

Abstract
The KvLQT1 gene encodes a voltage-gated potassium channel. Mutations in KvLQT1 underlie the dominantly transmitted Ward-Romano long QT syndrome, which causes cardiac arrhythmia, and the recessively transmitted Jervell and Lange-Nielsen syndrome, which causes both cardiac arrhythmia and congenital deafness. KvLQT1 is also disrupted by balanced germline chromosomal rearrangements in patients with Beckwith-Wiedemann syndrome (BWS), which causes prenatal overgrowth and cancer. Because of the diverse human disorders and organ systems affected by this gene, we developed an animal model by inactivating the murine Kvlqt1. No electrocardiographic abnormalities were observed. However, homozygous mice exhibited complete deafness, as well as circular movement and repetitive falling, suggesting imbalance. Histochemical study revealed severe anatomic disruption of the cochlear and vestibular end organs, suggesting that Kvlqt1 is essential for normal development of the inner ear. Surprisingly, homozygous mice also displayed threefold enlargement by weight of the stomach resulting from mucous neck cell hyperplasia. Finally, there were no features of BWS, suggesting that Kvlqt1 is not responsible for BWS.
Introduction
The human KvLQT1 gene is known to be important in normal development of the myocardium and the inner ear. Mutations in this gene cause either prolongation of the electrocardiographic QT interval leading to fatal arrhythmias, deafness, or both (1–4). In addition, KvLQT1 is the target of balanced germline chromosomal rearrangements in Beckwith-Wiedemann syndrome (BWS) (5), which causes prenatal overgrowth and predisposition to childhood cancers. This gene is preferentially expressed from the maternal allele. There are multiple isoforms, including a nonimprinted isoform expressed in the heart and an imprinted isoform that is expressed ubiquitously, suggesting that KvLQT1 is important in more than cardiac and auditory tissues (5).
KvLQT1 encodes a voltage-gated potassium channel, which enables a potassium current after electrical depolarization of the cell membrane (1–3). Such channels are important in a wide variety of cellular functions, including synaptic transmission and muscle excitability. To determine how KvLQT1 might be important in normal physiology, we developed a knockout mouse. This mouse might serve as a useful model of hereditary deafness or arrhythmia, but also might address the role of KvLQT1 in BWS. We and others have predicted that the chromosomal rearrangements within KvLQT1 act in a novel way by disrupting the expression of nearby imprinted genes rather than through loss of the transcript itself (5–7). The absence of features of BWS in a mouse lacking Kvlqt1 would support this positional effect model.
Methods
Generation of Kvlqt1 knockout mice.
We disrupted Kvlqt1 exon 1, an exon shared among all the known isoforms of the gene. Comparison between the human and mouse Kvlqt1 gene sequence revealed a potential intron-exon boundary for the mouse Kvlqt1 gene. Two pairs of primers were synthesized to amplify exon 1 and exon 2 from 129/SvEv mouse genomic DNA. These primers were then used to PCR screen a 129/SvEv genomic DNA BAC library. One BAC clone containing exons 1 and 2 was isolated. A 6-kb EcoRI-EcoRV fragment containing exon 1 was subcloned into pBluescript and the insert was sequenced, confirming the presence of the exon 1 and the intron-exon boundary. The PCR primers were designed to introduce Sal I and Xho I restriction sites into the exon, and a 1.6-kb Sal I-Xho I fragment containing the neo gene with the TK promoter was inserted into exon 1, disrupting Kvlqt1 by insertion and creating multiple stop codons. This construct, termed pMLQT1, was validated by sequencing and transfected into embryonic stem (ES) cell line E14. DNA was isolated from 200 G418-resistant clones and analyzed by genomic Southern blot. This analysis identified two clones that had Kvlqt1 disrupted by homologous recombination. Cells from homologous recombinants were individually expanded and injected into 3.5-day blastocysts of C57BL/6 mice. The blastocysts containing ES cells were transplanted back to a pseudopregnant host mother (F1 derived from C57BL/6 × BALB/c). Five chimeras were identified by a 50–90% white coat color. Chimeras were backcrossed to C57BL/6 mice. Germline transmission was determined by DNA typing of tail DNA. Heterozygotes were then intercrossed to obtain mice null for Kvlqt1. All mice analyzed were from F2 and F3.
Acoustic brainstem response testing.
Four wild-type (Kvlqt1+/+), four heterozygotes (Kvlqt1+/–) and five homozygotes (Kvlqt1–/–) mice were selected for acoustic brainstem response (ABR) studies to measure hearing thresholds using methods described previously (8). Mice ranged in age from 6 weeks to 3 months. Mice were anesthetized using intraperitoneal injections of a 3.5% chloral hydrate solution (0.008 mg/kg) and xylazine hydrochloride (0.006 mg/kg). A ground needle electrode and recording needle were placed subcutaneously in the scalp. An electrostatic speaker coupled to a hollow ear bar was placed inside the pinna and calibrated with a Bruel & Kjaer (Norcross, Georgia, USA) microphone to measure the signal level in dB SPL (sound pressure level). Clicks (n = 1,000) of 100 microseconds duration and alternating polarity were presented in the animal’s ears in 10 dB increments, starting from a signal of 0 dB SPL and ending at 95 dB SPL. Evoked potentials were recorded for 15 milliseconds in response to each click and then averaged for each intensity level. Hearing threshold was determined by examining the first two consecutive waveforms occurring within 8 milliseconds of stimulus presentation that had peak to trough amplitudes of 1.5 μV/ms for positive waveforms or –1.5 μV/ms for negative waveforms. Statistical analysis comparing the three groups of mice was performed using a Student’s Newman-Keuls test.
Inner ear histology.
The histology of the cochlea and vestibular organs of the inner ears was studied in a Kvlqt1+/+ mouse of approximately 4 months of age and in a Kvlqt1–/– mouse of the same age. After removal, the inner ears were prepared for Araldite embedding as described previously (9) and then sectioned. The mice were given a lethal dose of sodium pentobarbital and transcardially perfused with a solution of 0.1 M cacodylate buffer, followed by a fixative solution of 2.0% glutaraldehyde and 2.0% paraformaldehyde. After the temporal bones were harvested, the inner ears were gently perfused with fixative through the oval and round windows and then postfixed overnight. The temporal bones were decalcified in a 0.1 M EDTA solution for approximately 3 weeks and then dehydrated in graded ethanol solutions followed by propylene oxide. The inner ears were infiltrated with increasing concentrations of Araldite, starting with a 1:1 and then a 2:1 Araldite/polypropylene oxide mixture. The cochleae were then degassed for 2 hours in a desiccator. Cochleae were embedded in 100% Araldite and allowed to cure for 3 days at 60°C. The inner ears were then sectioned using a tungsten carbide knife on a rotary microtome at a thickness of 20 μm. Sections were mounted on glass slides, stained with Toluidine blue and coverslipped.
Cardiac electrophysiology.
Mice were lightly anesthetized with inhaled Metofane (Mallinckrodt Veterinary, Mundelein, Illinois, USA) and placed on a warming blanket. Body temperature was monitored with a rectal probe. An unfiltered electrocardiographic (ECG) limb lead was acquired, digitized with 12-bit precision at 1,000 samples per second, and stored on removable magnetic media for offline analysis, with a multichannel data acquisition system (Biopac Systems Inc., Santa Barbara, California, USA) connected to a 486-based computer. Two to four consecutive 180-second epochs of ECG data were obtained in mouse. The lead was selected based on the most optimal T-wave morphology.
Basic ECG intervals (cycle length, PR, QRS duration) were measured offline. An average of ten cycles were measured after 2 minutes of recording to ensure the heart rate had reached steady state. Two to three 2-minute epochs were recorded from each animal. Heart rate and heart rate variability were measured using a QRS detection algorithm, and the algorithm for QT interval measurement was performed as described previously (10). An evenly sampled heart rate and QT interval time series was constructed from the sequence of RR and QT intervals respectively as described previously (11), using a sampling frequency of 10 samples per second. Data exhibiting any extrasystoles were excluded from analysis.
All data are expressed as mean ± SD. An unpaired t test was used to compare variables between groups. Post hoc subgroup comparisons were made with unpaired t tests with Bonferroni correction. Correlation between continuous variables was tested by linear regression. Statistical significance for all tests was accepted at the P < 0.05 level.
Gastric histology and gene expression.
Immunohistochemistry was performed for Ki-67 using the MIB-1 clone from Immunotech (Westbrook, Maine, USA) at 1:50 dilution for H/K+ ATPase using a rabbit polyclonal antibody from A. Smolka (Medical University of South Carolina, Charleston, South Carolina, USA) at 1:1,000 dilution, for spasmolytic polypeptide using a murine monoclonal IgM antibody from N. Wright (Hammersmith Hospital, London, United Kingdom), and for intrinsic factor using a rabbit polyclonal antibody from D. Alpers (Washington University, St. Louis, Missouri, USA). Gastrins assays were performed as described elsewhere (12). RNA extraction was performed as described (13, 14) from serial sectioned paraffin-embedded specimens. Laser capture microdissection was performed using a PixCell LCM (Arcturus Engineering, Mountain View, California, USA) according to the manufacturer’s protocol from fresh frozen specimens embedded in TissueTek OCT medium (VWR Scientific Products, West Chester, Pennsylvania,USA). Real-time PCR was performed on an ABI Prism 7700 Sequence Detection System (PE Applied Biosystems, Foster City, California, USA) according to the manufacturer’s recommendation, using the following primers (5′ to 3′) TGCCTCACTTCAGTGTCCTTGT and GACCACATATTCTGTC CCAAAGAA and detected with the TaqMan probe VIC-CCACCGGGACCCTCTTCTGGATGGATG-TAMRA, with rodent GAPDH as a control for total RNA.
Results
Disrupting the Kvlqt1 gene.
A bacterial artificial chromosome (BAC) clone containing mouse Kvlqt1 exons 1 and 2 was isolated by screening a BAC library derived from mouse 129/SvEv genomic DNA. A restriction enzyme map was established for the genomic DNA flanking exon 1 (Figure 1a). Knockout vector pMLQT1 was transfected into ES cells, and two homologous recombinants were identified by genomic Southern blot using a probe outside pMLQT1 (Figure 1a, probe). Both ES clones were used to generate chimeric mice by injection into mouse blastocysts. The chimeras were mated to C57BL/6 mice, and germline transmission of the mutant allele was confirmed by genomic Southern analysis of tail DNA from F1 progeny. Mating between heterozygous male and female mice generated F2 progeny with all three genotypes, homozygous mutant, heterozygous mutant, and wild-type mice (Figure 1b). The wild-type allele generates a 4.2-kb Pvu II fragment (Figure 1b, lanes 3, 6, and 9), and the mutant allele generates a 3.9-kb Pvu II fragment (Figure 1b, lanes 1, 4, and 7). The heterozygous mice contain both fragments (Figure 1b, lanes 2, 5, and 8).
Figure 1.

Mutational inactivation of the mouse Kvlqt1 gene. (a) Mouse Kvlqt1 genomic locus and knockout construct. Top: restriction map of exons 1 and 2; middle: 6-kb EcoRV-EcoRI fragment subcloned into pBluescript. The open box denotes the insertion of the neo gene in exon 1. The neo cassette was inserted after A345 as marked by the arrow in the following sequence: CCACTATTGA↓GCAGTATGCC. The nucleotide sequences are based on U70068. The KvLQT1 is translated from the nucleotide 104. The exon 1 is shared by all isoforms, so targeting at exon 1 inactivates all KvLQT1 isoforms. The detailed description of isoforms and their organization can be found in ref. 5. The dashed lines mark the targeted region for homologous recombination. Bottom: restriction map of the Kvlqt1 locus after recombination. A diagnostic Pvu II site generates a mutant-specific 3.9-kb fragment using the probe indicated, in contrast to a 4.2-kb Pvu II fragment in the wild-type mouse locus. (b) Genotyping of transgenic mice. Genomic DNA was digested with Pvu II and hybridized with the probe shown in a. The +/+ indicates wild-type as demonstrated by a single 4.2-kb Pvu II fragment, +/– indicates heterozygous mice as demonstrated by the presence of both 4.2- and 3.9-kb fragments, and –/– indicates homozygous mutant as demonstrated by a single 3.9-kb mutant Pvu II fragment. This is also consistent with typing by PCR (data not shown) and phenotype. (c) Presence or absence of KvLQT1 expression in wild-type and mutant mice, as measured by RT-PCR using primers spanning an intron-exon boundary. The forward primer, mLQT111 (GTGTTTCGTGTACCACTTCACCGTCTT), in exon 1a and exon 1 (across the junction) is upstream to the neo insertion site, and reverse primer, mLQT211 (TACCATTGGCTACGGGGATAAGGTACC) in exon 6 is downstream to the neo insertion site. The presence of a 1.6-kb insertion in homozygous mutant mice prevents the efficient amplification in RT-PCR reaction.
Absence of expression in the F2 homozygous mutant mice was confirmed by RT-PCR analysis (Figure 1c) and by real-time quantitative PCR (data not shown). There were 38 homozygous mutant mice (30%), 62 heterozygous mice (49%), and 27 wild-type mice (21%), indicating that the mutant mice develop normally and are viable. However, by 4 weeks, the mutant mice behaved dramatically differently from their heterozygous and wild-type littermates, exhibiting hyperactivity, with repetitive running, circling, nodding, and wobbling behaviors.
Kvlqt1 mice are deaf.
The abnormal mouse behaviors suggested a possible defect in hearing and/or balance. We therefore measured ABR in four Kvlqt1+/+, four Kvlqt1+/–, and five Kvlqt1–/– mice (Figure 2). The wild-type mice showed an average hearing threshold of 59 dB (±7.7 dB), and heterozygotes showed a threshold of 49 dB (±12.5 dB). This difference was not statistically significant. All four homozygotes for the Kvlqt1 mutation exhibited no response by ABR to the maximum level (>95 dB) of acoustic stimulation (Figure 2).
Figure 2.

ABR of Kvlqt1 knockout mice. Representative ABR hearing threshold measurements from the left ears of wild-type (a) and homozygous knockout (b) mice. The numbers to the right of each tracing represent decibels (dB) in sound pressure level (SPL) delivered to the ear. Representative ABR waveforms enabling the determination of threshold are marked in brackets. In all animals, thresholds were the same for both ears within a given mouse. Stimulus artifact generated at high decibels (>90 dB) is noted by an S.
We selected one of the homozygous mutant mice for detailed histological analysis of the inner ear. Severe abnormalities were observed in both the auditory and vestibular end organs, whereas the inner ear anatomy was normal in a hearing heterozygous mouse. Within the cochlea, Reissner’s membrane was collapsed onto the tectorial membrane, resulting in a severe contraction of the scala media, particularly in the basal end (Figure 3). Hair cells and supporting cells of the organ of Corti were absent, replaced by fibrous adhesions between the tectorial membrane and the basilar membrane. Degeneration was more severe at the basal end compared with the apical segments. Within the vestibular end organs, there was a reduction in the volume of the endolymphatic compartment of the vestibule and semicircular canals, associated with contraction and fibrosis of the membranous lining. The otolithic membrane of the utricle and saccule was separated from the neuroepithelium, and the neuroepithelia of the maculae and cristae were intact but contained fewer hair cells than did the wild type (Figure 4).
Figure 3.
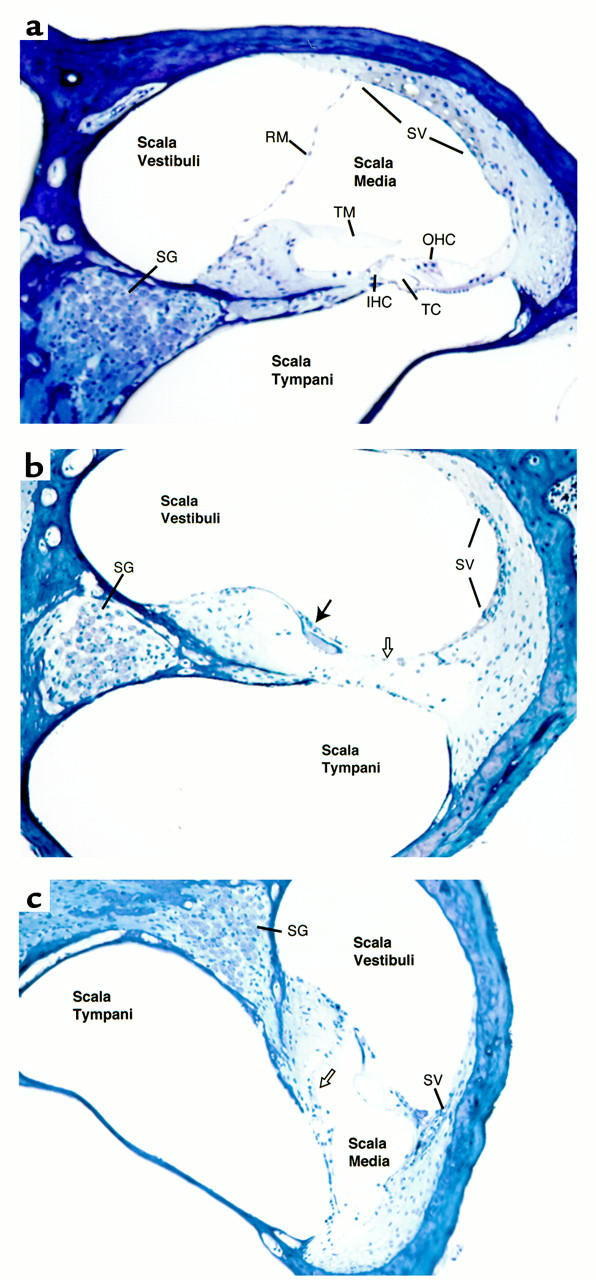
Cochlear histopathology of Kvlqt1 knockout mouse. (a) Normal histology of unaffected heterozygous mouse for comparison. (b and c) Base (b) and apex (c) of affected homozygous knockout mouse, showing complete loss of hair cells and supporting cells from the organ of Corti, which is replaced by fibrosis between the tectorial membrane and the basilar membrane (open arrow). The cell density in the spiral ganglion (SG) is decreased in the base but normal in the apex. There is marked degeneration of the stria vascularis (SV) throughout the cochlea, with more dramatic loss seen in the basal and middle half-turns. Reissner’s membrane is adherent to the spiral ligament and the tectorial membrane in the basal regions of the cochlea, resulting in the obliteration of the scala media (filled arrow in b). The reduction of the scala media volume is more severe in the base than in the apex. IHC, inner hair cells; OHC, outer hair cells; RM, Reissner’s membrane; TM, tectorial membrane; TC, tunnel of Corti.
Figure 4.

Vestibular histopathology of Kvlqt1 knockout mouse. (a) Normal histology of the utricle of unaffected heterozygous mouse for comparison. (b) Affected homozygous knockout mouse, showing marked reduction in the size of the endolymphatic compartment, associated with adhesions between the membranous envelope, the inner surface of the otic capsule and the otolithic membrane. The otolithic membrane shows irregular borders and is separated from the neuroepithelium (filled arrows). There is also a loculated collection of granular material (open arrows) in close association with the otolithic membrane. There is a marked reduction in hair cell number (identified by the presence of a white halo around the nucleus) in the neuroepithelium of the utricle in the homozygous mutant mouse (asterisk). These cells are particularly prominent in the center of the neuroepithelium in the heterozygous mutant mouse in which type I cells with large chalice-like nerve endings predominate.
Gastric hyperplasia in Kvlqt1 mutant mice.
Both homozygous and heterozygous knockout mice showed no visible external abnormality. Gross internal examination was also unremarkable, except that by 3 months of age, there was a threefold enlargement by weight of the stomach in homozygotes, compared with age-matched heterozygotes and wild-type mice. Mucosal hyperplasia (increase in the number of epithelial cells) accounted for the increased gastric wall thickness of the mutant mice. Although both the antral and fundic mucosa showed hyperplasia, the changes were more striking in the fundus (Figure 5). The majority of the fundic hyperplasia occurred in the middle (isthmic) portion of the mucosa and in the gastric neck (proliferative compartment) immediately above it. The proliferative compartment of the mucosa, as demonstrated by immunohistochemical labeling for the proliferation marker Ki-67, was expanded in both the antrum and fundus of the mutant mice (Figure 5b). The isthmic portion of the mucosa is normally occupied by parietal cells and mucous neck cells (Figure 5c). This region was markedly disorganized in the mutant mice, and normal appearing parietal cells were decreased in number (Figure 5d). While PAS-staining mucin cells in the normal mucosa were confined to the surface and mucous neck cells (Figure 5c), the gastric glands of the knockout mice were dominated by PAS-positive cells located in the mid- to basal regions of the glands (Figure 5d). Surface cells in the mutant mice appeared relatively depleted of PAS-positive mucin (Figure 5d). The isthmic regions of the glands were dilated and appeared populated by mucin-secreting cells and vacuolated parietal cells (Figure 5, e and f). The vacuoles did not appear to be immunoreactive for H/K-ATPase and were also not immunoreactive for a canalicular marker, ezrin (data not shown).
Figure 5.

Gastric pathology in Kvlqt1 knockout mice. Compared with paraffin sections of wild-type fundic mucosa (a), Kvlqt1 knockout mouse fundic mucosa (b) showed increased epithelial cell proliferation as measured by Ki67 indirect immunoperoxidase. In both cases, however, the proliferative zone was properly placed in the isthmic region of the glands. This zone was expanded in the knockout mouse, and the overall mucosal thickness was increased owing to increased numbers of epithelial cells in the deeper compartments. PAS staining in gastric fundus of wild-type mice (c) and homozygous mutant mice (d) was largely confined to the apical portions of normal foveolar epithelial cells. In mutant mice, a broad zone of less intensely staining cells was present in the midportion of the mucosa, and the PAS staining of surface mucous cells was reduced (d). Hematoxylin and eosin staining of cells in the neck region of the mutant mice (e) showed dilated glands with many mucinous cells and vacuolated parietal cells. Immunostaining for H/K-ATPase (f) demonstrated staining in the cytoplasm of these parietal cells without staining of the intracellular vacuoles. Immunostaining of mucous neck cells for spasmolytic polypeptide in wild-type mice (g) showed staining of a small number of cells in the isthmic region. Prominent spasmolytic polypeptide staining was observed in knockout mouse hyperplastic mucous cells (h) along the entire length of the gland. Immunostaining for intrinsic factor in wild-type (i) and knockout (j) mice showed decreased numbers of chief cells in mutant mice. Bar, 40 μm (a, b, i, and j), 20 μm (c and d), 10 μm (e and f), and 60 μm (g and h).
The expanded mucin-secreting cell population was not due to foveolar hyperplasia, but rather represented an expansion of mucous neck cells. Thus, although normal mucosa contains only a small number of spasmolytic polypeptide-expressing mucous neck cells (Figure 5g), the fundic glands of Kvlqt1 knockout mice were dominated by spasmolytic polypeptide-expressing mucous neck cells deep to the proliferative zone (Figure 5h). In knockout mice intrinsic factor-expressing chief cells appeared smaller and were present in decreased numbers. These results suggest that the Kvlqt1 knockout has altered the normal cellular repertoire of lineage maturation in the gastric mucosa.
To confirm that Kvlqt1 is expressed in the normal stomach, we used real-time quantitative PCR (RTQ-PCR) analysis using an ABI Prism 7700 Sequence Detection System. RTQ-PCR was used to examine the expression of Kvlqt1 in serial cross sections of formalin fixed stomach. Kvlqt1 was expressed in all tissue layers examined of normal mice, including esophagus, antrum, fundus, body, and duodenum, and there was no expression in the stomach in the homozygous mutant (data not shown). Furthermore, Kvlqt1 was expressed in both parietal and chief cells at approximately equal levels, as measured by laser capture microdissection of fresh frozen stomach mucosa from unaffected littermates, followed by RTQ-PCR.
Given the strikingly abnormal morphological appearance of the gastric parietal cells, the cells that secrete gastric acid, we hypothesized that acid production might be impaired in the mutant mice. Indeed, the homozygous mice were hypochlorhydric (gastric lumen pH 6–7) compared to the normal wild-type and heterozygous counterparts (pH 1–2). Consistent with the observed hypochlorhydria, the mutant mice had elevated serum gastrin levels (90, 200 pM) compared with age-matched heterozygous littermates (32, 21 pM) suggesting a functional deficit in parietal cell secretion.
Normal cardiac electrophysiology in knockout mice.
Electrocardiographic analysis of the mice included measurement of baseline intervals, HR and QT variability. KvLQT1 is a component of the delayed rectifier implicated in the chromosome 11 form of the congenital long QT syndrome (15). KvLQT1 and the accessory protein minK comprise the slow component of the delayed rectifier current (IKs). Expression of IKs in the adult mouse heart is controversial. We examined the ECGs of the knockout mice, control littermates, and heterozygotes. The T waves were flat or obscured by the subsequent P wave, precluding quantitative analysis of repolarization in both groups of animals (three of 12 control and heterozygotes, seven of 11 knockout mice). ECG assessment of repolarization revealed no significant difference between the wild-type, heterozygotes, and knockout mice in the QT interval and QT variability when T waves were apparent.
It is notable that the minK protein appears to be enriched in the specialized conducting system of the mouse heart (16). Similar localization of KvLQT1 is presumed but has not been demonstrated. Sinus node function and autonomic innervation were assessed by measurement of heart rate and heart rate variability. Under conditions of light anesthesia, the heart rate and heart rate variability was not different among the three groups of mice. Heart rate variability did not significantly differ between male and female mice in any of the groups (see supplemental figure at http://www.jci.org/cgi/content/full/106/12/1447/DC1). There was no significant difference in atrioventricular conduction as assessed by the PR interval at comparable heart rates. The PR intervals of the controls and heterozygotes was 68 ± 12 (n = 12) compared with 60 ± 10 milliseconds (n = 11, P = 0.08) for the knockouts, with mean heart rates of 409 ± 36 bpm and 412 ± 52 bpm, respectively. There is no evidence for intraventricular conduction delay in the knockout mice; the QRS duration (24 ± 2 milliseconds) is similar to that of wild-type and heterozygous mice (24 ± 2 milliseconds; P = 0.52). Similarly, there is no significant difference in the PR and QRS duration at comparable heart rates among male and female mice of any group.
No features of BWS in mutant mice.
Ten homozygous knockout mice, from 2 days to 3 months of age were examined with complete gross and histopathological examination and compared to age-matched heterozygous or wild-type littermates, except for one female at 3 months of age who was compared to an aged-matched, nonlittermate female. Birth weights were identical, and no abnormalities were seen in any organ system other than the inner ear and stomach, as already noted here.
Discussion
The most obvious feature of the Kvlqt1 knockout mouse was deafness, with no measurable audiological response to sounds at maximum levels of stimulation (>95 dB). This deafness is associated with a loss of the sensory epithelium, atrophy of the stria vascularis, and collapse of the endolymphatic space in an adult mouse. The Kvlqt1-encoded potassium channel is expressed in the stria vascularis of mice (4), where it may play an important role in the ionic maintenance of endolymph, the potassium-rich fluid bathing neuroepithelia of the inner ear. The strial dysfunction resulting from the Kvlqt1 mutation may be responsible for the accelerated collapse and degeneration of the scalia media and its contents, or the abnormal development of the inner ear. The Kvlqt1 knockout mouse shows a similar pattern of cochlear degeneration to deaf mice with mutations of the Isk gene, which produces a protein that coassembles with the Kvlqt1 channel (17). The gradual disruption of normal cochlear anatomy within the first days of life in Isk mutant mice suggests that the Kvlqt1 mutation may also affect the normal maintenance of inner ear structure and function by the stria vascularis, rather than its primary organogenesis. However, understanding the spectrum of strial degeneration will require additional developmental studies in progress.
As in the homozygous Kvlqt1 knockout mouse, an abnormal KvLQT1 gene is also associated with deafness in the Jervell and Lange-Nielsen syndrome (JLNS) in humans (4). In JLNS, as in the Kvlqt1 knockout mouse, collapse of the membranous labyrinth and degeneration of sensory neuroepithelia involve all vestibular end organs and the cochlea, in association with severe atrophy of the stria vascularis.
The gastric hyperplasia in the Kvlqt1 knockout mouse was remarkable and surprising, as Kvlqt1 was not known to play a role in stomach development or function. Previous detailed studies by Karam and Leblond (18) have demonstrated that fundic glandular lineages are derived from second-order stem cells (presurface, preparietal, and preneck cells) that differentiate from a common precursor located near the gland lumen. Alterations in the differentiation of second-order stem cells can lead to marked changes in the mucosal cell lineage repertoire within gastric glands (19). The Kvlqt1 knockout causes an alteration in the lineage repertoire in the fundic mucosa, with mucous neck cell hyperplasia and production of vacuolated, nonfunctional parietal cells. Hypergastrinemia in these mice is consistent with the observed profound hypochlorhydria. These results along with the observed vacuolation of the parietal cells suggest a functional deficit in gastric parietal cells. Parietal cells are also known to secrete a number of critical growth factors such as TGF-α and HB-EGF, so it is possible that some of the sequelae of lineage changes (e.g., mucous neck cell hyperplasia) are secondary to other losses in parietal cell function. Alternatively, the changes in mucosal lineage production may be a direct result of the loss of the channel protein in mucosal stem cells.
The phenotypic pattern in the Kvlqt1 knockout mice differs from most of the previously reported models of achlorhydria-associated gastropathy. For example, mice infected with Helicobacter felis also exhibit mucous neck cell hyperplasia in the fundus; however, there is a profound loss of other fundic glandular components, including parietal and chief cells, and the antrum appears normal (20). Mice with deletion of another ion channel, the NHE-2 exchanger, show a marked reduction in parietal cell numbers and the remaining parietal cells also are vacuolated (21), but in that model, there is a decrease in mucosal height and stomach size. However, there are intriguing similarities between the Kvlqt1 knockout and a mouse model created by deletion of the β subunit of the H/K-ATPase (22). These mice similarly develop gastric hyperplasia at maturity, are achlorhydric, have high serum gastrin levels, and demonstrate parietal cell vacuolization. One key difference is that the striking mucous neck cell expansion seen in our Kvlqt1 knockouts was apparently not noted in the H/K-ATPase knockout, though the results of PAS and spasmolytic polypeptide stains that would help identify such cells were not reported in the latter study (22). Nonetheless, given the phenotypical and morphological similarities of these knockouts, we speculate that Kvlqt1 may be required for normal acid secretion, perhaps by maintaining intracellular potassium at a relatively low level to permit H/K exchange. Interestingly, Kvlqt1 was recently postulated to interact with a novel subunit (KCNE3) to produce a constitutively open potassium channel in the intestine (23), suggesting that Kvlqt1 may serve diverse roles in different tissues depending on the interacting subunit.
KvLQT1 and min K underlie the slow component of the delayed rectifier current (IKs) in human heart. Mutation of either component produces autosomal dominant and recessive forms of the inherited arrhythmia known as the long QT syndrome. The delayed rectifier currents are a crucial component of the repolarization machinery in human ventricle; however, the role of the delayed rectifier potassium currents in the electrophysiology of the adult mouse heart is controversial. Cellular electrophysiological studies suggest that IKr and to a lesser extent IKs play a role in repolarization of the fetal and neonatal ventricle (24–27), but Ito supplants both of these currents in the adult heart. In fact IKs is not detectable in the adult murine heart (27). Targeted deletion of min k has produced divergent electrocardiographic phenotypes; in one knockout mouse rate dependent QT prolongation was observed, but there was no prolongation of the cellular action potential duration (28). In another min k knockout mouse the ECG phenotype was not different from wild-type (16).
Although min K and KvLQT1 combine to form IKs, the effects of homozygous deletion of min k on ventricular repolarization are not comparable to knockout of Kvlqt1. Several lines of evidence suggest that min K modulates other K currents in the heart. Kupershmidt et al. found that IKr is significantly reduced and its deactivation slowed in neonatal ventricular myocytes isolated from min k knockout mice (16). E-4031, a specific blocker of IKr (encoded by MERG + MiRP1) consistently prolonged repolarization in min k+/+ but not in min k–/– mice. By contrast, the chromanol compound 293B, a specific blocker of KvLQT1 K channels, produced comparable effects on repolarization in min k–/– and min k+/+ mice (28). Heterologous expression studies of min K with HERG (29) and reduction in the density of IKr in AT-1 cells by antisense knock down of min K provide further evidence for the interaction of the min k gene product with ERG (30).
We observed no ECG phenotype in the Kvlqt1 knockout mice. The basal heart rate, heart rate variability, and ECG intervals were not different from wild-type and heterozygous littermates. In a limited number of animals a robust, typically inverted T wave was recorded. Analysis of the QT intervals and QT variability in these animals revealed no differences between controls and knockouts. The absence of QT prolongation is consistent with the developmental downregulation of IKs in the murine ventricle (26–28).
It is notable that the min K protein appears to be enriched in the specialized conducting system of the mouse heart (16). Similar localization of KvLQT1 is presumed but has not been demonstrated. Interestingly transgenic studies have suggested a role for IKs in regulation of heart rate and impulse propagation through the atrioventricular node (28, 31). These data suggested to us the possibility that instead of repolarization, activation of the heart might be altered in the Kvlqt1 knockout mouse. We found no evidence for a difference in heart rate or heart rate variability in the knockout mice. Atrioventricular conduction as assessed by the PR interval on the ECG was not different among the groups and displayed similar behavior in response to different heart rates. The PR intervals in this study are somewhat longer than those measured by Berul et al.; however, the average HR at which the measurements were made was 100 bpm slower (32). Finally the activation time of the ventricle, the QRS duration, was the same in all groups. In addition when these parameters were compared among gender-matched animals there were no differences between knockout and control mice (data not shown).
Finally, we were interested in clarifying the relationship between KvLQT1 and BWS, as we have shown that this gene is the frequent target of germline balanced chromosomal rearrangements in BWS. These rearrangements could act directly on KvLQT1 or inactivate a nearby gene through a position effect (5). As expected, there was no phenotype in the heterozygous knockout mice, as unlike in humans, Kvlqt1 is only imprinted early in development in the mouse (33, 34). However, even the homozygous knockout mice showed no stigmata of BWS. This absence of a BWS phenotype is of great interest, and it suggests that KvLQT1 itself plays no direct role in BWS, which is also consistent with absence of KvLQT1 mutations in BWS patients (M.P. Lee et al., unpublished observations). It may be the chromosomal rearrangements exerting a position effect on other nearby genes. These genes could be p57KIP2, which is mutated in a small number of patients with BWS (35, 36), LIT1, a novel antisense transcript within KvLQT1 that undergoes loss of imprinting in most patients with BWS (37), or TSSC5, which shows rare mutations in cancers (38). LIT1 would be unaffected by this knockout, as it is separated by 200 kb and is on the opposite strand. Although additional mouse models and human studies will be needed to resolve the molecular pathology of BWS, the model described here demonstrates a critical role for Kvlqt1 in the inner ear, and it suggests a novel role for potassium channels in the regulation of gastric acid secretion in the stomach. The generation of this knockout mouse will also allow a genetic approach to the study of potassium channel physiology, by crossing these animals with mice deficient for other channel subunits.
Acknowledgments
This work was supported by NIH grants to A.P. Feinberg. We thank G. Dockray (University of Liverpool, Liverpool, United Kingdom) for assistance with the Gastrin assays, D. Sidransky for assistance with laser capture microdissection, and H. Uejima for assistance with Figure 5.
References
- 1.Wang Q, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 2.Schulze-Bahr E, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267–268. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- 3.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 4.Neyroud N, et al. A novel mutation in the potassium channel gene KvLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–189. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- 5.Lee MP, Hu R-J, Johnson LA, Feinberg AP. Human KvLQT1 shows tissue-specific imprinting and is physically disrupted by Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat Genet. 1997;15:181–185. doi: 10.1038/ng0297-181. [DOI] [PubMed] [Google Scholar]
- 6.Feinberg AP, Kalikin LM, Johnson LA, Thompson JS. Loss of imprinting in human cancer. Cold Spring Harb Symp Quant Biol. 1994;59:357–364. doi: 10.1101/sqb.1994.059.01.040. [DOI] [PubMed] [Google Scholar]
- 7.Mannens M, Wilde A. KvLQT1, the rhythm of imprinting. Nat Genet. 1997;15:113–114. doi: 10.1038/ng0297-113. [DOI] [PubMed] [Google Scholar]
- 8.Doucet JR, Ryugo DK. Projections from the ventral cochlear nucleus to the dorsal cochlear nucleus in rats. J Comp Neurol. 1997;385:245–264. [PubMed] [Google Scholar]
- 9.Nadol JB, Jr, Ketten DR, Burgess BJ. Otopathology in a case of multichannel cochlear implantation. Laryngoscope. 1994;104:299–303. doi: 10.1288/00005537-199403000-00010. [DOI] [PubMed] [Google Scholar]
- 10.Berger RD, et al. Beat-to-beat QT interval variability: novel evidence for repolarization lability in ischemic and nonischemic dilated cardiomyopathy. Circulation. 1997;96:1557–1565. doi: 10.1161/01.cir.96.5.1557. [DOI] [PubMed] [Google Scholar]
- 11.Berger RD, Akselrod S, Gordon D, Cohen RJ. An efficient algorithm for spectral analysis of heart rate variability. IEEE Trans Biomed Eng. 1986;33:900–904. doi: 10.1109/TBME.1986.325789. [DOI] [PubMed] [Google Scholar]
- 12.Varro A, Voronina S, Dockray GJ. Pathways of processing of the gastrin precursor in rat antral mucosa. J Clin Invest. 1995;95:1642–1649. doi: 10.1172/JCI117839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Argani P, et al. Olfactory neuroblastoma is not related to the Ewing family of tumors: absence of EWS/FLI1 gene fusion and MIC2 expression. Am J Surg Pathol. 1998;22:391–398. doi: 10.1097/00000478-199804000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Argani P, Zakowski MF, Klimstra DS, Rosai J, Ladanyi M. Detection of the SYT-SSX chimeric RNA of synovial sarcoma in paraffin-embedded tissue and its application in problematic cases. Mod Pathol. 1998;11:65–71. [PubMed] [Google Scholar]
- 15.Wang Q, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 16.Kupershmidt S, et al. Replacement by homologous recombination of the minK gene with lacZ reveals restriction of minK expression to the mouse cardiac conduction system. Circ Res. 1999;84:146–152. doi: 10.1161/01.res.84.2.146. [DOI] [PubMed] [Google Scholar]
- 17.Vetter DE, et al. Inner ear defects induced by null mutation of the Isk gene. Neuron. 1996;17:1251–1264. doi: 10.1016/s0896-6273(00)80255-x. [DOI] [PubMed] [Google Scholar]
- 18.Karam SM, Leblond CP. Dynamics of epithelial cells in the corpus of the mouse stomach. III. Inward migration of neck cells followed by progressive transformation into zymogenic cells. Anat Rec. 1993;236:297–313. doi: 10.1002/ar.1092360204. [DOI] [PubMed] [Google Scholar]
- 19.Goldenring JR, et al. Overexpression of transforming growth factor-alpha alters differentiation of gastric cell lineages. Dig Dis Sci. 1996;41:773–784. doi: 10.1007/BF02213134. [DOI] [PubMed] [Google Scholar]
- 20.Fox JG, et al. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology. 1996;110:155–166. doi: 10.1053/gast.1996.v110.pm8536852. [DOI] [PubMed] [Google Scholar]
- 21.Schultheis PJ, et al. Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. J Clin Invest. 1998;101:1243–1253. doi: 10.1172/JCI1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scarff KL, Judd LM, Toh BH, Gleeson PA, Van Driel IR. Gastric H(+),K(+)-adenosine triphosphatase beta subunit is required for normal function, development, and membrane structure of mouse parietal cells. Gastroenterology. 1999;117:605–618. doi: 10.1016/s0016-5085(99)70453-1. [DOI] [PubMed] [Google Scholar]
- 23.Schroeder BC, et al. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–199. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- 24.Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. J Physiol. 1994;479:265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies MP, et al. Developmental changes in ionic channel activity in the embryonic murine heart. Circ Res. 1996;78:15–25. doi: 10.1161/01.res.78.1.15. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Duff HJ. Identification and characteristics of delayed rectifier K+ current in fetal mouse ventricular myocytes. Am J Physiol. 1996;270:H2088–H2093. doi: 10.1152/ajpheart.1996.270.6.H2088. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Feng ZP, Kondo CS, Sheldon RS, Duff HJ. Developmental changes in the delayed rectifier K+ channels in mouse heart. Circ Res. 1996;79:79–85. doi: 10.1161/01.res.79.1.79. [DOI] [PubMed] [Google Scholar]
- 28.Charpentier F, Merot J, Riochet D, Le Marec H, Escande D. Adult KCNE1-knockout mice exhibit a mild cardiac cellular phenotype. Biochem Biophys Res Commun. 1998;251:806–810. doi: 10.1006/bbrc.1998.9554. [DOI] [PubMed] [Google Scholar]
- 29.McDonald TV, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr) Nature. 1997;388:289–292. doi: 10.1038/40882. [DOI] [PubMed] [Google Scholar]
- 30.Yang T, Kupershmidt S, Roden DM. Anti-minK antisense decreases the amplitude of the rapidly activating cardiac delayed rectifier K+ current. Circ Res. 1995;77:1246–1253. doi: 10.1161/01.res.77.6.1246. [DOI] [PubMed] [Google Scholar]
- 31.Demolombe S, et al. Atrio-venticular block and long QT in KvLQT1 deficient transgenic mice. Circulation. 1999;100:351. [Google Scholar]
- 32.Berul CI, Aronovitz MJ, Wang PJ, Mendelsohn ME. In vivo cardiac electrophysiology studies in the mouse. Circulation. 1996;94:2641–2648. doi: 10.1161/01.cir.94.10.2641. [DOI] [PubMed] [Google Scholar]
- 33.Jiang S, Hemann MA, Lee MP, Feinberg AP. Strain-dependent developmental relaxation of imprinting of an endogenous mouse gene, Kvlqt1. Genomics. 1998;53:395–399. doi: 10.1006/geno.1998.5511. [DOI] [PubMed] [Google Scholar]
- 34.Gould TD, Pfeifer K. Imprinting of mouse Kvlqt1 is developmentally regulated. Hum Mol Genet. 1998;7:483–487. doi: 10.1093/hmg/7.3.483. [DOI] [PubMed] [Google Scholar]
- 35.Hatada H, et al. An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet. 1996;14:171–173. doi: 10.1038/ng1096-171. [DOI] [PubMed] [Google Scholar]
- 36.Lee MP, DeBaun M, Randhawa GS, Reichard BA, Feinberg AP. Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am J Hum Genet. 1997;61:304–309. doi: 10.1086/514858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee MP, et al. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci USA. 1999;96:5203–5208. doi: 10.1073/pnas.96.9.5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee MP, et al. Somatic mutation of TSSC5, a novel imprinted gene from human chromosome 11p15.5. Cancer Res. 1998;58:4155–4159. [PubMed] [Google Scholar]