as mean±SEM, from the results of three to six independent experiments. The intensity of bands from Western blots was scanned with densitometry and digitally analyzed. Statistical significance was tested with Student’s t test or ANOVA with post hoc analysis when appropriate. p<0.05 was considered statistically significant.
3. Results
3.1. Perturbation of oxidative phosphorylation complex activities in diabetic myocardium
Oxidative phosphorylation is driven by interlinked steps of chain reactions through OXPHO complexes in the inner membrane of mitochondria. To investigate whether mitochondrial oxidative phosphorylation is altered in streptozotocin-diabetic mice, we have analyzed specific complex activities. Citrate synthase activity was initially used to serve as a control for mitochondria prep, because citrate synthase was commonly used for this purpose. However, we soon discovered that citrate synthase was mildly altered in diabetic myocardium. Therefore, we decided to use the content of mitochondria porin (by western blots) to normalize OXPHO complex activity, and the results were shown in Fig. 1. Complex I+III activities were moderately reduced by 19%, and complex V activities were significantly lowered by 36%. Complex IV activities were marginally lowered in the diabetic myocardium, however, this was not statistically significant. These data indicate that there was significant perturbation of myocardial oxidative phosphorylation complex activities in this model of diabetes.
Fig. 1.
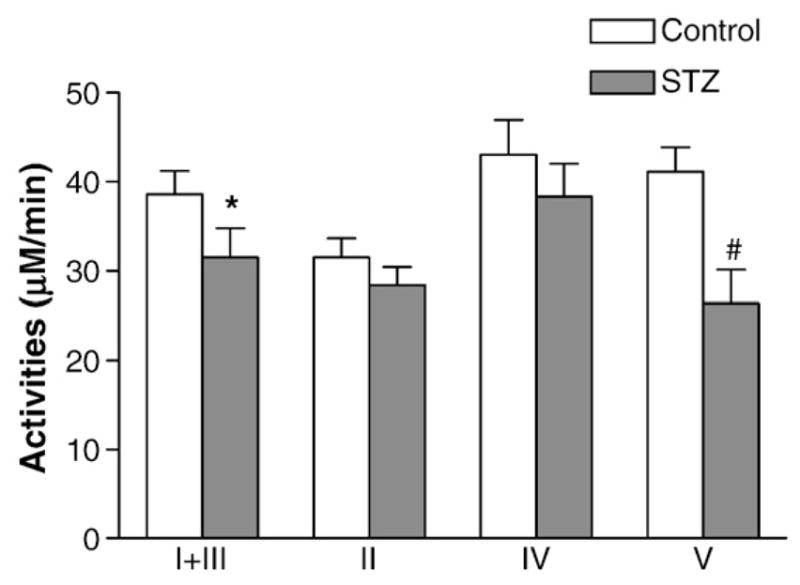
Perturbation of OXPHO complex activities in the myocardium of STZ-DM mice. Diabetes was induced with streptozotocin injection (160 mg/kg of body weight) (STZ). Myocardium was isolated 10 days after the onset of diabetes. Blood glucose levels were significantly higher in the STZ mice (453.8±51.7 mg/dL) than the control group (166± 5.5 mg/dL) (p<0.01). OXPHO complex activities were normalized with the contents of porin in each mitochondria sample. Data represent mean±SEM. (*p<0.05, vs. control; #p<0.005, vs. control; ##p<0.001, vs. control).
3.2. The abundance of OXPHO complex V was not reduced in STZ-DM myocardium
To determine whether reduction of complex V activity can be explained by the protein abundance of complex V, we surveyed the content of complex V subunits in the normal and STZ-DM myocardium. First, equal amounts of mitochondria proteins from control and STZ-DM mice were pulled down with complex V capturing Kit (MitoSciences), resolved with SDS-PAGE and silver-stained. As shown in Fig. 2A, the abundance of myocardial complex V subunits visible on the gel was not different between the control and the diabetic mice. Next we used specific antibodies against four key complex V subunits to immunoblot mitochondria proteins and the results showed no reduction of these subunit proteins in the mitochondria isolated from diabetic mice (Fig. 2B). These experiments suggest that the decreased complex V activity in diabetic myocardium was not secondary to a reduction of complex V proteins.
Fig. 2.
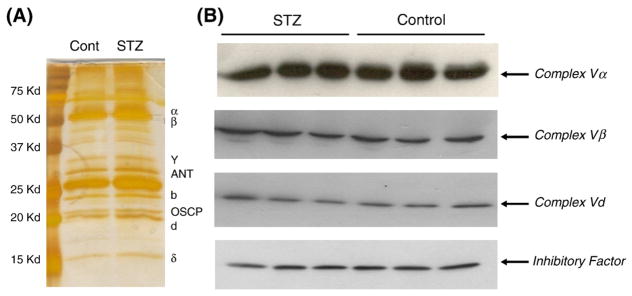
The abundance of complex V subunits did not change in STZ-DM myocardium. (A) Immunocaptured complex V subunits from normal and diabetic myocardium. Equal amounts of mitochondria proteins from control and STZ-DM mice were pulled down with complex V capturing kit, resolved with SDS-PAGE and visualized with silver stain. The photo is a representative gel, the staining time was extended to visualize all bands. (B) Immunoblots of mitochondrial proteins from control and STZ-DM myocardium with complex V subunit antibodies. Equal amounts of mitochondria proteins from each mouse were loaded to each lane.
3.3. Reduction of complex V activity in STZ-DM myocardium: hyperglycemia vs. insulin deficiency
This series of experiments were designed to further understand the cause of complex V dysfunction in diabetic myocardium. Diabetes was induced in C57BL/6 mice by streptozotocin injection as outlined above. STZ-DM is associated with hyperglycemia and insulin deficiency. In order to dissect the independent effects of hyperglycemia and insulin deficiency, subsets of diabetic mice were treated with insulin (insulin glargine) or phlorizin to correct hyperglycemia. Phlorizin inhibits Na-glucose co-transporter in renal tubule, promotes glucosuria, and thus corrected hyperglycemia without correcting insulin deficiency. Plasma glucose levels were 147±20 mg/dL (control), 443±16 mg/dL (DM), 180±47 mg/dL (DM+insulin), and 213±47 mg/dL (DM±Phlorizin). Mitochondria OXPHO V activities were measured in the control, STZ-DM, insulin-treated DM, and phlorizin-treated DM mice (Fig. 3). Insulin treatment improved complex V activity while phlorizin did not. These results suggest that insulin deficiency, not hyperglycemia, is the culprit of complex V dysfunction in this model of diabetes.
Fig. 3.
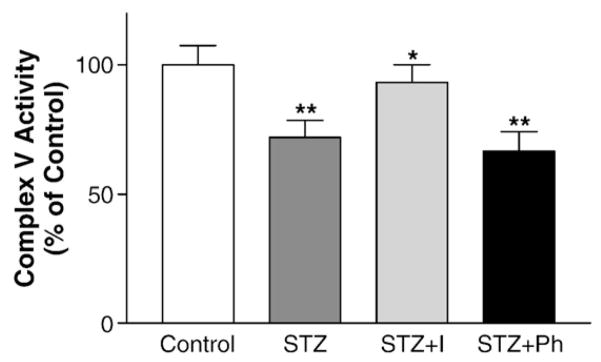
Insulin improved mitochondria complex V activity in diabetic myocardium. Mitochondria were prepared and equal amounts of proteins were analyzed for complex V activity. Diabetes was induced with streptozotocin injection. 2 days after streptozotocin injection, the diabetic mice were treated with insulin (I) or Phlorizin (Ph) for 8 days when indicated. The activity was normalized by the content of mitochondria porin (by immunoblots). Data were calculated as percent of mean control activity and presented as mean±SEM from 6 mice in each group. (*p<0.05 vs. STZ; **p<0.01 vs. Control).
3.4. Mitochondria abundance was reduced in diabetic myocardium
In order to determine the abundance of mitochondria in diabetic myocardium, STZ-DM mice were used for this series of study. Myocardial mitochondria abundance was determined by the relative copy number of mitochondria DNA to nuclear DNA. The results showed 21% reduction in mitochondria DNA contents in the diabetic myocardium and insulin treatment restored mitochondria DNA contents (Fig. 4A). However, phlorizin treatment could not restore mitochondria DNA content in the diabetic mice despite hyperglycemia was significantly improved in the phlorizin-treated diabetic mice. Therefore insulin deficiency, not hyperglycemia, was responsible for reduction of mitochondria abundance in diabetic myocardium. Together with the results on complex V activities, these data rendered evidence that insulin deficiency, not hyperglycemia, is a major cause of myocardial mitochondria dysfunction in this model of insulin-deficient (Type 1) DM.
Fig. 4.

The effects of insulin treatment on mitochondria biogenesis and total myocardial complex activities in diabetic myocardium. (A) Mitochondria abundance. The abundance of mitochondria DNA was measured by quantitative real-time PCR to determine the copy number of ND5 and β-actin. 2 days after streptozotocin injection, the diabetic mice were treated with insulin (I) or Phlorizin (Ph) for 8 days when indicated. Data represent mean±SEM from 6 mice in each group. (B) Total myocardial complex V activities adjusted by the content of mitochondria. Total myocardial complex V activities per myocardial unit were calculated with the following formula: total complex V activity=(complex V activity per mitochondria unit)×(mitochondria content per myocardial unit). STZ-DM mice were treated with insulin (I) and phlorizin (Ph) when indicated. (**p<0.01 vs. STZ; #p<0.005 vs. control; ##p<0.001 vs. control; &p<0.0005 vs. control).
3.5. The effect of mitochondria abundance on mitochondria OXPHO complex activities in diabetic myocardium
We have analyzed mitochondria OXPHO activities per mitochondria unit thus far. However, myocardial OXPHO complex activity would have been altered if the abundance of mitochondria per myocardial unit was changed. To this end, we have calculated mitochondria complex V activities per myocardial unit and the results were shown in Fig. 4B. The data indicated 47% reduction of total complex V activity in the STZ-DM myocardium and insulin treatment improved total myocardial complex V activity. Thus, myocardial oxidative phosphorylation is impaired to a greater extent in diabetic mice when the reduction of mitochondria abundance is taken into consideration.
3.6. Insulin induced acute Akt translocation to mitochondria
Our data suggested that insulin may directly modulate mitochondria function. In order to determine whether insulin receptor signaling can reach mitochondria in cardiac muscle, C57BL/6 mice were fasted overnight and injected with insulin via inferior vena cava as we previously described [16]. The results are shown in Fig. 5, insulin treatment acutely induced translocation of phospho-Akt to mitochondria within a few minutes (Fig. 5A). Myocardial mitochondria preps may contain non-mitochondria proteins therefore the preps were further digested with proteinase K to remove residual cytoplasmic proteins and surface-exposed proteins on the mitochondria outer membrane as previously described [19]. The mitochondria inner membrane and matrix proteins remained intact under this protocol. TOM 20, a protein exposed on the outer membrane of mitochondria, was removed by proteinase K, but Akt translocation remained visible upon insulin stimulation. Porin was embedded inside the outer membrane and could not be digested by proteinase K [19]. We also investigated whether insulin receptor could be translocated to mitochondria, however, insulin receptor protein could not be identified in the mitochondria prep after proteinase K digestion. Insulin similarly increased accumulation of phospho-Akt in the mitochondria in primary cardiomyocytes. In contrast, insulin did not induce phospho-Erk translocation to mitochondria (Fig. 5B). Both Akt protein and phospho-Akt translocated into mitochondria in the insulin-stimulated myocardium (Fig. 5C).
Fig. 5.

Insulin stimulates phosphorylation and translocation of Akt to mitochondria in cardiac muscle. (A) Insulin increased accumulation of p-Akt in myocardial mitochondria. Proteinase K digestion did not alter p-Akt translocation to mitochondria. Myocardial mitochondria preps were digested with proteinase K as described in the Research design and methods section to remove non-mitochondria proteins. TOM 20, a mitochondria protein, served as a control for proteinase K digestion. Immunoblotting with anti-insulin receptor β subunit antibodies showed that crude mitochondria preps were contaminated with insulin receptors (Ins-R) and were removed after proteinase K digestion. (B) Time-course of insulin stimulation on phospho-Akt translocation in primary cardiomyocytes. Cardiomyocytes were incubated with insulin (10−7 M) after overnight fasting. Equal protein amounts of proteinase K-treated mitochondria preparations were used for western blots. (C) Dose–response of insulin stimulation on phospho-Akt translocation in myocardium in vivo. Mice were overnight-fasted and injected with insulin, myocardial mitochondria were isolated and equal protein amounts of proteinase K-treated mitochondria preps were loaded to each lane. In the phospho-Akt blot, the lower band represents p-Akt and the upper band is a non-specific band. The last lane (C) represents total myocardial lysates from insulin-stimulated mice, as positive control. Immunoblot with anti-porin antibodies served as loading control.
3.7. Insulin-stimulated Akt translocation was altered in diabetic myocardium
To determine whether insulin receptor signaling to mitochondria is altered in diabetic myocardium, two different models of diabetes were used in this series of experiments. STZ-DM mice were used as insulin-deficient (Type 1) DM model. To produce a murine model of diet-induced (Type 2) DM, C57BL/6 mice were fed with a high fat-high fructose diet (HFF). High fat diet and high fructose diet had been used to generate animal models featuring obesity, insulin resistance and hyperglycemia [21–23]. The control group was fed with a standard murine chow diet. After six weeks, the HFF group showed fasting hyperglycemia and weight gain (FBS 115 vs. 191 mg/dL, p<0.01; BW 23 vs. 27 g, p<0.01). Insulin was injected via inferior vena cava after overnight fasting and myocardial mitochondria were prepared and treated with proteinase K. The results showed that insulin-induced phospho-Akt translocation to mitochondria was significantly altered in the STZ-DM myocardium and in the diet-induced Type 2 DM model (Fig. 6). In STZ-DM mice, insulin-induced Akt phosphorylation was significantly augmented in the myocardial mitochondria as compared to the non-diabetic mice (Fig. 6). At basal, there were less Akt proteins in the mitochondria in the STZ-DM myocardium. Upon insulin stimulation, the magnitude of Akt protein translocation was enhanced in the STZ-DM myocardium. The effect of insulin on Akt translocation was significantly reduced in the HFF myocardium, and insulin-stimulated Akt phosphorylation in mitochondria was reduced in parallel (Fig. 6). Insulin-stimulated Akt phosphorylation per Akt protein unit (stoichiometry of Akt phosphorylation) in mitochondria was increased in the STZ-DM myocardium and reduced in the HFF myocardium (Fig. 6E). Diet-induced Type 2 diabetes such as HFF model is associated with insulin resistance whereas STZ-DM features enhanced insulin receptor signaling to PI3K/Akt pathway [21,22,24]. Therefore, insulin-activated Akt phosphorylation in mitochondria paralleled myocardial insulin sensitivity in these two models of diabetes.
Fig. 6.

Mitochondria translocation of phospho-Akt in insulin-deficient diabetes and diet-induced diabetes. Control and diabetic mice were overnight-fasted and injected with insulin (10 min). STZ-DM was used as insulin-deficient diabetes model (A). Mice fed with high fat-high fructose (HFF) diet were used as Type 2 DM model (B). Proteinase K-treated mitochondria preps were used for western blots to determine Akt translocation. (A) Insulin-stimulated Akt translocation to mitochondria in the myocardium of STZ-DM mice. Lane 1 — control basal, lane 2 — insulin-stimulated control, lane 3 — STZ-DM basal, lane 4 — insulin-stimulated STZ-DM. In the phospho-Akt blot, the lower band represents p-Akt and the upper band is a non-specific band. (B) Insulin-stimulated Akt translocation to mitochondria in the HFF myocardium. Equal protein amount of proteinase K-treated mitochondria prep was used in each lane. Lane 1 — control basal, lane 2 — insulin-stimulated control, lane 3 — HFF-DM basal, lane 4 — insulin-stimulated HFF-DM. In the phospho-Akt blot, the lower band represents p-Akt and the upper band is a non-specific band. (C) Phospho-Akt translocation was enhanced in the STZ-DM myocardium. Equal protein amount of proteinase K-treated mitochondria prep was used in each lane. The bar graphs represent the data summarized from multiple experiments, these data were normalized to the contents of porin in each prep. The lower graph compared the magnitude of insulin-stimulated translocation between the control and STZ-DM myocardium. *p<0.05 vs. control basal; &p<0.05 vs. DM basal; ##p<0.001 vs. insulin-stimulated control. (D) Phospho-Akt translocation was reduced in the HFF-DM myocardium. The bar graphs represent the data summarized from multiple experiments, these data were normalized to the content of porin in each prep. The lower graph compared the magnitude of insulin-stimulated translocation between the control and HFF-DM. ##p<0.001 vs. control basal, *p<0.05 vs. HFF basal, ###p<0.00001 vs. insulin-stimulated control. (E) Stoichiometry of Akt phosphorylation in diabetic myocardium. The ratios of Akt phosphorylation to Akt protein abundance were calculated in the normal, STZ-DM, and HFF-DM mice injected with insulin. Equal amounts of myocardial mitochondria proteins were respectively immunoblotted with anti-p-Akt and anti-Akt antibodies and the relative ratios of p-Akt to Akt were calculated.
3.8. Acute insulin effect on myocardial OXPHO complex V was mediated by PI3K–Akt pathway and insulin-activation of complex V altered in diabetes myocardium
To determine whether the acute effect of insulin on complex V is dependent on activation of Akt, the mice were pretreated with LY294002 prior to insulin stimulation (Fig. 7). Insulin-activation of Akt phosphorylation in mitochondria was inhibited by LY294002, and the effect of insulin on complex V was also significantly blunted by LY294002. This experiment indicated that the acute effect of insulin on myocardial complex V requires activation of PI3K–Akt pathway.
Fig. 7.
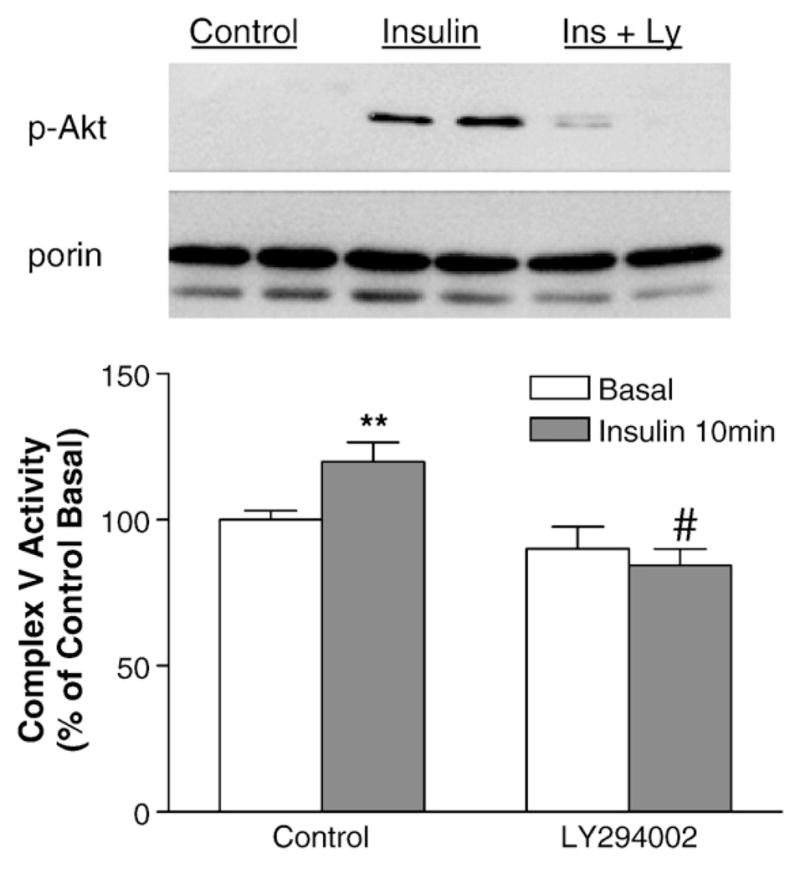
In vivo inhibition of Akt signaling blunted insulin-stimulated complex V activities in myocardium. Mice were overnight-fasted and anesthetized. When indicated, LY294002 (Ly) were injected (40 mg/kg BW, i.p.) 20 min prior to insulin injection. Myocardium was harvested after insulin injection, mitochondria were isolated and mitochondria proteins were used for immunoblotting. Insulin-stimulated p-Akt translocation was inhibited by LY294002 in myocardium (upper panel). The effect of insulin on complex V activity was blunted in the LY294002-pretreated myocardium (lower panel). #p<0.005 vs. insulin-treated control, **p<0.01 vs. control basal.
Since insulin-stimulated p-Akt translocation to mitochondria is altered in the diabetic myocardium, we next studied the acute effect of insulin on myocardial complex V activity. The results showed that acute insulin stimulation indeed increased complex V activities in the control mice (Fig. 8). Interestingly, insulin-stimulation of complex V activities was blunted in the HFF myocardium and enhanced in the STZ-DM myocardium. The acute effect of insulin on complex V activity mirrored the changes of insulin receptor signaling to Akt in these two models.
Fig. 8.

The effect of acute insulin injection on myocardial complex V activity. The mice were overnight-fasted and myocardium was harvested 10 min after insulin injection. Equal amounts of mitochondria proteins from each sample were used for complex V assay. The effects of acute insulin injection were compared in the control, STZ-DM, and HFF-DM mice. Complex V activity is expressed as percentage of control basal (A) or respective basal in each group (B). (A) *p<0.05 vs. control basal, &p<0.0005 vs. control basal, ##p<0.001 vs. STZ basal. (B) @p<0.05 vs. control.
4. Discussion
Impaired myocardial mitochondria function has been characterized in several models of animal diabetes, accompanied by reduced energy formation, increased oxidative stress, and activation of apoptosis signaling [2,4,5]. Previous efforts had focused on characterization of mitochondria dysfunction in diabetic myocardium, but little is known regarding the mechanisms underlying such mitochondria dysfunction. The results presented in this paper outlined a unique paradigm coupled to insulin signaling and Akt translocation, this paradigm may help further understand how mitochondria function is regulated in the normal myocardium and dysregulated in the diabetic myocardium.
The maintenance of normal myocardial function requires adequate energy production from mitochondria. ATP production in mammalian tissues should be coupled to the presence of substrates (carbohydrates and fatty acids) and energy demands of tissues and organs. Cellular energy homeostasis should not be maintained in a steady state, rather, in order to meet the dynamic physiological cues and changing physical activities, extracellular mechanisms must exist to signal and coordinate mitochondrial oxidative phosphorylation [25,26]. Here we presented an intriguing observation in an insulin-responsive tissue that allowed insulin to modulate energy production in response to carbohydrate intake.
OXPHO complexes collectively form the major functional module in mitochondria. There are multiple potential phosphorylation sites on most OXPHO complexes [26]. Whether these phosphorylation sites can be regulated by intracellular signaling largely remains unknown. However, some studies began to shed light on the potential role of hormone signaling. For example, cytochrome c oxidase can be phosphorylated by PKA at serines 115/116 of subunit I, threonine 52 of subunit IV, and serine 40 of subunit Vb [27]. Translocation of PKCδ, Shp-2 and Src family kinases to the mitochondria also have been described in the literatures [26,28,29], but their substrates in mitochondria have not been identified.
Complex V (F0–F1 ATP synthase) is a multisubunit enzyme located in the inner membrane of mitochondria. Complex synthesizes ATP from ADP and inorganic phosphate using the energy provided by the electrochemical gradient across inner membrane. This gradient is maintained by the respiratory chains and the chemical compositions of membranes. Our laboratory had previously shown that the beneficial effect of insulin-like growth factor 1 (IGF-1) on the mitochondria electrochemical gradient was dependent on the PI3K–Akt pathway in cardiomyocytes, and the electrochemical gradient could be protected by a constitutively active PI3K and disrupted by a dominant negative Akt [3]. Insulin and IGF-1 receptors share similar signaling pathways. The data presented in this study extend our previous findings and expand PI3K–Akt pathway into regulation of mitochondria bioenergetics.
Insulin receptor number is upregulated and its receptor signaling to PI 3-kinase/Akt is exaggerated in the myocardium of the insulin-deficient STZ-DM model [24]. In contrast, Type 2 diabetes models are associated with insulin resistance and down-regulation of insulin receptor signaling [21,22,25]. Insulin-stimulated phospho-Akt translocation to mitochondria was augmented in the STZ-DM model and blunted in the HFF-DM model. The magnitudes of Akt translocation in these two models mirrored alterations of insulin receptor signaling to PI3K/Akt in each model, thereby suggesting dysregulation of insulin receptor signaling to mitochondria reflected the underlying insulin sensitivity. Collectively, these findings indicated that insulin deficiency (Type 1 DM) and insulin resistance (Type 2 DM) could lead to dysregulation of oxidative phosphorylation through inadequate insulin effect on mitochondria.
Considerable efforts in the past focused on how hyperglycemia induced metabolic dysfunction in diabetic myocardium, and numerous clinical trials have shown that improvement of glucose control with intensive insulin therapy improved the outcomes of those patients with acute heart diseases [10]. The results of this study suggest that hyperglycemia is not the cause of complex V dysfunction in the STZ-DM myocardium. Rather, insulin deficiency is the culprit of reduced complex V activities. Modulation of mitochondria function and reactive oxygen species had been implicated in pre-conditioning of myocardial ischemia and myocardial protection [30]. It is tempting to speculate that inadequate insulin effect on mitochondria might contribute to modulation of myocardial protection. However, whether our animal study is applicable to human diabetic cardiomyopathy will require further study.
Mitochondria have been implicated in the pathogenesis of Type 2 diabetes and metabolic syndrome [31,32]. Certain mtDNA mutations are associated with diabetes mellitus [1]. In Type 2 diabetes patients, reduced mitochondrial electron transport activity has been described in the skeletal muscle of Type 2 diabetic patients, accompanied by reduced mitochondria DNA abundance [33]. However, the reduction of electron transport activity was more profound than the changes of mitochondria abundance [34]. Pancreatic β cells require high energy support, impaired oxidative phosphorylation and subsequent oxidative stress may precipitate apoptosis [35]. On the other hand, impaired mitochondria function in muscle and adipocytes can lead to accumulation of reactive oxygen species, inhibition of insulin receptor signaling, and development of insulin resistance [34]. The results of this paper suggested an interesting signaling pathway in an insulin sensitive tissue that directly linking insulin signaling to mitochondria through translocation of Akt. Evidently mitochondria are not only modulators of insulin receptor signaling, but also targets of insulin receptor signaling.
Acknowledgments
This work is supported in part by research grant from the American Heart Association (to PHW). The authors would like to thank Dr. Douglas C. Wallace and Samuel E. Schriner for their assistance with OXPHO complex assays, and Ying-Pu Tien for her excellent technical assistance.
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai HC, Liu TJ, Ting CT, Yang JY, Huang L, Wallace D, et al. Regulation of IGF-I receptor signaling in diabetic cardiac muscle: dysregulation of cytosolic and mitochondria HSP60. Am J Physiol Endocrinol Metab. 2007;292(1):E292–7. doi: 10.1152/ajpendo.00189.2006. [DOI] [PubMed] [Google Scholar]
- 3.Lai H, Liu J, Ting C, Sharma P, Wang PH. Insulin-like growth factor-1 prevents loss of electrochemical gradient in cardiac muscle mitochondria via activation of PI 3 kinase/Akt pathway. Mol Cell Endocrinology. 2003;205:99–106. doi: 10.1016/s0303-7207(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 4.Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 2008 Feb;114(3):195–210. doi: 10.1042/CS20070166. [DOI] [PubMed] [Google Scholar]
- 5.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107(24):3040–6. doi: 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- 6.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112(17):2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 7.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115(7):909–17. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM, Jr, Klein JB, et al. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2004;287(5):E896–905. doi: 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- 9.Ovide-Bordeaux S, Grynberg A. Docosahexaenoic acid affects insulin deficiency-and insulin resistance-induced alterations in cardiac mitochondria. Am J Physiol Regul Integr Comp Physiol. 2004;286(3):R519–27. doi: 10.1152/ajpregu.00303.2003. [DOI] [PubMed] [Google Scholar]
- 10.Deedwania P, Kosiborod M, Barrett E, Ceriello A, Isley W, Mazzone T, et al. American Heart Association Diabetes Committee of the Council on Nutrition, Physical Activity, and Metabolism. Hyperglycemia and acute coronary syndrome: a scientific statement from the American Heart Association Diabetes Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2008;117(12):1610–9. doi: 10.1161/CIRCULATIONAHA.107.188629. [DOI] [PubMed] [Google Scholar]
- 11.Retnakaran R, Zinman B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371(9626):1790–9. doi: 10.1016/S0140-6736(08)60767-9. [DOI] [PubMed] [Google Scholar]
- 12.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocytes insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285(3):H1261–9. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 13.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 14.White MF. Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol. 2006;84(7):725–37. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- 15.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen HS, Shan YX, Yang TL, Lin HD, Chen JW, Lin SJ, et al. Insulin deficiency downregulated heat shock protein 60 and IGF-1 receptor signaling in diabetic myocardium. Diabetes. 2005;54(1):175–81. doi: 10.2337/diabetes.54.1.175. [DOI] [PubMed] [Google Scholar]
- 17.Shan Y, Liu T, Su H, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. 2003;35:1135–43. doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- 18.Su H, Samsamshariat A, Fu J, Shan Y, Chen Y, Piomelli D, et al. Oleylethanolamide activates Ras–Erk pathway and improves myocardial function in doxorubicin-induced heart failure. Endocrinology. 2006;147:827–34. doi: 10.1210/en.2005-1098. [DOI] [PubMed] [Google Scholar]
- 19.Krimmer T, Rapaport D, Ryan MT, Meisinger C, Kassenbrock CK, Blachly-Dyson E, et al. Biogenesis of porin of the outer mitochondrial membrane involves an import pathway via receptors and the general import pore of the TOM complex. J Cell Biol. 2001;152(2):289–300. doi: 10.1083/jcb.152.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrientos A. In vivo and in organello assessment of OXPHO activities. Methods. 2002;26(4):307–16. doi: 10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 21.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118(2):789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450(7170):712–6. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakagawa T, Tuttle KR, Short RA, Johnson RJ. Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol. 2005;1(2):80–6. doi: 10.1038/ncpneph0019. [DOI] [PubMed] [Google Scholar]
- 24.Wang PH, Almahfouz A, Giorgino F, McCowen KC, Smith RJ. In vivo insulin signaling in the myocardium of streptozotocin-diabetic rats: opposite effects of diabetes on insulin stimulation of glycogen synthase and c-Fos. Endocrinology. 1999;140(3):1141–50. doi: 10.1210/endo.140.3.6595. [DOI] [PubMed] [Google Scholar]
- 25.Devenish RJ, Prescott M, Rodgers AJ. The structure and function of mitochondrial F1F0-ATP synthases. Int Rev Cell Mol Biol. 2008;267:1–58. doi: 10.1016/S1937-6448(08)00601-1. [DOI] [PubMed] [Google Scholar]
- 26.Salvi M, Brunati AM, Toninello A. Tyrosine phosphorylation in mitochondria: a new frontier in mitochondrial signaling. Free Radic Biol Med. 2005;38:1267–77. doi: 10.1016/j.freeradbiomed.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 27.Fang JK, Prabu SK, Sepuri NB, Raza H, Anandatheerthavarada HK, Galati D, et al. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 2007;58:11302–10. doi: 10.1016/j.febslet.2007.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, et al. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094–100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Protein kinase Cdelta targets mitochondria, alters mitochondrial membrane potential, and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol Cell Biol. 1999;19:8547–58. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, et al. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87(6):460–6. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 31.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54(1):8–104. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 34.Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102(4):401–14. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–10. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]