

Abstract
Atherogenic dyslipidemia (AD) refers to elevated levels of triglycerides (TG) and small-dense low-density lipoprotein and low levels of high-density lipoprotein cholesterol (HDL-C). In addition, elevated levels of large TG rich very low-density lipoproteins, apolipoprotein B and oxidised low-density lipoprotein (LDL), and reduced levels of small high-density lipoproteins plays a critical role in AD. All three elements of AD per se have been recognised as independent risk factor for cardiovascular disease. LDL-C/HDL-C ratio has shown excellent risk prediction of coronary heart disease than either of the two risk markers. Asian Indians have a higher prevalence of AD than western population due to higher physical inactivity, low exercise and diet deficient in polyunsaturated fatty acids (PUFA). The AD can be well managed by therapeutic lifestyle changes with increased physical activities, regular exercise, and diets low in carbohydrates and high in PUFA such as omega-3-fatty acids, as the primary intervention. This can be supplemented drug therapies such as statin monotherapy or combination therapy with niacin/fibrates. Rosuvastatin is the only statin, presently available, to effectively treat AD in diabetes and MS patients.
Keywords: Atherogenic dyslipidaemia, cardiovascular disease, small-dense low-density lipoprotein statins
INTRODUCTION
In 1990, Austin and colleagues first explained Atherogenic Dyslipidemia (AD) as a clinical condition[1] characterized by elevated levels of serum triglyceride (TG) levels and small-dense low-density lipoprotein (sdLDL) particles with low levels of high-density lipoprotein cholesterol (HDL-C), highlighting its atherogenic lipoprotein phenotype.[2] Additionally, elevated levels of large TG rich very low-density lipoproteins (VLDL) and apolipoprotein B (Apo B) and reduced levels of small high-density lipoproteins plays critical role in AD.[3] It is often observed in patients with metabolic syndrome (MS), obesity, insulin resistance and type 2 diabetes mellitus; hence also referred as either diabetic dyslipidemia or dyslipidemia of metabolic syndrome and is considered as an important CVD (Cardiovascular disease) risk factor in these patients.[4,5]
The present review strives to underscore the role of AD in enhancing the risk of CVD. It also deals with the management of oxidized LDL integral to optimum CVD pharmacotherapy.
PATHOPHYSIOLOGY OF ATHEROGENIC DYSLIPIDAEMIA
In AD, there is impaired insulin signalling which increases lipolysis i.e. conversion of TG into free fatty acids (FFA) in adipocytes. These FFA are transported to liver and muscles via blood.[6] Majority of FFA are re-esterified to TG which together with posttranslational stabilization of ApoB enhances the assembly and secretion of VLDL particles. The VLDL production is further augmented by elevated plasma glucose concentrations. Two types of VLDL are synthesized by liver- VLDL-1, the large TG-rich VLDL, and VLDL-2, the smaller TG-poor VLDL.[7] Predominantly, overproduction of VLDL-1 in the liver is seen in patients with AD, insulin resistance and type 2 diabetes; determining atherogenicity of plasma lipoproteins.
Increased secretion of VLDL-1 led to increase in sdLDL production and decrease in HDL; substantially influencing the development of atherosclerosis.[8,9] VLDL-1 production involves 3 steps: 1) Lipidation of Apo B100 in hepatocytes by utilising microsomal enzyme transfer protein in the endoplasmic reticulum, leading to production of nascent pre-VLDL particle; 2) lipidation of nascent pre-VLDL particle produce VLDL-2; and 3) lipidation of VLDL-2 produce VLDL-1. The synthesis of sdLDL from TG-rich VLDL-1 is a 2 step process: 1) Transfer of TG from VLDL1 to LDL by cholesteryl ester transfer protein (CETP) and 2) Conversion of TG rich LDL to sdLDL by hepatic lipase (HL). Though all LDLs are reported as atherogenic but sdLDLs are more atherogenic and serves as a better predictor of cardiovascular risk than LDL-C.[5,9] They have an increased ability to penetrate arterial intima, susceptibility to retention in the extracellular matrix by binding to arterial proteoglycans,[10] and have decreased antioxidant capacity.[3,4] Increase in sdLDL generation has been noted when TG levels are >1.5 mmol/L. Since these particles have lower affinity to LDL-receptors on hepatocytes, there is decrease uptake and clearance of sdLDL, leading to their increased presence in the systemic circulation. Based on the size of LDL-C particles in the blood, there are 2 types of LDL patterns- A and B. LDL A has a particle size >25.5 nm while LDL B has particle size ≤25.5nm.[5] Each VLDL, IDL, and LDL particle contains 1 Apo B100.
The LDL-C reflects the amount of cholesterol carried by an LDL particle. There are significant inter-individual variation in cholesterol content of LDL particles and varies as high as two times between individuals and change with lipid altering treatments. Therefore, total LDL particle (LDL-P) or Apo B is considered as an effective method to quantify LDL. In many patients, the levels of LDL-C and LDL-P were observed similar but in many others, due to variability in the cholesterol content of LDL particles both LDL-C and LDL-P levels were found discordant. Discordance was also reported in terms of LDL-C and LDL-P in patients receiving statin therapy as stains considerably lowers LDL-C than LDL-P.[11] In AD, patients were reported to have a greater increase in LDL-P concentration at a given level of HDL-C.
Besides sdLDL, oxidized LDL also plays a significant role in AD. Oxidized LDL is generated via LDL or sdLDL. In vitro oxidation of LDL by metal ions occurs in three phases: 1) Initial lag phase- consumption of endogenous antioxidant; 2) propagation phase- rapid oxidation of unsaturated fatty acids to lipid hydroperoxides; and 3) decomposition phase-formation of reactive aldehydes. These aldehydes react with lysine residues in apoB-100 resulting in oxidized LDL. Circulating oxidized LDL does not originate from extensive metal ion-induced oxidation in the blood but from mild oxidation in the arterial wall by cell-associated LOX and/or myeloperoxidase.[12,13] The amount of polyunsaturated fatty acids and antioxidant varies significantly within individuals, resulting in a great variation in susceptibility to LDL oxidation. The oxidized-LDL further interacts with scavenger receptors present on endothelial cells, macrophages, and smooth muscle cells causing endothelial dysfunction resulting in a huge build up of cholesterol within the blood vessel leading to atherosclerosis.[14] Other functions of oxidized-LDL are inhibition of endothelial nitric-oxide synthase (eNOS) expression, adhesion molecule induction, facilitation of monocyte adhesion and infiltration, smooth muscle cell migration and proliferation, including the release of cytokine and growth factor from endothelial and smooth muscle cells.[15,16,17]
Besides production of sdLDL, CETP and HL acts on VLDL1 to produce small HDL. This small HDL has a high clearance from the circulation leading to decrease in the plasma level of HDL-C and apolipoprotein A-I. Hence, the metabolic disturbance that began with increased production of VLDL-TG ended in atherogenic reduction of HDL, intravascular remodelling, and reduced reverse cholesterol transport from peripheral tissues, hepatocytes and macrophages to liver, further aggravating atherosclerosis [Figure 1].
Figure 1.
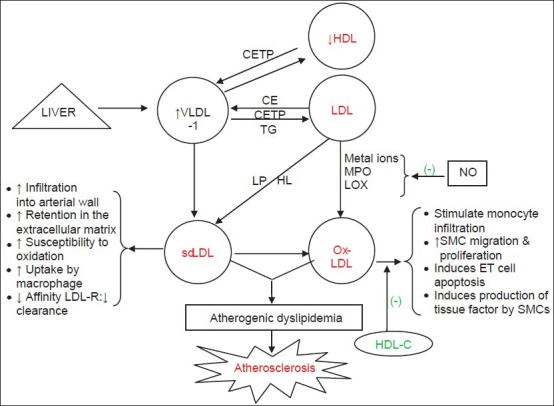
Pathophysiology of atherogenic dyslipidemia. Generation of small dense-LDL particles (pattern B) and oxidation of LDL particle are key elements in atherosclerosis develomany of the atherogenic activitiespment. HDL-C antagonizes (-) many of the atherogenic activities of ox-LDL.[2] TG rich LDL is a substrate for hepatic lipase. CE: Cholesteryl esters, CETP: Cholesteryl ester transfer protein, ET cells: Endothelial cells, HDL-C: High-density lipoprotein cholesterol, HL: Hepatic lipase, LOX: Lipoxygenase, LP: Lipoprotein lipase, MPO: Myeloperoxidase, Ox-LDL: Oxidised low-density lipoprotein, SMCs: Smooth muscle cells
In 2003, Kontush et al.[18] and Smith in 2010[19] reported pro-atherogenicity of HDL in many patients with coronary artery disease (CAD). They explained proatherogenity of HDL due to following alterations in the HDL structure and function: 1) Changes in its protein composition, 2) decrease in Apo A1 and CETP, 3) changes in HDL-associated lipids (lipid peroxidase interferes with HDL antioxidant, antiinflammatory and cholesterol acceptor activities), and 4) post-translational modification of Apo A1. This proatherogenic nature of HDL originated due to structural and functional heterogeneity of HDL particles. It was found that the antioxidant activity of HDL increased with increment in density i.e., in the following order HDL2b < HDL2a < HDL3a < HDL3b < HDL3c; HDL3c would have higher antioxidant activity as compared to HDL2b.[19]
There have been marked racial differences in AD. Various studies support increased prevalence of AD in Asian Indian populations compared to western populations which may be due to their less physical activity and consumption of carbohydrate rich and low polyunsaturated fatty acid (PUFA) diet.[20,21,22] In comparison to western populations, Asian Indians possessed significantly lower levels of HDL and LDL-C with hypertriglyceridemia; had 53.2% prevalence of sdLDL than 29.9% and 18.9% prevalence in whites and blacks, respectively; and significantly higher proportion of Indians (66%) had TG/HDL ratio >3 than 21.1% whites and 13.7% blacks. Besides being more dyslipidaemic, Indians had higher intra-abdominal visceral fat which increased their insulin resistance and CVD risk than western populations. In the comparison between whites and blacks, blacks had less likelihood of AD due to significant lower levels of total cholesterol, LDL-C, LDL pattern B, and triglycerides than whites.[20,23] They have lower HL activity, resulting in more buoyant and less atherogenic LDL particles than whites, explaining less prevalence of AD in their population. However, the HL gene has not been well studied/elucidated in Indian population.
EVIDENCE TO GO BEYOND LDL-C IN IDENTIFYING CVD RISK
Several major statin trials have reported the existence of significant residual CVD risk, despite increased reductions in LDL-C level. Recent evidence suggested important contribution of lipid parameters other than LDL-C, including high TG, low HDL-C, and more recently the sdLDL particles in residual CVD risk.[24,25,26] A number of studies have reported that sdLDL predict cardiovascular endpoints comparably to if not better than LDL-C.[27] They have 1) Greater susceptibility to oxidation than larger particles; 2) trigger inflammatory processes in vascular endothelium that underlie atherosclerotic disease; 3) had high affinity to arterial proteoglycans and were more accessible to the arterial wall; and 4) had relatively lower affinity for the LDL receptor compared to mid-size particles. All these characteristics lead to decreased cellular uptake of sdLDL and increase their time spent circulating in the bloodstream, prolonging their influence on the atherosclerotic process. The risk of coronary diseases elevates to around two to three fold in patients with sdLDL and considered as a new marker for coronary artery stenosis.[29]
The data from Framingham Heart Study (FHS) showed low HDL-C levels to pose substantial risk despite presence of very low LDL-C levels. As HDL-C decreases, it contributes significantly to coronary heart disease (CHD) risk at all levels of LDL-C and even when LDL-C levels were optimal (<100 mg/dL), lower HDL-C level correlated with higher risk of CHD.[30] The study also suggested that non-HDL-C (i.e., total cholesterol minus HDL-C) served as a stronger predictor of CHD risk than LDL-C, irrespective of TG levels.[31] Other non-HDL-C potential diagnostic markers which were considered proatherogenic and more predictive of CVD risk than LDL-C alone[32] are depicted in Table 1.
Table 1.
Potential diagnostic markers of atherogenic dyslipidemia

An international multicenter trial, PROVE-IT TIMI 22 tested the correlation of reduced risk of CVD events with lowering of absolute LDL-C level in patients with acute coronary syndrome (ACS) and evaluated relative efficacy and safety of aggressive LDL-C lowering. This was the first study that evaluated the clinical equivalence of two statins, pravastatin (40 mg) and atorvastatin (80 mg). The results showed significantly lowering of CVD events in patients with TGs < 150 mg/dL than patients with TGs ≥ 150 mg/dL.[33] The PROCAM (Prospective Cardiovascular Münster) study consisted of >20000 volunteers, aged 16-65, from 52 companies and government offices in the Münster and northern Ruhr areas of Germany. The study imparted valuable clinical information on the importance of low HDL-C and elevated TG levels, in addition to the raised LDL-C level and novel risk factors such as lipoprotein(a). The result of the trial demonstrated that TGs was an independent predictor of CHD events; patient with TGs >200 mg/dL and LDL-C/HDL-C ratio >5.0 had six times higher risk of CHD.[34] Several epidemiological studies reported LDL-C/HDL-C ratio as an excellent predictor of CHD risk and an excellent monitor for the effectiveness of lipid-lowering therapies.[35] In a clinical trial with >4,000 middle-aged men with dyslipidemia, the LDL-C/HDL-C ratio was reported to have a more prognostic value than LDL-C or HDL-C alone.[36] This ratio accurately predicted the risk of CHD in patients with elevated levels of TGs. Along with fasting TG concentration, the LDL-C/HDL-C ratio was used to identify a subgroup who achieved over 70% reduction in the CHD risk with gemfibrozil (a lipid-lowering agent). This finding suggested that relatively simple laboratory measurements can be used to identify a small group of people that most likely benefits from long-term drug intervention. The PROSPER (Prospective Study of Pravastatin in the Elderly at Risk) study was a randomised, double blind, placebo-controlled trial involving 5804 patients aged 70-82 years with a history of vascular diseases or cardiovascular risk factors and were treated with pravastatin 40 mg/day or placebo. The study reported LDL-C/HDL-C ratio as the most powerful measure of CVD risk in the elderly people.[37] Individuals with LDL-C/HDL-C ratio >3.3 were qualified for statin initiation. The PROCAM study also reported a continuous and graded relationship between the LDLC/HDL-C ratio and CVD mortality.[34] The deaths due to CAD abruptly increased when the LDL-C/HDL-C ratio was between 3.7 and 4.3. One unit increase in the LDL-C/HDL-C ratio was associated with 53% increase in risk of MI.[38] Another study reported 75% increase in the risk of MI with one-unit increase in LDL-C/HDL-C ratio.[39] The FHS also reported LDL-C/HDL-C ratio to be a stronger predictor of CVD than the levels of LDL-C or HDL-C alone.[40] According to the recent NCEP guidelines, the LDL-C/HDL-C ratio of 2.5 governs the starting point of statin therapy and increased CVD deaths were reported at the LDL-C/HDL-C ratio of 3.3-3.7.[41]
ROLE OF HDL IN REGRESSION OF ATHEROSCLEROTIC PLAQUE
The FHS showed HDL-C as the most potent lipid predictor of CHD risk in men and women aged >49 years. Every 1 mg/dl increment in HDL-C was associated with 2 and 3% decreased risk of CHD in men and women, respectively.[42] Though anti-atherogenic activities of HDL-C are not well elucidated, its effects are attributed to reverse cholesterol transport and host of other protective activities. The reverse cholesterol transport involves transfer of excess cholesterol from lipid-laden macrophages (foam cells) present in peripheral tissues to the hepatocytes via HDL for metabolism or excretion into bile. The other protective activities of HDL-C against atherosclerosis are due to its antioxidant (counteracting LDL oxidation), anti-inflammatory, antithrombotic/profibrinolytic (reducing platelet aggregation and coagulation), and vasoprotective (facilitating vascular relaxation and inhibiting leukocyte chemotaxis and adhesion) effects. Collectively, HDL and its components (including ApoA-I, paraoxonase, platelet activating factor acetylhydrolase, and other antioxidant enzymes) exert an array of effects that may help to prevent atherosclerosis, acute coronary syndromes, and restenosis after coronary angioplasty.[43]
MANAGEMENT OF ATHEROGENIC DYSLIPIDAEMIA
Atherosclerosis in AD is significantly expedited in the presence of increased sdLDL and oxidized LDL-C particles. Statin mono- and/or combination therapy (statin/niacin and statin/fibrates), agents that increases HDL-C levels, nutritional diet, and strict adherence to therapeutic lifestyle changes (TLCs) have been highly successful in reversing the deleterious effects of raised blood LDL-C levels and restoring normal health. Though, clinical data on their routine use remains inconclusive, omega-3 fatty acids, vitamin E and carnitine have been used as add-on therapy.
Statins
Small-dense LDL particles, characteristic of AD, have higher susceptibility to oxidation, leading to the formation of oxidized LDL particles which acts as ligand for novel lectin-like oxidized LDL receptor-1, increases its expression, causes apoptosis in endothelial cells, phagocytose aged and worn-out cells, suppresses constitutive nitric oxide synthase activity and increases cell adhesion.[44,45] In the year 2001, Li and coworkers reported inhibitory activities of statins-simvastatin and atorvastatin on the expression of lectin-like oxidized LDL receptor-1, showing its beneficial effects in atherosclerosis.[46] Besides simvastatin and atorvastatin, rosuvastatin also demonstrated benefits in hypercholesterolaemia in patients with AD. In the COMETS (Comparative study with rosuvastatin in subjects with METabolic Syndrome) trial, a first large multinational double-blind, randomized trial, the efficacy of rosuvastatin was compared with atorvastatin in patients with the MS. The trial enrolled 397 adults and randomized them to rosuvastatin 10 mg, atorvastatin 10 mg, or placebo for six weeks. The primary end point of the study was the percent change from baseline in LDL-C. Rosuvastatin lowered LDL-C by 42.7% which was significantly higher than atorvastatin (36.6%) and increased HDL-C by 9.3% compared 4.8% increase by atorvastatin in patients with MS. Hence, rosuvastatin was found more effective than atorvastatin in reducing LDL-C levels, in achieving lipid goals and in improving other aspects of atherogenic lipid profile in patients with MS.[47] The STELLAR (Statin Therapies for Elevated Lipid Levels compared across doses to Rosuvastatin) trial compared rosuvastatin with other statins. This was a six week randomized, open-label study that compared the effects of rosuvastatin (doses: 10, 20, and 40 mg) with atorvastatin (doses: 10, 20, 40, and 80 mg), simvastatin (doses: 10, 20, 40, and 80 mg) and pravastatin (doses: 10, 20, and 40 mg) on plasma lipids in hypercholesterolemic patients.[48] Of all statins, rosuvastatin at doses 20 and 40 mg showed the most favorable effect on the atherogenic lipid profile of MS. Another trial, SOLAR (Satisfying Optimal LDL-C ATP III goals with Rosuvastatin) was a randomized, open-label multicenter trial conducted in managed care patients at high risk for CHD. After 12 weeks of treatment, 76% of patients who took rosuvastatin reached the LDL-C target of <100 mg/dL in comparison to 58% with atorvastatin and 53% with simvastatin.[49] Another open-label study compared oral doses of rosuvastatin (40 mg/daily) with atorvastatin (80 mg/day) with an objective to observe the reduction in sdLDL-C levels over six week duration in 271 hyperlipidemic patients. The post-hoc analysis of the data reported rosuvastatin to be more effective than atorvastatin in lowering sdLDL-C while lowering of TG levels were similar with both the statins.[50] In a recent multicenter open labeled, randomized trial, Park and his colleagues compared the effects of rosuvastatin (10 mg/day) with atrovastatin (10 mg/day) on lipid and glycaemic control in nondiabetic Korean patients with MS. The results again supported rosuvastatin to be significantly more effective than atorvastatin in lowering LDL-C levels in these patients.[51]
Agents raising HDL-C
Low levels of HDL-C constitute a strong, independent, and inverse predictor of the risk of premature development of atherosclerosis and CVD.[52] Low HDL-C levels are a hallmark of type 2 diabetes as well as mixed or combined dyslipidemia, renal and hepatic insufficiency states, and autoimmune diseases. These disease states also feature a moderate or marked degree of hypertriglyceridemia. Only few options are available for elevating low HDL-C levels. Though HDL-C levels may be increased by up to 10% by implementing TLC, including weight reduction, exercise, smoking cessation, and moderate alcohol consumption, many patients still require pharmacological intervention. But still there is no clinching evidence available today to directly correlate elevated HDL-C level alone with CVD prevention.
CETP Inhibitors were regarded as the most potent HDL-raising agents. The first CETP inhibitor, torcetrapib, was prematurely terminated in December 2006 due to excess cardiovascular and noncardiovascular mortality in the active treatment group. Torcetrapib considerably raised aldosterone level and blood pressure and incurred changes in serum electrolytes, indicative of mineralocorticoid excess. The recent failure of Phase 3, Dalcetrapib Outcomes (dalOUTCOME) trial, planned in 16,000 stable coronary heart disease patients with recent acute coronary syndrome (ACS), was further a blow in pursuing CETP inhibitor, dalcetrapib, as HDL raising agents as it was associated with lack of benefit in terms of lower cardiovascular events, inability to lower LDL levels, and not effective enough to reduce morbidity and mortality. Though 30% increase in HDL was observed with dalcetrapib, >100% increase in HDL and 35-40% reduction in LDL was observed with the use of evacetrapib and anacetrapib.(Merck, Whitehouse Station, NJ) Hence, there is still a hope that the more potent CETP inhibitors would improve outcomes in patients with ACS.[53] In addition, Apo A1 mimetic peptides which are not only active in cellular cholesterol efflux may also exert anti-inflammatory effects and show promises.[53]
Nutritional diets and therapeutic lifestyle changes
Therapeutic lifestyle changes received considerable attention in the third National Cholesterol Education Program Adult Treatment Panel (NCEP-ATP)[41] guidelines since lifestyle risk factors like overweight/obesity, physical inactivity and atherogenic diet have been considered as important contributors to the CHD risk. This guideline popularized a multifaceted life-habit approach. Hypertriglyceridemia is caused by several factors such as obesity, physical inactivity, cigarette smoking, excess alcohol consumption, high-carbohydrate diets (>60% of total energy), other diseases such as type 2 diabetes mellitus, chronic renal failure, nephrotic syndrome, and genetic predisposition.[54]
Dietary carbohydrate is considered as the major determinant of raised TG levels in AD.[55] Approximately 3-5% reduction in LDL-C was reported with increase use of viscous fiber from 5 to 10 g/day, 6-15% reduction with 2 g/day plant stanols/sterols, and 20-30% reduction with use of low saturated fat and cholesterol intake with therapeutic diet, including weight loss. Other dietary modifications rich in omega-3 fatty acids like inclusion of soy protein and nuts in diet potentially reduced the levels of LDL-C, due to its excellent antioxidant activities in patients with hypertriglyceridemia.[41] In general, simple sugars and rapidly hydrolyzed starches have a greater glyceridaemic effect than more complex carbohydrates and those consumed in conjunction with high-fiber intake. It was reported that over all carbohydrate restriction improved atherogenic lipid states in the absence of weight loss or in the presence of higher saturated fat. While at the same time, low fat diets seemed to require weight loss for effective improvement in atherogenic dyslipidemia.[56]
The recommended level of dietary fat is 25 to 35% of calories. Within this range, complex carbohydrates and high-fiber diet facilitates TG lowering, increases HDL-C levels and produces larger buoyant LDL particles.[57]
Apart from dietary carbohydrate, weight management and increased physical activity have played an important role in lowering the LDL-C levels. Weight loss showed positive effect on HDL-C and was also suggested as a primary goal to lower TG levels in patients with AD. Physical activity potentially influenced most of the atherosclerotic risk factors. Individuals who jogged for six months, participated in regular exercise for eight months and had three weeks of diet and brisk walking, revealed higher lowering of LDL-C and larger buoyant LDL particles.
A significant increase in plasma HDL-C from 4 to 22% (absolute increase from 2 to 8 mg/dL) was observed with >12 weeks of exercise training with strict adherence. In addition, there was a decrease of HDL2b and increase of HDL 3b levels. Physical activity for more than an hour/day for five to seven days in a week was recommended for patients who are required to achieve weight loss. It was assumed that if a patient gave proper emphasis on nutrition and had strict adherence to exercise, then he may more likely lose body weight and would require less treatment therapy.[58] Given the apparent benefits of physical activities and or exercise training in dyslipidaemia, it is the most appropriate strategy towards managing dyslipidemia.
CONCLUSION
Atherogenic dyslipidemia is a part of complex cluster of abnormalities called the metabolic syndrome which has a direct correlation with CVD events. In recent years, Indian population have increasing incidence of AD and CVD as compared to western population, which may be due to adverse life style changes such as physical inactivity, diet deficient in PUFA and a higher genetic predisposition. Despite large reduction in LDL-C levels, significant residual cardiovascular risk due to low HDL-C, high TG and non-HDL-C levels exists. Their management with the therapeutic lifestyle changes with statins or statins combination with niacin and/or fibrates has considerably reduced the incidences of CVD events. Apo B/ApoA-1 ratio has been considered as an accurate predictor of CVD risk, however several studies have reported LDL-C/HDL-C ratio to be more accurate.
Studies such as COMET, STELLAR and SOLAR have successfully established rosuvastatin to be significantly more effective in lowering LDL-C, TGs, and in improving other aspects of atherogenic lipid profile in patients with MS and diabetes.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Austin MA, King MC, Vranizan KM, Krauss RM. Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation. 1990;82:495–6. doi: 10.1161/01.cir.82.2.495. [DOI] [PubMed] [Google Scholar]
- 2.Reaven GM, Chen YD, Jeppesen J, Maheux P, Krauss RM. Insulin resistance and hyperinsulinemia in individuals with small, dense low density lipoprotein particles. J Clin Invest. 1993;92:141–6. doi: 10.1172/JCI116541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinik AI. The metabolic basis of atherogenic dyslipidemia. Clin Cornerston. 2005;7:27–35. doi: 10.1016/s1098-3597(05)80065-1. [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM. Hypertriglyceridemia, atherogenic dyslipidemia and the metabolic syndrome. Am J Cardiol. 1998;81:18B–25. doi: 10.1016/s0002-9149(98)00033-2. [DOI] [PubMed] [Google Scholar]
- 5.Musunuru K. Atherogenic dyslipidemia: Cardiovascular risk and dietary intervention. Lipids. 2010;45:907–14. doi: 10.1007/s11745-010-3408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miranda PJ, DeFronzo RA, Califf RM, Guyton JR. Metabolic syndrome: Definition, pathophysiology, and mechanisms. Am Heart J. 2005;149:33–45. doi: 10.1016/j.ahj.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Olofsson SO, Wiklund O, Boren J. Apolipoprotein A-1 and B: Biosynthesis, role in the development of atherosclerosis and targets for intervention against cardiovascular disease. Vas Health Risk Manag. 2007;3:491–2. [PMC free article] [PubMed] [Google Scholar]
- 8.Taskinen MR. Diabetic dyslipidaemia: From basic research to clinical practice. Diabetologia. 2003;46:733–49. doi: 10.1007/s00125-003-1111-y. [DOI] [PubMed] [Google Scholar]
- 9.Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–36. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 10.Griffin BA, Minihane AM, Furlonger N, Chapman C, Murphy M, Williams D, et al. Inter-relationships between small, dense low-density lipoprotein (LDL), plasma triacylglycerol and LDL apoprotein B in an atherogenic lipoprotein phenotype in free-living subjects. Clin Sci (Lond) 1999;97:269–76. [PubMed] [Google Scholar]
- 11.Davidson MH, Carlson DM, Guthrie RM, Kelly MT, Lele A, Setze CM, et al. ABT-335 in combination with rosuvastatin improves multiple lipid ratios in patients with mixed dyslipidemia. J Clin Lipidol. 2008;2:211–2. [Google Scholar]
- 12.Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991;88:1785–92. doi: 10.1172/JCI115499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: Does it hold for humans. Trends Cardiovasc Med. 2001;11:93–2. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- 14.Erl W, Weber PC, Weber C. Monocytic cell adhesion to endothelial cells stimulated by oxidized low density lipoprotein is mediated by distinct endothelial ligands. Atherosclerosis. 1998;136:297–3. doi: 10.1016/s0021-9150(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 15.Mehta A, Yang B, Khan S, Hendricks JB, Stephen C, Mehta JL. Oxidized low-density lipoproteins facilitate leukocyte adhesion to aortic intima without affecting endothelium-dependent relaxation. Role of P-selectin. Arterioscler Thromb Vasc Biol. 1995;15:2076–83. doi: 10.1161/01.atv.15.11.2076. [DOI] [PubMed] [Google Scholar]
- 16.Keaney JF, Jr, Hare JM, Balligand JL, Loscalzo J, Smith TW, Colucci WS. Inhibition of nitric oxide synthase augments myocardial contractile responses to beta-adrenergic stimulation. Am J Physiol. 1996;271:H2646–52. doi: 10.1152/ajpheart.1996.271.6.H2646. [DOI] [PubMed] [Google Scholar]
- 17.Pryor WA. Vitamin E and heart disease: Basic science to clinical intervention trials. Free Rad Biol Med. 2000;28:141–64. doi: 10.1016/s0891-5849(99)00224-5. [DOI] [PubMed] [Google Scholar]
- 18.Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol. 2003;23:1881–8. doi: 10.1161/01.ATV.0000091338.93223.E8. [DOI] [PubMed] [Google Scholar]
- 19.Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–5. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulukutla SR, Venkitachalam L, Marroquin OC, Kip KE, Aiyer E, Edmundowicz D, et al. Population variations in atherogenic dyslipidemia: A report from HeartSCORE and India SCRORE studies. J Clin Lipidol. 2008;2:410–7. doi: 10.1016/j.jacl.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Ramchandran A, Snehalatha C, Satyavani K, Sivasankari S, Vijay V. Metabolic syndrome in urban Asian Indian adults: A population study using modified ATP III criteria. Diabetes Res Clin Pract. 2003;60:199–4. doi: 10.1016/s0168-8227(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 22.Mohan V, Shanthirani S, Deepa R, Premalatha G, Sastry NG, Saroja R, et al. Intra-urban differences in the prevalence of the metabolic syndrome in Southern India: The Chennai urban population study. Diabet Med. 2001;18:280–7. doi: 10.1046/j.1464-5491.2001.00421.x. [DOI] [PubMed] [Google Scholar]
- 23.Sharma RK, Singh VN, Reddy HK. Thinking beyond low-density lipoprotein cholesterol: strategies to further reduce cardiovascular risk. Vas Health Risk Manag. 2009;5:793–9. doi: 10.2147/vhrm.s5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blake GJ, Otvos JD, Rifai N, Ridker PM. Low-density lipoprotein particle concentration and size as determined by nuclear magnetic resonance spectroscopy as predictors of cardiovascular disease in women. Circulation. 2002;106:1930–7. doi: 10.1161/01.cir.0000033222.75187.b9. [DOI] [PubMed] [Google Scholar]
- 25.Smith SC, Jr, Clark LT, Cooper RS, Daniels SR, Kumanyika SK, Ofili E, et al. American Heart Association Obesity, Metabolic Syndrome, and hypertension writing group. Discovering the full spectrum of cardiovascular disease: Minority Health Summit 2003: Report of the obesity, metabolic syndrome, and hypertension writing group. Circulation. 2005;111:e134–9. doi: 10.1161/01.CIR.0000157743.54710.04. [DOI] [PubMed] [Google Scholar]
- 26.Sharma RK, Singh VN, Reddy HK. Thinking beyond low-density lipoprotein cholesterol: Strategies to further reduce cardiovascular risk. Vasc Health Risk Manag. 2009;5:793–9. doi: 10.2147/vhrm.s5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alagona P., Jr Beyond LDL cholesterol: The role of elevated triglycerides and low HDL cholesterol in residual CVD risk remaining after statin therapy. Am J Manag Care. 2009;15:S65–73. [PubMed] [Google Scholar]
- 28.Rosenson RS, Otvos JD, Freedman DS. Relations of lipoprotein subclass levels and low-density lipoprotein size to progression of coronary artery disease in the pravastatin limitation of atherosclerosis in the coronary arteries (PLAC-I) trial. Am J Cardiol. 2002;90:89–94. doi: 10.1016/s0002-9149(02)02427-x. [DOI] [PubMed] [Google Scholar]
- 29.Kwon SW, Yoon SJ, Kang TS, Kwon HM, Kim JH, Rhee J, et al. Significance of small dense low density lipoprotein as a risk factor for coronary artery disease and acute coronary syndrome. Yonsei Med J. 2006;47:405–14. doi: 10.3349/ymj.2006.47.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castelli WP. Cholesterol and lipids in the risk of coronary artery disease-the Framingham Heart Study. Can J Cardiol. 1988;4(suppl A):5A–10. [PubMed] [Google Scholar]
- 31.Liu J, Sempos CT, Donahue RP, Dorn J, Trevisan M, Grundy SM. Non-high-density lipoprotein and very-low density lipoprotein cholesterol and their risk predictive values in coronary heart disease. Am J Cardiol. 2006;98:1363–8. doi: 10.1016/j.amjcard.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 32.Arsenault BJ, Boekholdt SM, Kastelein JJ. Lipid parameters for measuring risk of cardiovascular disease. Nat Rev Cardiol. 2011;8:197–6. doi: 10.1038/nrcardio.2010.223. [DOI] [PubMed] [Google Scholar]
- 33.Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E. Impact of triglyceride levels beyond low density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724–30. doi: 10.1016/j.jacc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 34.Cullen P, Schulte H, Assmann G. The Munster Heart Study (PROCAM) Total Mortality in Middle-Aged Men is increased at low total and LDL cholesterol concentrations in smokers but not in nonsmokers. Circulation. 1997;96:2128–36. doi: 10.1161/01.cir.96.7.2128. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez ML, Webb D. The LDL to HDL cholesterol ratio as a valuable tool to evaluate coronary heart disease risk. J Am Coll Nutr. 2008;27:1–5. doi: 10.1080/07315724.2008.10719668. [DOI] [PubMed] [Google Scholar]
- 36.Manninen V, Tenkanen L, Koskinen P, Huttunen JK, Manttari M, Heinonen OP, et al. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation. 1992;85:37–45. doi: 10.1161/01.cir.85.1.37. [DOI] [PubMed] [Google Scholar]
- 37.Packard CJ, Ford I, Robertson M, Shepherd J, Blauw GJ, Murphy MB, et al. Plasma lipoproteins and apolipoproteins as predictors of cardiovascular risk and treatment benefit in the PROspective Study of Pravastatin in the Elderly at Risk (PROSPER) Circulation. 2005;112:3058–65. doi: 10.1161/CIRCULATIONAHA.104.526848. [DOI] [PubMed] [Google Scholar]
- 38.Stampfer MJ, Sacks FM, Salvini S, Willett WC, Hennekens CH. A prospective study of cholesterol apolipoproteins and the risk of myocardial infarction. N Engl J Med. 1991;325:373–81. doi: 10.1056/NEJM199108083250601. [DOI] [PubMed] [Google Scholar]
- 39.Gaziano JM, Hennekens CH, O’Donnell CJ, Breslow JL, Buring JE. Fasting triglycerides, high-density lipoprotein, and risk of myocardial infarction. Circulation. 1997;96:2520–5. doi: 10.1161/01.cir.96.8.2520. [DOI] [PubMed] [Google Scholar]
- 40.Kannel WB. Risk stratification of dyslipidemia: Insights from the framingham study. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:187–93. doi: 10.2174/1568016054368250. [DOI] [PubMed] [Google Scholar]
- 41.Adult Treatment Panel III. The Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection evaluation, and treatment of high blood cholesterol in adults. J Am Med Assoc. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 42.Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986;256:2835–8. [PubMed] [Google Scholar]
- 43.Singh IM, Shishehbor MH, Ansell BJ. High-density lipoprotein as a therapeutic target: A systematic review. JAMA. 2007;298:786–98. doi: 10.1001/jama.298.7.786. [DOI] [PubMed] [Google Scholar]
- 44.Oka K, Sawamura T, Kikuta K, Itokawa S, Kume N, Kita T, et al. Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proc Natl Acad Sci USA. 1998;95:9535–40. doi: 10.1073/pnas.95.16.9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kume N, Murase T, Moriwaki H, Aoyama T, Sawamura T, Masaki T, et al. Inducible expression of lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ Res. 1998;83:322–7. doi: 10.1161/01.res.83.3.322. [DOI] [PubMed] [Google Scholar]
- 46.Mehta JL, Li DY. Identification and autoregulation of receptor for Ox-LDL in cultured human coronary artery endothelial cells. Biochem Biophys Res Commun. 1998;248:511–4. doi: 10.1006/bbrc.1998.9004. [DOI] [PubMed] [Google Scholar]
- 47.Stalenhoef AF, Ballantyne CM, Sarti C, Murin J, Tonstad S, Rose H, et al. A comparative study with rosuvastatin in subjects with metabolic syndrome: Results of the COMETS study. Eur Heart J. 2005;26:2664–72. doi: 10.1093/eurheartj/ehi482. [DOI] [PubMed] [Google Scholar]
- 48.Deedwania PC, Hunnighake DB, Bays HE, Jones PH, Cain VA, Blasetto JW. Effects of rosuvastatin, atrovastatin, simvastatin and pravastatin on atherogenic dyslipidemia in patients with characteristics of the metabolic syndrome. Am J Cardiol. 2005;95:360–6. doi: 10.1016/j.amjcard.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 49.Insull W, Jr, Ghali JK, Hassman DR, Y As JW, Gandhi SK, Miller E, et al. Achieving low-density lipoprotein cholesterol goals in high-risk patients in managed care: Comparison of rosuvastatin, atorvastatin, and simvastatin in the SOLAR trial. Mayo Clin Proc. 2007;82:543–50. doi: 10.4065/82.5.543. [DOI] [PubMed] [Google Scholar]
- 50.Ai M, Otokozawa S, Asztalos BF, Nakajima K, Stein E, Jones PH, et al. Effects of maximal doses of atorvastatin versus rosuvastatin on small dense low-density lipoprotein cholesterol levels. Am J Cardiol. 2008;101:315–8. doi: 10.1016/j.amjcard.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 51.Park JS, Kim YJ, Chai JY, Kim YN, Hong TJ, Kim DS, et al. Comparative study of low doses of rosuvastatin on lipid and glycemic control in patients with metabolic syndrome and hypercholesterolemia. Korean J Intern Med. 2010;25:27–35. doi: 10.3904/kjim.2010.25.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooney MT, Dudina A, De Bacquer D, Wilhelmsen L, Sans S, Menotti A, et al. HDL cholesterol protects against cardiovascular disease in both genders, at all ages and at all levels of risk. Atherosclerosis. 2009;206:611–6. doi: 10.1016/j.atherosclerosis.2009.02.041. [DOI] [PubMed] [Google Scholar]
- 53.Navab M, Reddy ST, Van Lenten BJ, Anantharamaiah GM, Fogelman AM. The role of dysfunctional HDL in atherosclerosis. J Lipid Res. 2009;50:S145–9. doi: 10.1194/jlr.R800036-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kontush A, Guérin M, Chapman MJ. Spotlight on HDL-raising therapies: Insights from the torcetrapib trials. Nat Clin Pract Cardiovasc Med. 2008;5:329–36. doi: 10.1038/ncpcardio1191. [DOI] [PubMed] [Google Scholar]
- 55.ESC/EAS Guidelines for the management of dyslipidaemia. The Task Force for the management of dyslipidemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) Eur Heart J. 2011;32:1769–818. doi: 10.1093/eurheartj/ehr158. [DOI] [PubMed] [Google Scholar]
- 56.Fletcher B, Berra K, Ades P, Braun LT, Burke LE, Durstine JL, et al. Managing abnormal blood lipids: A collaborative approach: Cosponsored by the councils on cardiovascular nursing; Arteriosclerosis, thrombosis, and vascular biology; basic cardiovascular sciences; cardiovascular disease in young; clinical cardiology; epidemiology and prevention; nutrition, physical activity, and metabolism; and stroke; and the preventive cardiovascular nurses association. Circulation. 2005;112:3184–9. doi: 10.1161/CIRCULATIONAHA.105.169180. [DOI] [PubMed] [Google Scholar]
- 57.Parks EJ, Hellerstein MK. Carbohydrate induced hypertriglyceridemia: Historical perspective and review of biological mechanisms. Am J Clin Nutr. 2000;71:412–33. doi: 10.1093/ajcn/71.2.412. [DOI] [PubMed] [Google Scholar]
- 58.Feinman RD, Volek JS. Low carbohydrate diets improve atherogenic dyslipidemia even in the absence of weight loss. Nutr Metab (Lond) 2006;3:24. doi: 10.1186/1743-7075-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]