

Abstract
Ageing is associated with a blunted response to sympathetic stimulation and an increased risk of arrhythmia and sudden cardiac death. Aberrant calcium (Ca2+) handling is an important contributor to the electrical and contractile dysfunction associated with ageing. Yet, the specific molecular mechanisms underlying abnormal Ca2+ handling in ageing heart remain poorly understood. In this study, we used ventricular myocytes isolated from young (5–9 months) and old (4–6 years) rabbit hearts to test the hypothesis that changes in Ca2+ homeostasis are caused by post-translational modification of ryanodine receptors (RyRs) by mitochondria-derived reactive oxygen species (ROS) generated in the ageing heart. Changes in parameters of Ca2+ handling were determined by measuring cytosolic and intra-sarcoplasmic reticulum (SR) Ca2+ dynamics in intact and permeabilized ventricular myocytes using confocal microscopy. We also measured age-related changes in ROS production and mitochondria membrane potential using a ROS-sensitive dye and a mitochondrial voltage-sensitive fluorescent indicator, respectively. In permeablized myocytes, ageing did not change SERCA activity and spark frequency but decreased spark amplitude and SR Ca2+ load suggesting increased RyR activity. Treatment with the antioxidant dithiothreitol reduced RyR-mediated SR Ca2+ leak in permeabilized myocytes from old rabbit hearts to the level comparable to young. Moreover, myocytes from old rabbits had more depolarized mitochondria membrane potential and increased rate of ROS production. Under β-adrenergic stimulation, Ca2+ transient amplitude, SR Ca2+ load, and latency of pro-arrhythmic spontaneous Ca2+ waves (SCWs) were decreased while RyR-mediated SR Ca2+ leak was increased in cardiomyocytes from old rabbits. Additionally, with β-adrenergic stimulation, scavenging of mitochondrial ROS in myocytes from old rabbit hearts restored redox status of RyRs, which reduced SR Ca2+ leak, ablated most SCWs, and increased latency to levels comparable to young. These data indicate that an age-associated increase of ROS production by mitochondria leads to the thiol-oxidation of RyRs, which underlies the hyperactivity of RyRs and thereby shortened refractoriness of Ca2+ release in cardiomyocytes from the ageing heart. This mechanism probably plays an important role in the increased incidence of arrhythmia and sudden death in the ageing population.
Key points
Ageing is associated with increased risk of sudden cardiac death due to malignant arrhythmias.
Shortened refractoriness of Ca2+ release due to increased activity of Ca2+ release channels (RyRs) is recognized as an important contributor to cardiac-triggered arrhythmias. However, molecular mechanisms of RyR dysfunction and its contribution to arrhythmias in ageing remain to be examined.
Using ventricular myocytes isolated from old rabbit hearts we demonstrate that age-associated increase in rate of production of reactive oxygen species (ROS) by mitochondria leads to the thiol-oxidation of RyRs, which underlies the hyperactivity of the channels and thus shortened refractoriness of Ca2+ release in cardiomyocytes from the ageing heart. Mitochondria-specific scavenging of ROS in old myocytes restored the redox status of RyRs, reducing SR Ca2+ leak and arrhythmogenic spontaneous Ca2+ waves.
We conclude that increased ROS production by mitochondria contributes to age-associated increased risk of stress-induced arrhythmia and sudden cardiac death through thiol-modifications of RyRs.
Introduction
Ageing is associated with increased incidence of cardiac arrhythmia and sudden cardiac death (Kannel et al. 1987; Lakatta, 1993). Previously, in a rabbit model, we have shown that ageing alters the structure and the electrical and mechanical activity of the heart causing both systolic and diastolic dysfunction, slowing conduction velocity, and altering conduction anisotropy, which provides a pro-arrhythmic substrate that increases the risk of malignant arrhythmia (Cooper et al. 2012). Although much is known regarding the substrate that predisposes and sustains arrhythmias in the ageing heart, the underlying age-associated molecular trigger remains to be thoroughly examined. At the cellular level, aberrant Ca2+ handling is recognized as an important contributor to the electrical dysfunction associated with ageing (Xiao et al. 1994; Lakatta & Sollott, 2002; Dibb et al. 2004; Zhu et al. 2005; Howlett, 2010; Janczewski & Lakatta, 2010). However, the specific molecular mechanisms underlying age-associated abnormal Ca2+ handling in the heart have yet to be fully elucidated.
Diminished capacity to maintain redox balance has been proposed to be an underlying factor leading to abnormal cardiac function characteristic of ageing (Squier, 2001; Lakatta & Sollott, 2002; Chaudhary et al. 2011). In aged rat and rabbit ventricles, Morita et al. showed that exposure to oxidative stress led to early afterdepolarizations (EADs) in myocytes and EAD-mediated tachyarrhythmias at the tissue level (Morita et al. 2009). Recent studies identified the SR Ca2+ release channel, the ryanodine receptor (RyR), as a protein target that is sensitive to oxidation, and post-translational modifications of RyRs by reactive oxygen species (ROS) that destabilize interdomain interactions within RyRs (Mochizuki et al. 2007) have been implicated in alterations of Ca2+ homeostasis in conditions accompanied by oxidative stress, such as heart failure or myocardial infarct (Giordano, 2005; Zima & Blatter, 2006; Györke & Carnes, 2008; Terentyev et al. 2008; Belevych et al. 2009, 2011b, 2012). Specifically, accelerated leak of [Ca2+]SR via oxidized RyRs was shown to result in diminished systolic Ca2+ release that determines strength of contraction and an enhanced propensity to generate pro-arrhythmic, diastolic Ca2+ waves during β-adrenergic stimulation (Terentyev et al. 2008; Belevych et al. 2011b, 2012). Yet, the role of redox-mediated alterations of Ca2+ handling (RyRs in particular) with regard to ageing and Ca2+-triggered arrhythmias remains to be investigated. Moreover, it is well established that mitochondria, one of the main sources of ROS in myocytes, if damaged or dysfunctional, could increase ROS production rate up to 10-fold (Grivennikova et al. 2010). Dysfunctional mitochondria have been implicated as a mechanism for ventricular arrhythmia by altering ion channel function, action potential heterogeneity, and cell excitability (O’Rourke et al. 1994; Brown et al. 2010; Brown & O’Rourke, 2010), and mitochondria-derived ROS has recently been connected to the oxidation of RyRs (Ho et al. 2011; Bovo et al. 2012).
The goal of this study was to test the hypothesis that changes in Ca2+ homeostasis associated with ageing are caused by post-translational modification of RyRs by mitochondria-derived ROS generated in the old heart. Importantly, we sought to examine the mechanisms and effects of ROS generation and to determine whether exposure of cardiomyocytes from old rabbits to a reducing agent and a mitochondria-specific ROS scavenger reverses the age-related pro-arrhythmogenic cellular phenotype. Our results show that the age-associated increase in rate of ROS production by mitochondria leads to the thiol-oxidation of RyRs, which underlies the hyperactivity of RyRs and thereby shortened refractoriness of Ca2+ release in cardiomyocytes from the ageing heart. We conclude that this mechanism contributes to age-associated increased risk of stress-induced arrhythmia and sudden death.
Methods
Animal ethical statement
All animal work was performed in accordance with the local guidelines of the institutions and only after approval by the Institutional Animal Care and Use Committee in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Cardiomyocyte isolation
Ventricular myocytes were isolated from young (5–9 months) and old (4–6 years) female, New Zealand White rabbits (RSI Farms, Mocksville, NC, USA) as previously described (Brunner et al. 2008). The rabbits were anaesthetized with ketamine (60 mg kg−1) and xylazine (15 mg kg−1) (i.m.), buprenorphine (0.03 mg kg−1s.c.) and sodium pentobarbital (150 mg kg−1, i.v.). Hearts were quickly excised after thoracotomy and retrogradely perfused on a Langendorff apparatus with solution containing collagenase I (Roche Applied Science, Indianapolis, IN, USA) at 37°C.
Cellular electrophysiology
Ionic currents and action potentials were recorded at 36 ± 1°C using standard whole-cell patch clamp technique with an Axopatch-200B amplifier (Molecular Devices, Sunnyvale, CA). Intracellular solution contained (in mm): 120 KCl, 5 MgCl2, 0.36 CaCl2, 5 EGTA, 5 Hepes, 5 glucose, 5 K2-ATP, 5 Na2-CrP, 0.25 Tris-GTP (pH = 7.2). Pipette resistance was 2–4 MΩ. Capacitance and 70% of the series resistance were routinely compensated for. Action potentials and Ca2+ currents were recorded in Tyrode solution containing (in mm): 140 NaCl, 5.4 KCl, 0.33 NaH2PO4, 1 MgCl2, 1 CaCl2, 5 Hepes, and 7.5 glucose (pH = 7.4). Action potentials were recorded under current clamp using 1.2 × threshold current injection at 1 Hz. Ca2+ currents were recorded using voltage steps between −40 and 40 mV from a holding potential of −50 mV. E-4031-sensitive Rapid delayed rectifier (IKr), Chromanol 293B-sensitive Slow delayed rectifier (IKs), Transient outward (Ito) and inward rectifier (IK1) potassium currents were measured and analysed under voltage clamp using modified external solutions and voltage protocols as described in Brunner et al. (2008). Data were sampled at 2.5–5 kHz and filtered at 1 kHz. Current–voltage relationships are normalized to the cell capacitances and presented as mean ± SEM.
Confocal microscopy measurements of intracellular and intra-SR Ca2+ and Ca2+ sparks
Intracellular Ca2+ cycling, intra-SR Ca2+ cycling, and Ca2+ spark activity in isolated rabbit ventricular myocytes were monitored by a Leica SP2 confocal laser scanning system equipped with a ×60 1.4 NA oil-immersion objective in linescan and x–y mode using Ca2+-sensitive indicators Fluo-3, Fluo-5N and Fluo-4 (Invitrogen/Molecular Probes, Carlsbad, CA, USA), respectively.
For intracellular Ca2+ cycling measurements, cells were loaded with Fluo-3 for 12 min, and after 20 min de-esterification, the dye was excited with the 488 nm line of an argon laser. Emission was collected at 500–600 nm. Cardiomyocytes were studied in Tyrode solution (in mm: 140 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 10 Hepes and 5.6 glucose, pH = 7.3) at baseline and with 30 nm isoproterenol (isoprenaline; ISO), a β-adrenergic receptor agonist. Myocytes were paced via field stimulation at 1 Hz using extracellular platinum electrodes. To assess the SR Ca2+ load and decay kinetics, 20 mm caffeine was applied at the end of the experiments. Sodium–calcium exchanger (NCX) activity was estimated by measuring the rate of decay of caffeine-induced Ca2+ transients (kcaff), and SR (ER) Ca2+-ATPase (SERCA) activity was estimated via a derived rate of decay (kSR) by subtracting the rate of decay of caffeine-induced Ca2+ transients (kcaff) from the rate of decay of pacing-induced Ca2+ transients (Dibb et al. 2004; Belevych et al. 2011b).
To test the propensity for triggered activity, myocytes were paced at 1 Hz for 1 min and the latency between the last stimulus in the pacing train and the first SCW was calculated. To assess the effect of mitochondria-derived ROS on cardiomyocytes from old rabbits, myocytes were pretreated with the mitochondria-specific ROS scavenger (2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-o-xoethyl) triphenylphosphonium chloride (mito-TEMPO, 25 μm, Enzo Life Sciences, Farmingdale, NY, USA) for 10 min. The intracellular Ca2+ cycling measurements were performed under β-adrenergic stimulation with 30 nm ISO.
For Ca2+ spark recordings, myocytes were permeabilized with saponin (0.01% for ∼20 s). The intracellular solution contained (mm): 120 potassium aspartate, 20 KCl, 0.81 MgCl2, 1 KH2PO4, 0.5 EGTA, 3 MgATP, 10 phosphocreatine, 0.03 Fluo-4 pentapotassium salt, 20 Hepes (pH 7.2) and 5 U ml−1 creatine phosphokinase, 100 nm free [Ca2+]. To assess the SR Ca2+ load, 20 mm caffeine was applied at the end of the experiments. The dye was excited with the 488 nm line of an argon laser. Emission was collected at 500–600 nm. For intra-SR [Ca2+] measurements, cells were incubated with Fluo-5N for 2–3 h and were permeabilized with saponin (0.01% for ∼20 s), and the dye was excited with the 488 nm line of an argon laser in x–y mode. Emission was collected at 500–590 nm. SERCA activity was assessed in permeabilized myocytes with Fluo-5N entrapped in the SR as previously described in Belevych et al. (2011b). In brief, in these myocytes, SR Ca2+ was depleted via the application of 10 mm caffeine. Calcium was chelated with EGTA, and the SR Ca2+ uptake was measured by reapplying 250 nm Ca2+ in the presence of the RyR inhibitor Ruthenium Red (Belevych et al. 2007, 2011b). To assess the effect of a reducing agent on [Ca2+]SR, intra-SR [Ca2+] measurements were performed in permeabilized myocytes in the absence or presence of 1 mm dithiothreitol (DTT). The Fluo-5N signal was normalized to the minimum (10 mm caffeine) and maximum (20 μm cAMP + 40 μm Ruthenium Red) fluorescence. The Fluo-5N signal was converted to [Ca2+] using the formula: [Ca2+]SR=Kd(F –Fmin)(Fmax–F)−1, where Kd was 400 μm as previously described (Shannon et al. 2003; Belevych et al. 2011b).
To directly assess RyR-mediated SR Ca2+ leak in intact myocytes treated with ISO, cells were loaded with the membrane-permeable, low-affinity Ca2+ indicator Fluo-5N-AM, exposed to SERCA inhibitor thapsigargin (10 μm) after 30 s of field stimulation at 0.5 Hz, and fluorescence signal from Fluo-5N entrapped in the SR was monitored without stimulation using confocal microscopy. The time constant of decay of Fluo-5N signal was used as a measure of the leak (Belevych et al. 2011b).
Confocal microscopy measurements of intracellular ROS and mitochondrial membrane potential
ROS production was measured in isolated rabbit ventricular myocytes in Tyrode solution using the ROS-sensitive dye 5-(and-6)-chloromethyl-2′,7′-dichlo-rodihydrofluoroscein diacetate (DCFDA, 20 μm, incubated for 30 min) as previously described (Terentyev et al. 2008). The DCFDA dye was excited with the 488 nm line of an argon laser in x–y mode, and emission was collected at 500–530 nm. The rate of ROS production was measured at baseline and in the presence of 30 nm ISO after 3 min in cardiomyocytes from young and old rabbits and cardiomyocytes pretreated with mito-TEMPO from old rabbits.
Mitochondrial membrane potential was monitored with a voltage-sensitive fluorescent indicator, tetramethylrhodamine ethyl ester (TMRE) as previously described (Ho et al. 2011). In brief, isolated rabbit ventricular cardiomyocytes were loaded with 1 nm TMRE (10 min), and TMRE fluorescence was measured in x–y mode. TMRE was excited at 543 nm with a helium–neon laser, and the emission signals were collected at 570–650 nm. TMRE fluorescence was normalized to the minimum fluorescence signal obtained with the mitochondrial uncoupler carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP, 50 μm). Mitochondrial membrane potential was monitored at baseline and after application of 30 nm ISO with cardiomyocytes from young rabbits and with cardiomyocytes from old rabbits with and without mito-TEMPO (25 μm, 10 min preincubation).
RyR oxidation and Western blotting of Ca2+-handling proteins
The content of free thiols in RyRs was determined with the monobromobimane (mBB, EMD Millipore, Billerica, MA, USA) fluorescence method as previously described (Xu et al. 1998; Terentyev et al. 2008; Ho et al. 2011). In brief, myocytes were incubated with 20 mm mBB for 1 h in the dark at room temperature, and the proteins from cell lysates were acetone precipitated and resolved via SDS-PAGE. To compare relative oxidation between samples, RyR oxidation was normalized to minimum and maximum RyR thiol oxidation obtained with 10 mm DTT (a reducing agent) or 200 μm 2,2′-dithiodipyridine (DTDP, oxidizing agent), respectively.
The levels of calsequestrin (CASQ2, Thermo Fisher, Waltham, MA, USA), phospholamban (PLB, Thermo Fisher), SERCA (Thermo Fisher), NCX (Thermo Fisher), α1c subunit L-type calcium channel (LTCC, EMD Millipore), RyR (Thermo Fisher) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Thermo Fisher) proteins were determined by immunoblotting. Phosphorylation levels of PLB at its protein kinase A (PKA) site Ser-16 and Ca2+/calmodulin-dependent protein kinase II (CaMKII) site Thr-17 were assessed using phosphospecific antibodies from Upstate (Lake Placid, NY, USA) and Santa-Cruz (Santa-Cruz, CA, USA), respectively. Phosphorylation levels of RyRs were determined using custom-made anti-phospho-Ser-2031 (Yenzym, San Francisco, CA, USA); anti-phospho-Ser-28089 and Ser-2815 (Phosphosolutions, Aurora, CO, USA) antibodies as previously described (Belevych et al. 2011a). The levels of phosphorylation of RyRs and PLB at specific sites in myocytes at baseline conditions and after 3 min with 30 nm ISO were normalized to maximum phosphorylation achieved by incubation of cells for 10 min with 1 μm ISO plus 1 μm calyculin A, an inhibitor of Ser-Thr protein phosphatase 1 (PP1) and phosphatase 2A (PP2A). Tissue or cell homogenates proteins (20–30 μg) were resolved on a 4–20% gel via SDS-PAGE, transferred onto nitrocellulose membranes, and probed with mouse or rabbit antibodies specific for these proteins and subsequently probed with a donkey anti-mouse or mouse anti-rabbit secondary antibodies (Thermo Fisher). Blots were developed with SuperSignal West Pico and Femto (Thermo Fisher) and quantified and analysed by using GeneSnap (Syngene, Cambridge, UK) and ImageJ (US National Institutes of Health, Bethesda, MD, USA) softwares.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) of n measured cells or rabbits. Statistical comparisons between groups were performed with Student's t test (paired and unpaired) and one-way ANOVA where appropriate. Differences that are not statistically different are denoted as NS. Differences were considered statistically significant at P < 0.05.
Results
Preserved baseline cellular electrophysiology and Ca2+ homeostasis in myocytes from ageing hearts
First we examined the effect of ageing on basic cellular electrophysiology and Ca2+ homeostasis under basal conditions. The intracellular Ca2+ cycling in paced (1 Hz) cardiomyocytes isolated from young and old rabbits was monitored in the line scan mode of a confocal microscope using the Ca2+ indicator Fluo-3. The effects of ageing on cytosolic Ca2+ cycling at baseline conditions are illustrated in Fig. 1A and summarized in Fig. 1B–E. Ageing did not change Ca2+ transient and caffeine transient amplitudes nor did it change transient decay rates indicating that ageing does not alter NCX and SERCA activities at basal conditions. Importantly, ageing does not alter baseline cellular electrophysiology parameters, including action potential duration, Ca2+ and potassium currents, in single cardiomyocytes (see Fig. 1 in Supplemental material, available online). Moreover, with Western blots we did not observe any significant changes in expression levels of important Ca2+-handling proteins (Supplemental Fig. 2), including LTCC pore-forming subunit α1c, RyRs, NCX, SERCA and PLB.
Figure 1. Ageing reduces ability to maintain SR Ca2+ content by increasing RyR-mediated Ca2+ leak.
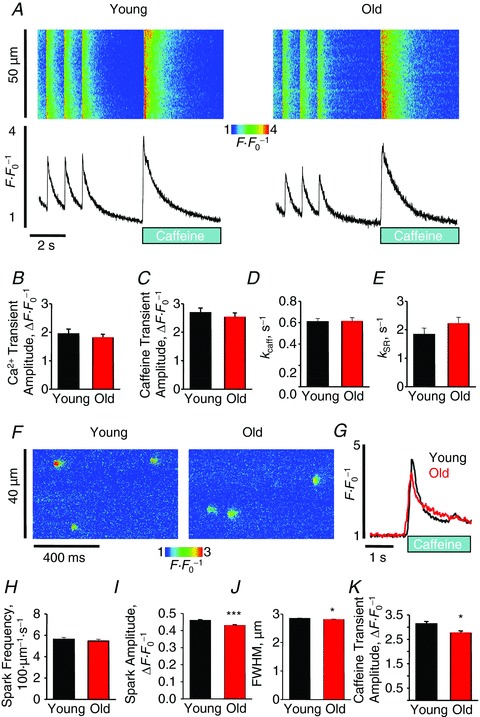
A, representative confocal line scan images and Fluo-3 F F0−1 profiles at 1 Hz. Graphs B–E depict mean data from Ca2+ transient amplitude (young, 1.94 ± 0.17 vs. old, 1.80 ± 0.13 ΔF F0−1), caffeine transient amplitude (2.69 ± 0.16 vs. 2.52 ± 0.16 ΔF F0−1), and decay rate constants (from exponential fit): kcaff: 0.61 ± 0.03 vs. 0.61 ± 0.04 s−1 and kSR: 1.83 ± 0.23 vs. 2.21 ± 0.23 s−1. For baseline Ca2+ transient, n = 12–23 cells from 3–4 heart preparations; all values P= NS and ± SEM. F, representative confocal line scan images with Fluo-4 of spark activity in saponin-permeabilized cardiomyocytes from young and old rabbits. G, representative Fluo-4 ΔF F0−1 profiles of caffeine-induced Ca2+ transients in permeabilized cardiomyocytes from young and old rabbits. Bar graphs H–K show mean data for spark frequency (young, (5.62 ± 0.18) × 100 μm−1 s−1 vs. old, (5.47 ± 0.17) × 100 μm−1 s−1, P = NS), spark amplitude (0.46 ± 0.004 vs. 0.43 ± 0.004 ΔF F0−1, ***P < 0.001), FWHM (2.85 ± 0.01 vs. 2.81 ± 0.01 μm, *P < 0.05), and caffeine transient amplitude (2.13 ± 0.10 vs. 1.76 ± 0.09 ΔF F0−1, *P < 0.05), respectively. For caffeine application, n = 20–35 cells, and for spark analysis n= 205–247 cells; n= 3431–4395 sparks from 3–4 heart preparations. All values are mean ± SEM.
Ageing alters spark parameters and SR load in cardiomyocytes
To assess the effect of ageing in a large animal model on RyR activity directly we employed a saponin-permeabilized cells experimental system to measure SR Ca2+ content and parameters of spontaneous local Ca2+ release events through RyR clusters, Ca2+ sparks (Fig. 1F–K). Ageing reduced spark amplitude and spark size (full width at half-maximum, FWHM) but had no significant effect on spark frequency. Parallel assessment of SR Ca2+ load via the amplitude of the caffeine-induced Ca2+ transients in saponin-permeabilized myocytes revealed small but significant reduction of SR Ca2+ content in old myocytes in comparison with myocytes from young rabbits (Fig. 2G and K). Preserved spark frequency under conditions of reduced SR Ca2+ load suggest that RyRs are more active in cardiomyocytes from old rabbits (Lukyanenko et al. 2001).
Figure 2. Increased thiol oxidation underlies increased RyR-mediated SR Ca2+ leak in cardiomyocytes from old rabbits.
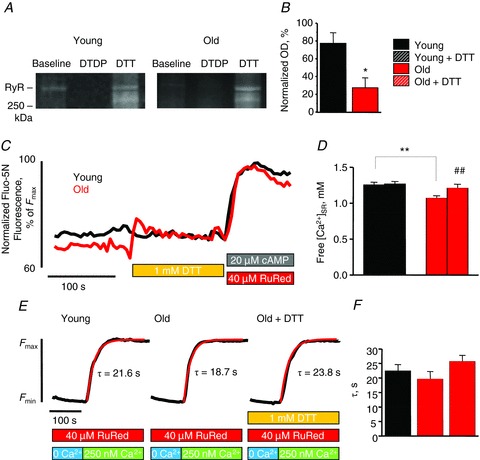
A and B, representative mBB fluorescence intensity signals of RyRs from cardiomyocytes from young and old rabbits and pooled data for free thiol contents normalized by DTT and DTDP: young, 77.89 ± 12.02 vs. old, 27.39 ± 11.09%. *P < 0.05. Cells were from 4–6 heart preparations. OD, optical density. C, representative tracing of time-dependent changes in luminal Fluo-5N signal in permeabilized cardiomyocytes from young and old rabbits after the addition of 1 mm DTT. Data are normalized to the minimum (10 mm caffeine) and maximum (20 μm cAMP + 40 mm Ruthenium Red (RuRed)) fluorescence. D, mean data for [Ca2+]SR before and after DTT application in cardiomyocytes from young and old rabbits. Fluo-5N signal was converted to [Ca2+] using the formula: [Ca2+]SR=Kd(F –Fmin)(Fmax–F)−1, where Kd was 400 μm. [Ca2+]SR values were as follows: young, 1.25 ± 0.04; young + DTT, 1.27 ± 0.04; old, 1.07 ± 0.03; and old + DTT 1.21 ± 0.06 mm. *P < 0.05 for young vs. old (unpaired analysis), #P < 0.05 for old vs. old + DTT (paired analysis), and *P = NS for young + DTT and old + DTT (unpaired analysis). n= 10–13 cells from 2–3 heart preparations. All values are ± SEM. E, representative tracing of time-dependent changes in luminal Fluo-5N signal in permeabilized cardiomyocytes from young and old rabbits and from permeabilized cardiomyocytes from old rabbits in the presence of 1 mm DTT after depletion of SR Ca2+, and chelating cytosolic Ca2+ ([Ca2+]cyt), blocking of RyRs, and reapplication of 250 nm of Ca2+. F, mean time constants (τ) from exponential fit of SR Ca2+ uptake for permeabilized cardiomyocytes: young, 22.51 ± 2.10; old, 19.69 ± 2.54; and old + DTT, 25.71 ± 2.13 s. P = NS for all comparisons. n = 9–21 cells from two heart preparations; all values are mean ± SEM.
Redox modification of RyRs with ageing
We directly examined the extent of RyR thiol modification in myocytes from young and old rabbits with an mBB fluorescence labelling assay, as depicted in Fig. 2A and summarized in Fig. 2B. The fraction of RyR free thiols was decreased significantly in cardiomyocytes from old rabbits indicating that ageing leads to increased RyR modification by reactive oxygen/nitrogen species (ROS/RNS). To investigate the role of redox status in the age-related increase of RyR activity and reduction of SR Ca2+ load, we examined the effect of antioxidant DTT, with a capacity to reverse glutathionylation, nitrosylation, and thiol oxidation, on SR Ca2+ content in Fluo-5N-loaded myocytes from young and old rabbits. The results are depicted in Fig. 2C and summarized in Fig. 2D. Consistent with the spark data, myocytes from old rabbits exhibit lower SR Ca2+ load, and application of 1 mm DTT restored the SR Ca2+ content in myocytes from old rabbits to the levels comparable to myocytes from young animals. Application of ascorbate (5 mm) to specifically reverse nitrosylation or glutaredoxin (1 U ml−1) plus glutathione (GSH, 0.5 mm) to reverse both nitrosylation and glutathionylation (Terentyev et al. 2008) produced no effects on SR Ca2+ content (Supplemental Fig. 3C). These data indicate that thiol oxidation and not nitrosylation or glutathionylation potentially causes enhanced RyR activity in ageing myocytes.
Preserved SERCA activity with ageing
Since enhanced RyR-mediated SR Ca2+ leak can effectively mask possible changes in SERCA function, SERCA-mediated Ca2+ uptake was measured in the absence of RyR-mediated Ca2+ leak as described above (Belevych et al. 2007). Figure 2E illustrates and Fig. 2F summarizes that ageing does not change SERCA activity, which is consistent with the above-mentioned kSR data from intracellular Ca2+ measurements in intact cells. Importantly, application of DTT had no effect on the rate of uptake in old myocytes.
Taken together, these results suggest that impairment of the ability of SR to retain Ca2+ in ageing is caused primarily by an abnormally high activity of thiol-oxidized RyRs while SERCA function remains largely preserved.
Age-related depolarization of mitochondria and increased ROS production
To further examine the mechanism of RyR oxidation in ageing myocytes, we performed mitochondrial membrane potential measurements with TMRE and ROS production rate experiments with the ROS-sensitive indicator DCFDA. Figure 3A shows representative images of mitochondrial membrane potential in young and old myocytes as measured by TMRE fluorescence at 0 s. As depicted in Fig. 3B and summarized in Fig. 3C, myocytes from old rabbits had more depolarized mitochondrial membrane at baseline and after β-adrenergic stimulation. Additionally, β-adrenergic stimulation slightly but significantly further depolarized mitochondrial membrane potential in both young and old myocytes by about 4% and 7%, respectively. Since ROS via mitochondrial dysfunction has been shown to be a source of protein oxidation in the heart (Giordano, 2005; Ho et al. 2011; Bovo et al. 2012), we investigated the effect of ageing on myocyte ROS production using DCFDA as shown in Fig. 4A and summarized in Fig. 4B and C. We observe that cardiomyocytes from old rabbits show a significantly higher rate of ROS production compared to young myocytes at baseline, which is exacerbated with β-adrenergic stimulation. Importantly, myocytes from old rabbit hearts that were preincubated with the mitochondria-specific ROS scavenger mito-TEMPO exhibited a markedly reduced rate of ROS production when compared to old myocytes at baseline and with β-adrenergic stimulation. Additionally, under β-adrenergic stimulation, we observe no statistically significant difference in the rate of ROS production between young myocytes and old myocytes pretreated with mito-TEMPO. These data further implicate the important role of age-associated mitochondrial dysfunction on increased ROS production and protein redox status in the heart.
Figure 3. Old cardiomyocytes have mild mitochondrial uncoupling, which is exacerbated in the presence of ISO.

A, representative images of mitochondrial membrane potential at 0 s measured by using TMRE. B and C, traces and mean data of normalized TMRE fluorescence at baseline and ISO (30 nm). Data are normalized to the minimum TMRE fluorescence in the presence of electron transport chain uncoupler FCCP (50 μm): young, 5.71 ± 0.55; young + ISO, 5.51 ± 0.53; old, 4.16 ± 0.42; and old + ISO, 3.87 ± 0.41. *P < 0.05 for young vs. old and for young + ISO vs. old + ISO (unpaired analysis), ##P < 0.01 for young vs. young + ISO (paired analysis), and ###P < 0.001 for old vs. old + ISO (paired analysis). n= 20–21 cells from 2–4 heart preparations; all values are mean ± SEM.
Figure 4. Rate of ROS production is higher in old cardiomyocytes and is attenuated with a mitochondrial ROS scavenger.
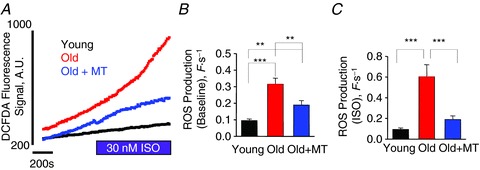
A, changes in DCFDA fluorescence signal at baseline and with ISO (30 nm) with cardiomyocytes from young and old rabbits and from cardiomyocytes pretreated with mito-TEMPO (MT, 25 μm) for 10 min from old rabbits. B, the mean data for rate of ROS production at baseline: young, 0.09 ± 0.01; old, 0.32 ± 0.04; and old + MT, 0.19 ± 0.03 F s−1. ***P < 0.001 for young vs. old and **P < 0.01 for young vs. old + MT and for old vs. old + MT. C, the mean data for rate of ROS production with 30 nm ISO: young, 0.09 ± 0.01; old, 0.60 ± 0.12; and old + MT, 0.19 ± 0.04 F s−1. ***P < 0.001 for young vs. old and for old vs. old + MT, and P = NS for young vs. old + MT. n= 8–20 cells from 2–4 heart preparations; all values are mean ± SEM.
Age-associated aberrant Ca2+ handling under β-adrenergic stimulation is reversed with mitochondria-specific ROS scavenger
To further investigate the role of ageing on aberrant Ca2+ dynamics and mitochondrial ROS, we examined intracellular Ca2+ cycling in paced (1 Hz) myocytes from young and old rabbits and myocytes from old rabbits that were pretreated with mito-TEMPO. Here, all myocytes were challenged with the β-adrenergic agonist ISO (30 nm). The effects of ageing on Ca2+ cycling under β-adrenergic stimulation are illustrated in Fig. 5A and summarized in Fig. 5B–E. Myocytes from old rabbit hearts had significantly lower Ca2+ transient amplitude and caffeine-induced transient amplitude when compared to young myocytes – reduced about 30% for both parameters. Moreover, myocytes from old rabbit hearts in the presence of the mitochondria-specific ROS scavenger mito-TEMPO exhibited significantly higher caffeine transient amplitudes when compared to untreated old myocytes. This increase brought caffeine transient amplitude to levels comparable to young cardiomyocytes indicating that treatment with mito-TEMPO restores SR Ca2+ load under these conditions. Analysis of decay kinetics of electrically evoked Ca2+ transients revealed no differences between groups indicating preserved SERCA activity. Interestingly, the decay of caffeine-induced Ca2+ transient that reflects NCX activity was significantly slower in old myocytes vs. young (Fig. 5D). This effect was completely abolished by mito-TEMPO in old myocytes.
Figure 5. Mitochondrial ROS scavenger mito-TEMPO reverses the effects of ageing with regard to SR Ca2+ release under β-adrenergic stimulation.

A, confocal line scan images and corresponding temporal Fluo-3 F F0−1 profiles at 1 Hz from cardiomyocytes from young and old rabbits, and from cardiomyocytes from old rabbits pretreated with mito-TEMPO (MT, 25 μm) for 10 min. All experiments were performed in the presence of 30 nm ISO. B, mean data for Ca2+ transient amplitude: young, 4.23 ± 0.24; old, 2.98 ± 0.26; and old + MT, 3.80 ± 0.42 ΔF F0−1. *P < 0.05 for young vs. old. C, mean data for caffeine transient amplitude: young, 4.65 ± 0.27; old, 3.25 ± 0.27; and old + MT, 4.44 ± 0.41 ΔF F0−1. *P < 0.05 for old vs. old + MT, **P < 0.01 for young vs. old. D, mean data for kcaff decay rate constant: young, 0.57 ± 0.04; old, 0.42 ± 0.02; and old + MT, 0.66 ± 0.07 s−1. *P < 0.05 for young vs. old. **P < 0.01 for old vs. old + MT. E, mean data for kSR rate constant: young, 5.09 ± 0.29; old, 5.93 ± 0.58; old + MT, 4.48 ± 0.72 s−1. P= NS. n = 13–17 cells from 3 heart preparations; all values are mean ± SEM.
Mito-TEMPO attenuates accelerated RyR-mediated leak and has no effects on ICa,L in ageing myocytes
Unchanged SERCA function and reduced NCX activity cannot account for the significantly diminished SR Ca2+ load observed in old cells treated with ISO. To test if altered responsiveness of L-type Ca2+ channels (LTCCs) to β-adrenergic stimulation plays a role in age-related changes in [Ca2+]SR, we performed voltage clamp experiments to measure ICa,L. Figure 6 demonstrates that augmentation of ICa,L amplitude by 30 nm ISO is ∼3-fold smaller in old myocytes in comparison to young cells, consistent with previous findings (Xiao et al. 1994; Cerbai et al. 1995). However, treatment with mito-TEMPO did not affect ICa,L amplitude in old myocytes (Fig. 6C), which indicates that a mito-TEMPO-mediated increase in SR Ca2+ content in old myocytes does not involve LTCCs and further implicates RyRs as the most probable target. Direct measurement of RyR-mediated leak was performed in intact myocytes. As shown in Fig. 7, preincubation of intact old myocytes with 25 μm mito-TEMPO for 10 min completely abolished age-related acceleration of SR Ca2+ leak, while application of 250 μm caffeine mimicked ageing phenotype in young cells. Taken together, our results further support the critical role of hyperactive RyRs, which are oxidized by mitochondria-derived ROS in the reduction of SR Ca2+ content in ageing cells.
Figure 6. Depressed β-adrenergic augmentation of ICa,L in old cardiomyocytes.
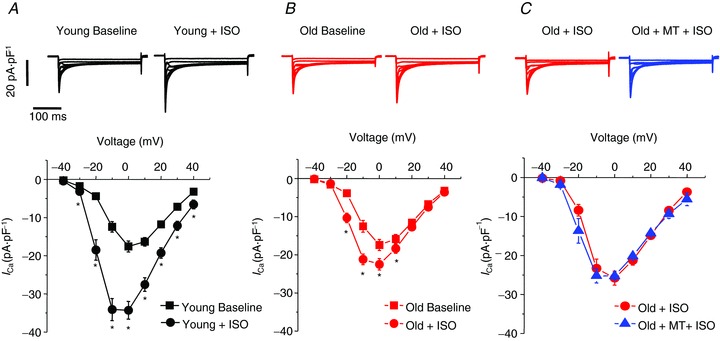
Representative Ca2+ current traces (top) and mean current–voltage relationships (bottom) are shown for baseline condition and after 30 nm ISO from young (A, n= 10) and old (B, n= 11) cardiomyocytes. Currents were normalized to cell capacitance. Calcium currents were activated with depolarizing membrane potentials between −40 and + 40 mV from a holding potential of −50 mV. *P < 0.05, paired Student's t test. Percentage increase of ICa,L at 0 mV in response to 30 nm ISO was 100.3 ± 13.5% in young myocytes vs. 34.6 ± 10.5%* in old cells, *P < 0.001. C, representative Ca2+ current traces (top) and mean current–voltage relationships (bottom) from old cardiomyocytes (n= 4) with 30 nm ISO (red) and >5 min after addition of 25 μm mito-TEMPO (blue). All values are mean ± SEM.
Figure 7. Ageing increases SR Ca2+ leak, which is reversed with mitochondrial ROS scavenging with mito-TEMPO.

A, representative F F0−1 time-dependent profiles of cell-averaged luminal Fluo-5N signals from intact cardiomyocytes from young and old rabbits as well as from cardiomyocytes from old rabbits pretreated with mito-TEMPO (MT, 25 μm) for 10 min, and from cardiomyocytes from young rabbits in the presence of 250 μm caffeine. Fluorescence signal was normalized to the minimum fluorescence with 10 mm caffeine. B, mean time constants (τ) from exponential fit of SR Ca2+ leak from intact cardiomyocytes: young, 481.98 ± 61.23; old, 246.31 ± 26.75; old + MT, 575.05 ± 35.52; and young + caffeine, 293.49 ± 34.98 s. *P < 0.05 for young vs. young + caffeine. **P < 0.01 for young vs. old and old + MT vs. young + caffeine. ***P < 0.001 for old vs. old + MT. n = 9–16 cells from 2 heart preparations; all values are mean ± SEM.
Mito-TEMPO restores RyR redox status but has no effects on phosphorylation patterns of RyR and PLB in ageing myocytes
The results of the mBB assay presented in Fig. 8 revealed that incubation of ISO-treated old myocytes with mito-TEMPO significantly decreases the amount of oxidized thiols on RyRs, achieving redox status similar to that of RyRs from young rabbit ventricular cells. To assess possible changes in phosphorylation levels of RyR and PLB in ageing and the effects of mito-TEMPO on the phosphorylation state of these polypeptides, we performed Western blot analysis using phospho-specific antibodies against RyR (PKA sites Ser-2031 and Ser-2809 and CaMKII site Ser-2815, and PLB (PKA site Ser-16 and CaMKII site Thr-17). Figure 9 demonstrates that ageing does not alter basal phosphorylation levels of RyRs and PLB at any tested site; however, ageing is associated with a 2-fold increase in the level of CaMKII phosphorylation at Ser-2815 and phosphorylation at Ser-2031 in the presence of 30 nm ISO. Nevertheless, analysis of samples obtained from old myocytes treated with mito-TEMPO did not uncover any effects of mitochondria-specific ROS scavenger on phosphorylation status of either RyRs or PLB. Therefore, mito-TEMPO restores SR Ca2+ content by reducing oxidized thiols on RyRs and not by altering RyR and PLB phosphorylation in ageing myocytes.
Figure 8. Scavenging of mitochondrial ROS restores decreased RyR free thiol content in ageing myocytes treated with the β-adrenergic agonist ISO.
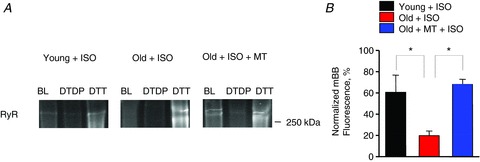
A, representative mBB-fluorescence of RyRs from young, old, and old myocytes incubated with mito-TEMPO (25 μm, 10 min) and exposed to ISO (30 nm, 3 min) in the absence and presence of the oxidizing agent DTDP (0.2 mm) and in the presence of the reducing agent DTT (5 mm). B, relative thiol content of RyRs from young myocytes, old myocytes, and old myocytes treated with mito-TEMPO myocytes. *P < 0.05. n= 4–6 heart preparations; all values are mean ± SEM.
Figure 9. Scavenging of mitochondrial ROS does not change phosphorylation status of RyRs and PLB in ageing myocytes under β-adrenergic stimulation.
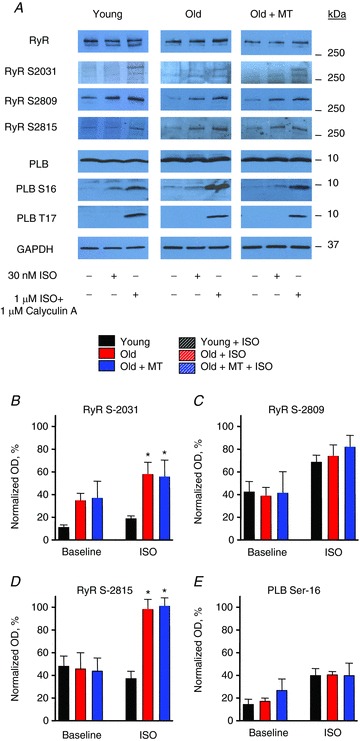
A, representative Western blots showing phosphorylation of RyRs at Ser-2809, Ser-2031 and Ser-2814 sites and PLB phosphorylation sites Ser-16 and Thr-17 in young, old and old myocytes treated with 25 μm mito-TEMPO for 10 min under baseline conditions and after treatment with 30 nm ISO for 3 min. B, pooled data for relative levels of phosphorylation of RyRs at Ser-2809, -2831 and -2814 and PLB monomers at PKA site Ser-16. Signals obtained from samples treated with anti-phospho site-specific antibodies were normalized to RyR or PLB levels measured in gels run in parallel and to maximum phosphorylation levels obtained by treatment of myocytes with 1 μm ISO and 1 μm calyculin A for 10 min. *P < 0.05. n= 3–6 heart preparations; all values are mean ± SEM.
Mitochondrial ROS scavenger attenuates age-associated increase in propensity for proarrhythmic spontaneous Ca2+ release
As portrayed in Fig. 10, the arrhythmogenic potential associated with ageing was examined by measuring spontaneous Ca2+ waves (SCWs) in myocytes in the presence of ISO after cessation of 1 Hz pacing for 1 min. As shown in Fig. 10B and C, we observed a ∼40% increase in the rate of occurrence of SCWs as well as an almost 4-fold reduction in SCW latency time in old myocytes when compared to young myocytes. A similar pattern was obtained in young myocytes in the presence of 250 μm caffeine, which activates RyRs. Furthermore, pretreatment with mito-TEMPO reversed this age-related effect: not only did mito-TEMPO pretreatment reduce the number of old myocytes that exhibited SCWs (comparable to the young myocytes), but also, mito-TEMPO pretreatment significantly increased refractoriness of RyR-mediated Ca2+ release in old myocytes as evidenced by an increase in SCW latency time. Thus, mito-TEMPO reversed the ageing cellular phenotype in cardiomyocytes elucidating the major role of mitochondrial ROS in aberrant Ca2+ dynamics and arrhythmogenesis in the ageing heart.
Figure 10. Mitochondrial ROS scavenger mito-TEMPO reverses the effects of ageing with regard to refractoriness of RyR-mediated Ca2+ release under β-adrenergic stimulation.
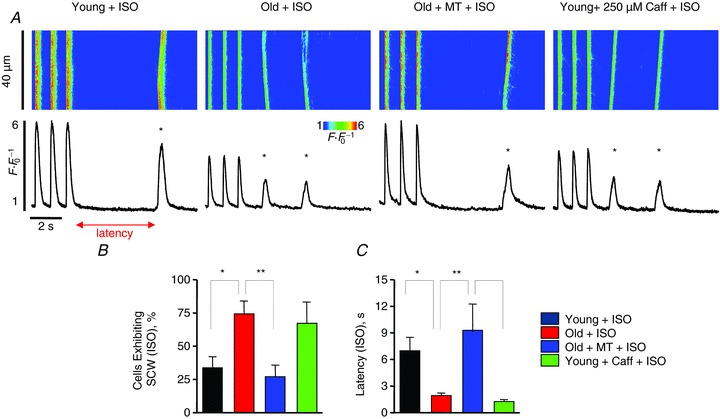
A, confocal line scan images and Fluo-3 F F0−1 profiles at the end of 1 min 1 Hz pacing train with cardiomyocytes from young and old rabbits and from cardiomyocytes pretreated with mito-TEMPO (25 μm) for 10 min from old rabbits with 30 nm ISO. Spontaneous Ca2+ waves (SCWs) are indicated by an asterisk. B, mean data for percentage of cells exhibiting SCWs: young, 33.33 ± 8.75; old, 73.68 ± 10.38; old + MT, 26.93 ± 8.87, and young + caffeine 66.67%. *P < 0.05 for young vs. old, **P < 0.01 for old vs. old + MT. n= 9–30 cells from 2–4 heart preparations. C, mean data for SCW latency: young, 6.94 ± 1.58; old, 1.88 ± 0.34; old + MT, 9.11 ± 3.15, and young + caffeine, 1.20 ± 0.28 s. *P < 0.05 for young vs. old and old + MT vs. young + caffeine. **P < 0.01 for old vs. old + MT. n= 6–14 cells from 2–4 heart preparations. All values are mean ± SEM.
Discussion
Here, we present a novel finding demonstrating a mechanism that is involved with normal age progression and its relation to alterations in Ca2+ cycling as it pertains to increased arrhythmogenic potential in the ageing heart. We showed that ageing significantly increased SR Ca2+ leak via post-translational thiol oxidation of RyRs caused by differential production of mitochondrial ROS. This led to diminished systolic Ca2+ release, accelerated RyR-mediated loss of [Ca2+]SR as well as the development of SCWs in the presence of the β-adrenergic agonist ISO. Thus, the major age-related differences in Ca2+ homeostasis occur at the level of the SR and manifest at sarcolemma as reduction in pacing-induced systolic Ca2+ release due to decreased SR Ca2+ content as well as potentially malignant SCWs under β-adrenergic stimulation. This mechanism probably underlies the enhanced molecular trigger leading to age-related, Ca2+-dependent arrhythmias.
Redox modification of RyR with age
Although it has been shown that ageing is associated with aberrant RyR function (Zhu et al. 2005), the molecular mechanism underlying the increase in RyR activity has yet to be thoroughly investigated. Thus, an important finding of this study is that ageing significantly increases ROS levels in rabbit cardiomyocytes leading to the oxidation of RyRs, rendering them hyperactive. It is known that the RyR is a highly cysteine-rich protein (Xu et al. 1998) and highly susceptible to oxidation (Zima & Blatter, 2006; Mochizuki et al. 2007; Terentyev et al. 2008; Belevych et al. 2011b, 2012; Donoso et al. 2011). Forms of reactive oxygen or nitrogen species have also been shown to increase the open probability of RyRs (Eager et al. 1997; Xu et al. 1998; Zima & Blatter, 2006; Terentyev et al. 2008; Donoso et al. 2011; Niggli et al. 2013); moreover, these modifications can be reversed by the reducing agent DTT (Eager et al. 1997; Zable et al. 1997; Haarmann et al. 1999; Terentyev et al. 2008). Here, we provide direct evidence of an age-associated increase in RyR thiol oxidation (Fig. 2), and we observe that the exposure of permeabilized myocytes to DTT reduced SR Ca2+ leak restoring SR Ca2+ content without effects on SERCA function in old myocytes (Fig. 2), which, when taken together, indicate that RyR oxidation via increased ROS is the mechanism of exacerbated SR Ca2+ leak in cardiomyocytes in ageing. Previous studies demonstrated that, depending on specific animal model or sex, the amplitude of Ca2+ transients under basal conditions can be increased (Dibb et al. 2004), reduced (Howlett, 2010) or unchanged (Xiao et al. 1994; Howlett, 2010). We show that age-related changes in RyR activity in myocytes isolated from ageing female rabbit hearts do not translate into readily detectable changes in parameters of Ca2+ transients under basal conditions, which bears similarity to other conditions associated with RyR hyperactivity including catecholaminergic polymorphic tachycardia, infarct or early stages of heart failure (Knollmann et al. 2006; Belevych et al. 2009, 2011b, 2012). Additionally, previous studies imply that the function of other important Ca2+-handling proteins, including SERCA, NCX and LTCC, can be effectively regulated by ROS (Eigel et al. 2004; Zima & Blatter, 2006; Zeng et al. 2008; Kuster et al. 2010; Lancel et al. 2010). In our present study, we show a lack of age-associated functional changes in parameters under basal conditions for ICa,L (Supplemental Fig. 1), SERCA-mediated SR Ca2+ uptake (Fig. 2E and 2F), and Ca2+ removal by NCX (Fig. 1D), which suggests that the age-associated increase in levels of ROS in our experimental model is not sufficient to modify their activities in old rabbit cardiomyocytes. Thus, our data elucidate RyRs as a primary highly sensitive target for modulation by ROS in the ageing heart within the cascade of Ca2+-induced Ca2+ release.
The role of mitochondrial ROS on increased arrhythmic potential with age
It has been well established by Harman et al. and others that mitochondria are a major source for age-related increase in ROS production and the primary source of ROS in cardiomyocytes (Harman, 1972; Miquel et al. 1980; Chaudhary et al. 2011). Also, previous studies have revealed that the distances between mitochondria and RyR SR Ca2+ release clusters are sufficiently short in cardiomyocytes to establish local Ca2+ communication (Ramesh et al. 1998; Szalai et al. 2000; Zhou et al. 2011). It would be expected that this close proximity would be permissive for retrograde transmission of signals from mitochondria in the form of transitory ROS that can effectively modify RyR function (Zhou et al. 2011). Although severe uncoupling of mitochondrial membrane potential (Δψm) leads to the cessation of oxidative phosphorylation and ROS production, it has been demonstrated that mild uncoupling of the mitochondria membranes leads to increased respiration and increased levels of ROS, such as superoxide and hydrogen peroxide (Johnson-Cadwell et al. 2007; Shabalina & Nedergaard, 2011). Here, we show that Δψm is reduced by ∼25% in myocytes from old rabbit hearts (Fig. 3) representing a mild uncoupling, which we posit as the likely mechanism leading to increased ROS production (Fig. 4), oxidation of RyRs (Figs 2A and B, and 8), and the subsequent proarrhythmic dysfunction described below in old cardiomyocytes.
Under β-adrenergic stimulation, when both LTCC-mediated Ca2+ influx and SERCA-mediated Ca2+ uptake are enhanced, hyperactivity of RyRs is directly linked to increased frequency of SCWs that (via an electrogenic NCX) translate into arrhythmogenic deflections of membrane potential in the form of delayed or early depolarizations (Bers et al. 2002; Pogwizd & Bers, 2002; Györke & Carnes, 2008; Belevych et al. 2011b). We show in myocytes from old rabbits that mitochondrial ROS increases oxidation of RyRs (Fig. 8) and enhances RyR functional activity as manifested by increased rate of SR Ca2+ leak (Fig. 7). Furthermore, reduced RyR refractoriness results in increased arrhythmic potential which is manifested in increased frequency of SCWs in the presence of ISO (Fig. 10). These data are in line with previous studies that show that redox modification of RyRs by mitochondrial ROS enhances RyR functional activity that results in increased frequency of SCWs (Ho et al. 2011; Bovo et al. 2012). Importantly, the cellular phenotype of old myocytes was mimicked by treatment of young myocytes with 250 μm caffeine to specifically increase RyR activity (Figs 7 and 10). Additionally, scavenging mitochondrial ROS during β-adrenergic stimulation resulted in a 3-fold reduction of rate of ROS generation (Fig. 4), and in these myocytes, RyR oxidation state was restored (Fig. 8), the Ca2+ transient amplitude was increased (Fig. 5B), the SR Ca2+ leak and the frequency of SCWs were reduced (Fig. 7 and Fig. 10, respectively), and the SR Ca2+ content, assessed by rapid application of 10 mm caffeine, was normalized to that comparable to young myocytes (Fig. 5C).
The effects of ageing on Ca2+ homeostasis under β-adrenergic stimulation are striking in the light of unchanged Ca2+ transients in intact cells and subtle effects on Ca2+ sparks in permeabilized myocytes under basal conditions (Fig. 1). This is probably due to the reduced ability of RyRs to deactivate at increased levels of luminal Ca2+ in conditions where inhibitory action of PLB on SERCA is countered by PKA phosphorylation. As previously shown, ROS modification results in increased sensitivity of RyRs to luminal Ca2+ (Terentyev et al. 2008); therefore, increased SR Ca2+ load in comparison to baseline in ISO-treated cells will further enhance SR Ca2+ leak via oxidized RyRs in old cardiomyocytes resulting in lowered SR Ca2+ content and Ca2+ transient amplitude. Our results are in line with findings demonstrating that increased activity of RyRs alone is insufficient to induce changes in Ca2+ spark frequency (Lukyanenko et al. 2001) or Ca2+ transient amplitude, or evoke SCWs (Venetucci et al. 2007), and enhanced SR Ca2+ uptake is necessary to unmask effects of dysfunctional RyRs on Ca2+ cycling (Eisner et al. 2013).
Reversible post-translational modifications of RyRs from ageing myocytes
In addition to thiol oxidation, post-translational control of RyR activity has been ascribed to reversible nitrosylation, glutathionylation, phosphorylation at RyR PKA sites Ser-2809 and Ser-2031, and CaMKII site Ser-2815 (Witcher et al. 1991; Xu et al. 1998; Rodriguez et al. 2003; Wehrens et al. 2004; Sanchez et al. 2005; Guo et al. 2006; Xiao et al. 2006; Zima & Blatter, 2006; Gonzalez et al. 2007; Sun et al. 2008; Donoso et al. 2011; Niggli et al. 2013). We did not find any functional effects of pharmacological agents that reverse glutathionylation and nitrosylation in ageing cells, suggesting there was no role for these modifications in ageing-related increase in RyR activity (Supplemental Fig. 3C). We also did not find any differences in basal levels of RyR phosphorylation at any of the three phosphorylation sites, or responses of PKA site Ser-2809 to β-adrenergic stimulation. Conversely, we detected a significant increase in RyR phosphorylation at CaMKII site Ser-2815 and site Ser-2031 from ageing myocytes treated with 30 nm ISO in comparison with controls (Fig. 9). Based on our previous studies, although initially identified as a PKA-specific site (Xiao et al. 2006), Ser-2031 is also sensitive to phosphorylation by CaMKII (Belevych et al. 2011a). Oxidative stress has been shown to increase CaMKII activity (Erickson et al. 2008), thereby indirectly affecting RyR function via enhanced phosphorylation at CaMKII sites. However, incubation of old myocytes with mito-TEMPO that reversed RyR oxidation and rescued aberrant Ca2+ handling in old myocytes did not affect altered RyR phosphorylation patterns (Fig. 9), suggesting additional changes in activity and/or expression levels of enzymes that control RyR phosphorylation with ageing. Furthermore, these results indicate that the restoration of thiol oxidation status of RyRs with mitochondria-specific ROS scavenger is sufficient to stabilize RyR function in ageing cardiomyocytes. Notably, inhibition of CaMKII also produced a significant decrease in the frequency of SCWs in old cells treated with ISO (Supplemental Fig. 5), confirming that simultaneous oxidation and CaMKII-mediated phosphorylation of RyRs produce much larger effects on RyR activity than either of these two modifications alone (Belevych et al. 2012).
Conclusions
These data reveal a novel mechanism underlying the development of age-associated proarrhythmic Ca2+ waves and decreased RyR refractoriness during β-adrenergic stimulation. A differential rate of mitochondrial ROS production with age along with sympathetic stimulation is critical in altering RyR redox status, leading to SR Ca2+ leak, and causing the development of proarrhythmic spontaneous Ca2+ waves. Since an age-related increase of mitochondrial ROS shifts the normal ROS balance into a deleterious range, maintenance of normal ROS production and mitochondrial integrity are important research and therapeutic targets that are paramount in preventing redox imbalance that leads to age-related cardiac dysfunction and Ca2+-dependent arrhythmias.
Acknowledgments
None declared.
Glossary
Abbreviations
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- DTDP
2,2′-dithiodipyridine
- DTT
dithiothreitol
- ISO
isoproterenol (isoprenaline)
- LTCC
L-type calcium channel
- mBB
monobromobimane
- NCX
sodium–calcium exchanger
- PKA
protein kinase A
- PLB
phospholamban
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SCW
spontaneous Ca2+ wave
- SERCA
SR (ER) Ca2+-ATPase
- SR
sarcoplasmic reticulum
- TMRE
tetramethylrhodamine ethyl ester
Additional information
Competing interests
None declared.
Author contributions
D.T. and G.K. contributed to the conception and design of the study. L.L.C., W.L., G.K. and D.T. worked on interpretation of data and writing the manuscript. L.L.C., W.L., R.T., J.C., Y.L. and D.T. performed experiments and analysed the results. All authors read and approved the final submission. All experiments were carried out in Cardiovascular Research Center of Rhode Island Hospital.
Funding
L.L.C. and W.L. were supported by NIH grant 5T32HL094300-05.
Supplemental material
Supplemental material
References
- Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Györke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Györke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PloS One. 2011a;6:e28324. doi: 10.1371/journal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Györke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Györke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011b;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Györke S. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl. 1):I36–42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, O’Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. 2010;88:241–249. doi: 10.1093/cvr/cvq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M, Koren G. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerbai E, Guerra L, Varani K, Barbieri M, Borea PA, Mugelli A. Beta-adrenoceptor subtypes in young and old rat ventricular myocytes: a combined patch-clamp and binding study. Br J Pharmacol. 1995;116:1835–1842. doi: 10.1111/j.1476-5381.1995.tb16671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary KR, El-Sikhry H, Seubert JM. Mitochondria and the aging heart. J Geriatr Cardiol. 2011;8:159–167. doi: 10.3724/SP.J.1263.2011.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LL, Odening KE, Hwang MS, Chaves L, Schofield L, Taylor CA, Gemignani AS, Mitchell GF, Forder JR, Choi BR, Koren G. Electromechanical and structural alterations in the aging rabbit heart and aorta. Am J Physiol Heart Circ Physiol. 2012;302:H1625–H1635. doi: 10.1152/ajpheart.00960.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibb KM, Rueckschloss U, Eisner DA, Isenberg G, Trafford AW. Mechanisms underlying enhanced cardiac excitation contraction coupling observed in the senescent sheep myocardium. J Mol Cell Cardiol. 2004;37:1171–1181. doi: 10.1016/j.yjmcc.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci (Landmark Ed) 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- Eager KR, Roden LD, Dulhunty AF. Actions of sulfhydryl reagents on single ryanodine receptor Ca2+-release channels from sheep myocardium. Am J Physiol Cell Physiol. 1997;272:C1908–C1918. doi: 10.1152/ajpcell.1997.272.6.C1908. [DOI] [PubMed] [Google Scholar]
- Eigel BN, Gursahani H, Hadley RW. ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H955–H963. doi: 10.1152/ajpheart.00721.2003. [DOI] [PubMed] [Google Scholar]
- Eisner D, Bode E, Venetucci L, Trafford A. Calcium flux balance in the heart. J Mol Cell Cardiol. 2013;58:110–117. doi: 10.1016/j.yjmcc.2012.11.017. [DOI] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikova VG, Kareyeva AV, Vinogradov AD. What are the sources of hydrogen peroxide production by heart mitochondria. Biochim Biophys Acta. 2010;1797:939–944. doi: 10.1016/j.bbabio.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Zhang T, Mestril R, Bers DM. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- Györke S, Carnes C. Dysregulated sarcoplasmic reticulum calcium release: Potential pharmacological target in cardiac disease. Pharmacol Ther. 2008;119:340–354. doi: 10.1016/j.pharmthera.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarmann CS, Fink RH, Dulhunty AF. Oxidation and reduction of pig skeletal muscle ryanodine receptors. Biophys J. 1999;77:3010–3022. doi: 10.1016/S0006-3495(99)77132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria. J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Ho HT, Stevens SC, Terentyeva R, Carnes CA, Terentyev D, Györke S. Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J Physiol. 2011;589:4697–46708. doi: 10.1113/jphysiol.2011.210005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett SE. Age-associated changes in excitation-contraction coupling are more prominent in ventricular myocytes from male rats than in myocytes from female rats. Am J Physiol Heart Circ Physiol. 2010;298:H659–H670. doi: 10.1152/ajpheart.00214.2009. [DOI] [PubMed] [Google Scholar]
- Janczewski AM, Lakatta EG. Modulation of sarcoplasmic reticulum Ca2+ cycling in systolic and diastolic heart failure associated with aging. Heart Fail Rev. 2010;15:431–445. doi: 10.1007/s10741-010-9167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Kannel WB, Cupples LA, Dagostino RB. Sudden death risk in overt coronary heart disease – the Framingham Study. Am Heart J. 1987;113:799–804. doi: 10.1016/0002-8703(87)90722-8. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuster GM, Lancel S, Zhang J, Communal C, Trucillo MP, Lim CC, Pfister O, Weinberg EO, Cohen RA, Liao R, Siwik DA, Colucci WS. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic Biol Med. 2010;48:1182–1187. doi: 10.1016/j.freeradbiomed.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev. 1993;73:413–467. doi: 10.1152/physrev.1993.73.2.413. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Sollott SJ. Perspectives on mammalian cardiovascular aging: humans to molecules. Comp Biochem Physiol A Mol Integr Physiol. 2002;132:699–721. doi: 10.1016/s1095-6433(02)00124-1. [DOI] [PubMed] [Google Scholar]
- Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Gαq-overexpressing mice. Circ Res. 2010;107:228–232. doi: 10.1161/CIRCRESAHA.110.217570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Györke I, Wiesner TF, Györke S. Potentiation of Ca2+ release by cADP-ribose in the heart is mediated by enhanced SR Ca2+ uptake into the sarcoplasmic reticulum. Circ Res. 2001;89:614–622. doi: 10.1161/hh1901.098066. [DOI] [PubMed] [Google Scholar]
- Miquel J, Economos AC, Fleming J, Johnson JE. Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–1732. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, Lin SF, Chen PS, Xie LH, Chen F, Qu Z, Weiss JN, Karagueuzian HS. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–H1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S, Polakova E, Shirokova N. Posttranslational modifications of cardiac ryanodine receptors: Ca2+ signaling and EC-coupling. Biochim Biophys Acta. 2013;1833:866–875. doi: 10.1016/j.bbamcr.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–966. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM. Na/Ca exchange in heart failure – Contractile dysfunction and arrhythmogenesis. Ann N Y Acad Sci. 2002;976:454–465. doi: 10.1111/j.1749-6632.2002.tb04775.x. [DOI] [PubMed] [Google Scholar]
- Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann N Y Acad Sci. 1998;853:341–344. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005;39:982–991. doi: 10.1016/j.yjmcc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Shabalina IG, Nedergaard J. Mitochondrial (‘mild’) uncoupling and ROS production: physiologically relevant or not. Biochem Soc Trans. 2011;39:1305–1309. doi: 10.1042/BST0391305. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Guo T, Bers DM. Ca2+ scraps – Local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- Squier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol. 2001;36:1539–1550. doi: 10.1016/s0531-5565(01)00139-5. [DOI] [PubMed] [Google Scholar]
- Sun JH, Yamaguchi N, Xu L, Eu JP, Stamler JS, Meissner G. Regulation of the cardiac muscle ryanodine receptor by O2 tension and S-nitrosoglutathione. Biochemistry. 2008;47:13985–13990. doi: 10.1021/bi8012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G, Csordas G, Hantash BM, Thomas AP, Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem. 2000;275:15305–15313. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res. 2007;100:105–111. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- Xiao BL, Zhong GF, Obayashi M, Yang DM, Chen KY, Walsh MP, Shimoni Y, Cheng HP, ter Keurs H, Chen SRW. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon β-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Spurgeon HA, O’Connor F, Lakatta EG. Age-associated changes in β-adrenergic modulation on rat cardiac excitation-contraction coupling. J Clin Invest. 1994;94:2051–2059. doi: 10.1172/JCI117559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- Zable AC, Favero TG, Abramson JJ. Glutathione modulates ryanodine receptor from skeletal muscle sarcoplasmic reticulum. Evidence for redox regulation of the Ca2+ release mechanism. J Biol Chem. 1997;272:7069–7077. doi: 10.1074/jbc.272.11.7069. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Zhou Q, Yao F, O’Rourke ST, Sun C. Endothelin-1 regulates cardiac L-type calcium channels via NAD(P)H oxidase-derived superoxide. J Pharmacol Exp Ther. 2008;326:732–738. doi: 10.1124/jpet.108.140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Aon MA, Liu T, O’Rourke B. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J Mol Cell Cardiol. 2011;51:632–639. doi: 10.1016/j.yjmcc.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XS, Altschafl BA, Hajjar RJ, Valdivia HH, Schmidt U. Altered Ca2+ sparks and gating properties of ryanodine receptors in aging cardiomyocytes. Cell Calcium. 2005;37:583–591. doi: 10.1016/j.ceca.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.