

Abstract
Objectives:
This paper describes the clinical diagnosis of Proteus syndrome (PS) in children referred for evaluation of asymmetric disproportionate overgrowth.
Materials and Methods:
Retrospective, descriptive, cross-sectional study conducted from January 1998 to December 2010.
Results:
During the study period, 2011 new patients were evaluated. Thirteen (0.65%) patients presented features suggestive of PS. These patients were formally evaluated based on the revised diagnostic criteria proposed by Biesecker. The mean age was 6.92 ± 5.1 years. Ten patients (76.9%) were females. All subjects had asymmetric disproportionate overgrowth. Other dysmorphic features were as follows: macrodactily (84.6%); linear epidermal nevus (41.6%); hemangioma (30.7%); and lipoma (23%). Six patients fulfilled the diagnostic criteria for PS.
Conclusions:
The diagnostic rate of only 46.1% of patients with PS confirms the diagnostic difficulties and the need for continuous monitoring and periodic review of these patients since the clinical manifestations of this syndrome become more evident with aging. Molecular tests may help the differential diagnosis of Proteus syndrome when they became commercially available.
Keywords: Asymmetric overgrowth, diagnostic criteria, proteus syndrome
INTRODUCTION
Proteus syndrome (PS) was first described in 1979. In 1983, Rudolf Wiedemann, a German Pediatrician, named it Proteus Syndrome for a Greek sea-God, who could change his shape at will assuming many forms to escape capture. This name was given to represent the variable clinical manifestations seen in the first patients identified with this syndrome and the morphological changes of its presentation and evolution.[1,2] It is a hamartomatous syndrome, with clinical manifestations that vary greatly and predominance of malformations and overgrowth of multiple organs. It is an extremely rare syndrome with an estimated prevalence of approximately 1:1.000.000, being more common among males at a ratio of 1:9:1.[1,2]
This disease is characterized by a mosaic distribution, sporadic occurrence, and progressive course, hyperplasia of connective tissue, vascular malformations, epidermal nevus, and hyperostosis.[1] Clinical features may be present at birth but typically develop over time, starting between 1 and 18 months of age. The postnatal, progressive, and asymmetrical overgrowth occurs in a mosaic pattern. Bone, connective tissue, fat, central nervous system, eye, spleen, thymus, and colon are commonly involved tissues.[1,2,3,4,5]
PS is probably caused by a post-zygotic mutation resulting in mosaicism, which possibly explains the variability of manifestations and the monozygotic twins discordant for the syndrome.[6,7] The association of PS with mutations in the PTEN suppressor gene is uncertain, being reported by Zhou et al.[7] and by Smith et al.[8] Nevertheless, Barker et al.[9] and Thiffault et al.[10] did not find mutations in the PTEN gene among the patients with PS included in their studies. Although there are reports of these mutations in PS, they are usually absent in the affected individuals.[8,11,12] A recent study, with 29 PS patients, revealed that an activating somatic mutation of the AKT1 oncogene kinase, an enzyme involved in cell proliferation, was present in 26 of these patients concluding that this mutation was the cause of PS.[13]
The rarity of the syndrome, the wide spectrum of presentation, the lack of an easily available diagnostic test, and the occurrence of syndromes with similar phenotypes contribute to the diagnostic challenge.[2] Due to the lack of commercially available genetic tests, knowledge of the multiple presentations and evolution of PS is still needed for proper clinical diagnosis.[14]
Although the use of diagnostic criteria has its flaws, the benefit of its use is justified because it classifies and defines, through specific criteria, a group of individuals who have similar clinical and prognostic features.[2]
Given the present difficulty in diagnosing PS, this study describes a series of patients with such diagnosis, using the criteria proposed by Biesecker et al.[1]
MATERIALS AND METHODS
Between January 1998 and December 2010, 2011 new patients were seen at the Genetics and Pediatric Endocrinology Outpatient Clinics of a University Hospital. The records of all cases referred due to asymmetric overgrowth and clinical suspicion of Proteus syndrome were identified and retrospectively analyzed—a retrospective chart review. Thirteen patients of the total number of patients seen in the study period (0.65%) had features compatible with the syndrome. These patients were evaluated according to the diagnostic criteria proposed by Biesecker et al.[1] being classified in two groups: General and specific criteria. The general criteria are mandatory and include mosaic distribution, sporadic occurrence, and progressive course. The specific criteria are classified into three subgroups named A, B, and C. One feature of subgroup A or two features of B, or else three features of C must be present to confirm the diagnosis [Table 1].
Table 1.

The present study was approved by our institution Research Ethics Committee. As the study involved review of medical records, patient informed consent was not required, with preservation of the confidentiality of data.
RESULTS
This study included 13 patients with an initial diagnostic suggestive of PS that is asymmetric overgrowth. The average age of the patient at first visit was 6.92 ± 5.1 years. Ten patients were females (76.9%). Only one patient was the son of consanguineous parents.
All 13 patients met the general diagnostic criteria proposed by Biesecker et al.[1]: Mosaic distribution, sporadic occurrence, and progressive course. Only when evaluated by the Biesecker specific criteria, the distinction between PS and non-PS was possible. All patients had asymmetric overgrowth, 11: Macrodactily (84.6%), 5: Epidermal nevus (41.6%), 4: Hemangioma (30.7%), 3: Lipoma (23%), 5: Linear epidermal nevus (41.6%) and 3: Connective tissue nevus (23%) [Table 2]. Of these 13 patients, the clinical diagnosis of PS was confirmed in 6 (46.1%): 6 (100%) patients fulfilled criteria 2 B, 3 (50%) patients fulfilled criteria 1A, and none of them fulfilled criteria 3C [Table 2].
Table 2.
Clinical findings in patients with a possible diagnosis of proteus syndrome
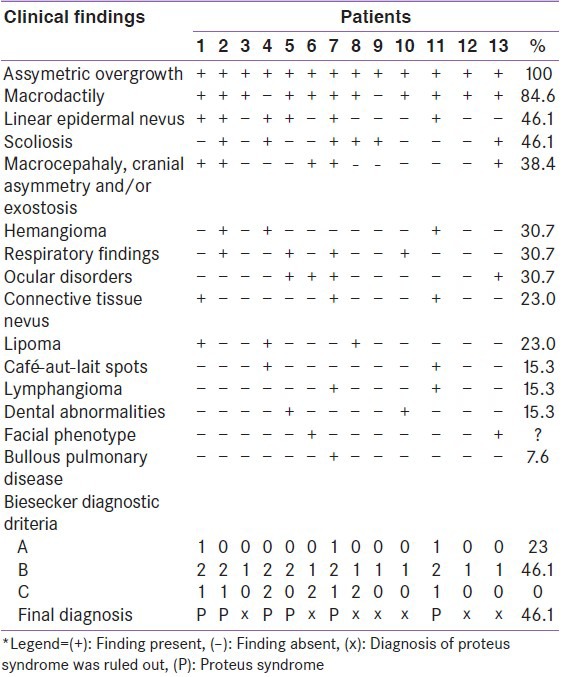
In the group where PS was not confirmed (53.8%): None of the patients met criteria 1A, all patients met criteria 1B (100%), and 2 met criteria 1 C (28.5%).
Linear epidermal nevus was present in all patients with a confirmed diagnosis of PS, whereas none of the patients in whom the diagnosis of PS was not confirmed had this clinical feature [Table 2].
DISCUSSION
The rarity of PS may be underestimated in some series, such as ours, due to young age of the patients and may be overestimated, in others series, due to the lack of stringent diagnostic criteria including other hamartomatous or hyperplastic disorders.[1,2,4,5] In this study, due to the young age of the children (6.92 ± 5.1 years), it is possible that greater number of cases of PS may be diagnosed in the future due to the progressive nature of the syndrome.
In 1999, Biesecker et al.[1] presented their recommendations for the diagnosis of PS in an attempt to reduce the number of false clinical diagnoses of the syndrome. Since then, this is the criteria most frequently used in clinical practice for the diagnosis of PS.[14,15,16,17] That's why it was selected for evaluating the patients of the present study.
All patients had asymmetric overgrowth since childhood, which is consistent with the literature.[2] The linear epidermal nevus was present in all six patients who met the diagnostic criteria for PS. This abnormality is described in the literature as a specific finding[2] though it has not been found in all cases according to Turner et al.[5] Little is known about the natural history of the cerebriform connective tissue nevus. Beachkofsky et al.[15] demonstrated that, in PS, this lesion tends to develop during childhood, affecting most commonly the sole of the feet, extending over the affected area or developing new lesions, and with a tendency to maintaining stable in adulthood. None of the patients in this series had this lesion.
In addition to the difficulty in confirming the diagnosis of PS, it is important to stress that there are also unusual features such as the presence of lung cyst.[16] This finding was observed in only one case reported in this study (case 7), whereas its prevalence, according to the literature, is approximately of 10%.[11] Although usually asymptomatic, lung cysts may present with respiratory failure. Other important findings of PS include central nervous system manifestations (40%), mental (30%), ophthalmological disabilities (42%), and urological abnormalities (9%).[4,5]
The differential diagnosis of PS must be made with other hamartomatous disorders such as Klippel-Trenaunay-Weber (asymmetry in one limb and hemangioma), Maffucci disease (enchondromatosis and hemangioma), Ollier's disease (enchondromatosis), neurofibromatosis type I (macrocephaly, café-au-lait spots, subcutaneous neurofibromas), Bannayan-Zonana syndrome (macrocephaly, craniofacial abnormalities), hemihyperplasia and multiple lipomatosis syndrome (HHML) and other disorders that present with hemihyperplasia.[17] The seven patients in this study who did not meet diagnostic criteria for PS did not fulfill criteria to be diagnosed with any of these disorders. This lack of diagnosis may be due to the young age of the patients.
The risk of tumor development appears to be higher in patients with PS than in the general population, so they should be assessed on a regular basis. Most tumors associated with PS are benign (e.g., monomorphic adenomas, bilateral ovarian cystadenomas), but 19% are malignant.[5]
The clinical features may worsen during the course of the patient's life. Although some authors believe that the progression of PS stops around 15-17 years, one reported case included a 23-year-old man.[11]
Patients are at increased risk of premature death, usually caused by deep vein thrombosis, pulmonary embolism, and pneumonia.[1,7] Premature death is more common among males (3.25:1) particularly boys aged less than 10 years.[3]
Once diagnosis is made, assessment must be provided by a multidisciplinary staff. Bone abnormalities and its accompanying asymmetries must be treated to preserve the functionality of the affected limb.
Although our paper has the limitation of being a retrospective study, the Biesecker diagnostic criteria were applied consistently trough information obtained in structured medical charts. PS was diagnosed in 6 of 13 (41%) patients evaluated with this diagnostic suspicion and in 6 of 2011 patients (0.3%) seen in the study period. These data confirms the rarity of PS and the highlights the importance of studies validating the clinical diagnosis even when of retrospective nature.
Since molecular diagnosis of PS were just recently reported and are not yet commercially available, the authors emphasize the importance of using strict clinical diagnostic criteria. With the establishment of clinical diagnosis, preventive and therapeutic procedures could be implemented early. Unconfirmed cases should be monitored lifetime because of the progressive course of the disease.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham JM, Jr, Viljoen DL, et al. Proteus syndrome: Diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389–95. doi: 10.1002/(sici)1096-8628(19990611)84:5<389::aid-ajmg1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Biesecker L. The challenges of Proteus syndrome: Diagnosis and management. Eur J Hum Genet. 2006;14:1151–7. doi: 10.1038/sj.ejhg.5201638. [DOI] [PubMed] [Google Scholar]
- 3.Cohen MM., Jr Causes of premature death in Proteus syndrome. Am J Med Genet. 2001;101:1–3. doi: 10.1002/ajmg.1311. [DOI] [PubMed] [Google Scholar]
- 4.Cohen MM., Jr Proteus syndrome: An update. Am J Med Genet C Semin Med Genet. 2005;137C:38–52. doi: 10.1002/ajmg.c.30063. [DOI] [PubMed] [Google Scholar]
- 5.Turner JT, Cohen MM, Jr, Biesecker LG. Reassessment of the Proteus syndrome literature: Application of diagnostic criteria to published Cases. Am J Med Genet A. 2004;130A:111–22. doi: 10.1002/ajmg.a.30327. [DOI] [PubMed] [Google Scholar]
- 6.Brockmann K, Happle R, Oeffner F, König A. Monozygotic twins discordant for Proteus syndrome. Am J Med Genet A. 2008;146A:2122–5. doi: 10.1002/ajmg.a.32417. [DOI] [PubMed] [Google Scholar]
- 7.Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. 2000;9:765–8. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 8.Smith JM, Kirk EP, Theodosopoulos G, Marshall GM, Walker J, Rogers M, et al. Germline mutation of the tumour suppressor PTEN in Proteus syndrome. J Med Genet. 2002;39:937–40. doi: 10.1136/jmg.39.12.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barker K, Martinez A, Wang R, Bevan S, Murday V, Shipley J, et al. PTEN mutations are uncommon in Proteus syndrome. J Med Genet. 2001;38:480–1. doi: 10.1136/jmg.38.7.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiffault I, Schwartz CE, Der Kaloustian V, Foulkes WD. Mutation analysis of the tumor suppressor PTEN and the glypican 3 (GPC3) gene in patients diagnosed with Proteus syndrome. Am J Med Genet A. 2004;130A:123–7. doi: 10.1002/ajmg.a.30335. [DOI] [PubMed] [Google Scholar]
- 11.Li CY, Chang YL, Chen WC, Lee YC. Pulmonary manifestations and management of Proteus syndrome. J Formos Med Assoc. 2010;109:397–400. doi: 10.1016/S0929-6646(10)60069-1. [DOI] [PubMed] [Google Scholar]
- 12.Di Stefani A, Gabellini M, Ferlosio A, Spagnoli LG, Chimenti S, Orlandi A. Cerebriform plantar hyperplasia: The clinical-pathological hallmark of Proteus syndrome. Acta Derm Venereol. 2011;91:580–1. doi: 10.2340/00015555-1087. [DOI] [PubMed] [Google Scholar]
- 13.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–19. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demir MK. Case 131: Proteus Syndrome. Radiology. 2008;246:974–9. doi: 10.1148/radiol.2463042155. [DOI] [PubMed] [Google Scholar]
- 15.Beachkofsky TM, Sapp JC, Biesecker LG, Darling TN. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol. 2010;63:799–804. doi: 10.1016/j.jaad.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim GY, Kim OH, Kim HW, Lee KS, Kang KH, Song HR, et al. Pulmonary manifestations in Proteus syndrome: Pulmonary varicosities and bullous lung disease. Am J Med Genet A. 2011;155:865–9. doi: 10.1002/ajmg.a.33926. [DOI] [PubMed] [Google Scholar]
- 17.Elsayes KM, Menias CO, Dillman JR, Platt JF, Willatt JM, Heiken JP. Vascular malformations and hemangiomatosis syndromes: Spectrum of imaging manifestations. AJR Am J Roentgenol. 2008;190:1291–99. doi: 10.2214/AJR.07.2779. [DOI] [PubMed] [Google Scholar]