

Introduction
Celiac disease is an intestinal inflammatory disease that is triggered by dietary gluten, a protein found in wheat, barley and rye in genetically susceptible individuals.1 Descriptions of a celiac disease-like phenotype can be traced back to the Greek physician Aretaeus in the 1st and 2nd century AD (reviewed in 2). Gluten was identified as the culprit of celiac disease by Dutch physicians who observed that, during the 1944–45 famine when wheat and rye were scarce, celiac children symptomatically improved.3 Subsequent studies characterized many features of celiac disease, and, while disease pathogenesis and pathophysiology remain incompletely understood, the disease is thought to arise from the interplay of genetic, environmental and immunological factors (Figure 1). Importantly, understanding of celiac disease pathophysiology, in which the trigger (wheat, rye and barley) is known, will undoubtedly reveal basic mechanisms that underlie other autoimmune diseases (e.g., type I diabetes) that share many common pathogenic perturbations. In this review, we describe seminal findings in each of the three domains of celiac disease pathogenesis: genetics, environmental triggers and immune dysregulation with a focus on newer areas of investigation such as non-HLA genetic variants, intestinal microbiome and the role of the innate immune system.
Figure 1. Factors involved in celiac disease pathophysiology.
Celiac disease is thought to arise from the interplay of genetic, environmental and immunological factors. This review highlights seminal findings in each of these domains.
Genetics
Celiac disease has a strong hereditary component. Epidemiological studies show that up to 20% of first-degree relatives are affected by the disease6 with concordance rates of 75–80% in monozygotic twins and 10% in dizygotic twins7. The strongest and best-characterized genetic susceptibility factors in celiac disease are human leukocyte antigen (HLA) class II genes known as HLA-DQ2 and HLA-DQ8, molecules responsible for presentation of antigens to immune cells. However, while HLA-DQ2 or DQ8 are necessary for disease to develop, they are not sufficient implicating other genetic or environmental factors in disease development. Approximately, 25–30% of individuals of European descent carry HLA-DQ2 susceptibility, but only about 4% of these individuals will develop celiac disease in their lifetime8 underscoring the role of additional factors. Recent large-scale genetics studies, called genome-wide association studies (GWAS), have identified a number of common non-HLA genetic factors (many in genes involved in immunity) associated with celiac disease which, on their own, contribute a small amount to overall risk but have great potential in discovering important and novel pathways involved in disease pathogenesis.
HLA-DQ2 & -DQ8 genetics and disease risk
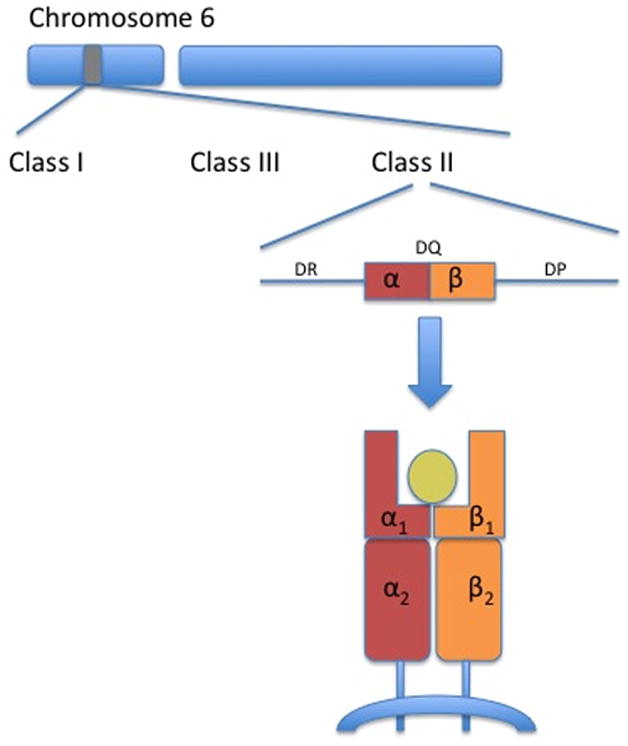
HLA is the name of the major histocompatibility complex (MHC) in humans.4 These genes reside on chromosome 6 and are divided into three classes (I–III) (Figure 2). HLA-DQ is a class II molecule on chromosome 6p21.3 responsible for presentation of peptides from outside cells (compared with class I molecules that present peptides from within cells and class III molecules that encode complement proteins). HLA-DQ is composed of a αβ heterodimer encoded by HLA-DQA1 and HLA-DQB1 genes respectively. The αβ heterodimer is a cell surface receptor located on APCs.
Figure 2. Class II HLA-DQ.
The genes encoding for HLA molecules are found in the major histocompatibility (MHC) complex on chromosome 6. HLA molecules involved in celiac disease are encoded in a region known as class II; by genes known as -DQ (other class II genes include -DR and –DP). Class II HLA-DQ genes encode for α- and β-chains that are associated as heterodimers on the surface of antigen presenting cells and form a cleft that binds antigens. HLA-DQA1 genes code for two α-chains (α1 & α2) and HLA-DQB1 genes code for two β-chains (β1 & β2).
The genetics of celiac disease is complex because the number, type and configuration of the DQA1 and DQB1 alleles determine disease risk. Current WHO nomenclature is HLA followed by a hyphen followed by the gene (e.g., DQA1, DQB1, etc), an asterisk (separator), allele group (field 1), colon (field separator), protein (field 2).5 The isoform encoded by a specific combination of DQA1 and DQB1 genes can be expressed as HLAx.y where x refers to DQB1 and y to DQA1. For example, DQ2.5 is the protein encoded by DQB1*02 and DQA1*05 inherited either in cis (on the same chromosome) or trans (on different chromosomes). When these alleles are located on the same chromosome (i.e., in cis), they are often inherited in a haplotype with another class II molecule DRB1*03 (DR3).
Figure 3 highlights HLA configurations associated with celiac disease. The highest risk group are those that carry DQB1*02 on both chromosomes (i.e. homozygotes) known as gene dose effect.6,7 DQB1*02 homozygosity (carrying two DQB1*02 alleles) has an estimated prevalence of 2% in the population but represents 25% of all celiac patients due to an estimated five-fold increased risk of celiac disease compared to heterozygotes (carrying one DQB1*02 allele).8,9 Studies have shown that the CD4+ helper T cell response from DQB1*02 homozygous individuals is stronger than the response from heterozygous individuals.6,10 Moreover, DQB1*02 homozygosity may be associated with younger age of onset11,12 and more complicated clinical course including refractory sprue. 13 DQ2.5 (DQB1*02/DQA1*05) heterozygotes are the most common HLA configurations and represent up to 50% of the HLA types found in celiac disease patients. 9 While DQ2.2 (DQB1*02/DQA1*02) is highly homologous to DQ2.5, it alone carries little risk of celiac disease due to decreased stability of bound peptides.14
Figure 3. HLA configurations in celiac disease. A) HLA-DQ2 homozygotes, heterozygotes and half-heterodimers, and B) HLA-DQ8 homozygotes, heterozygotes and DQ8/DQ2.
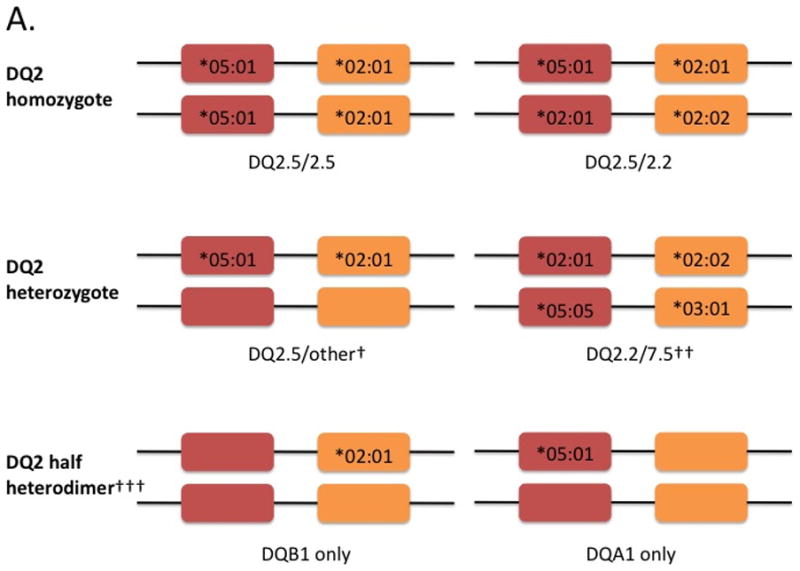
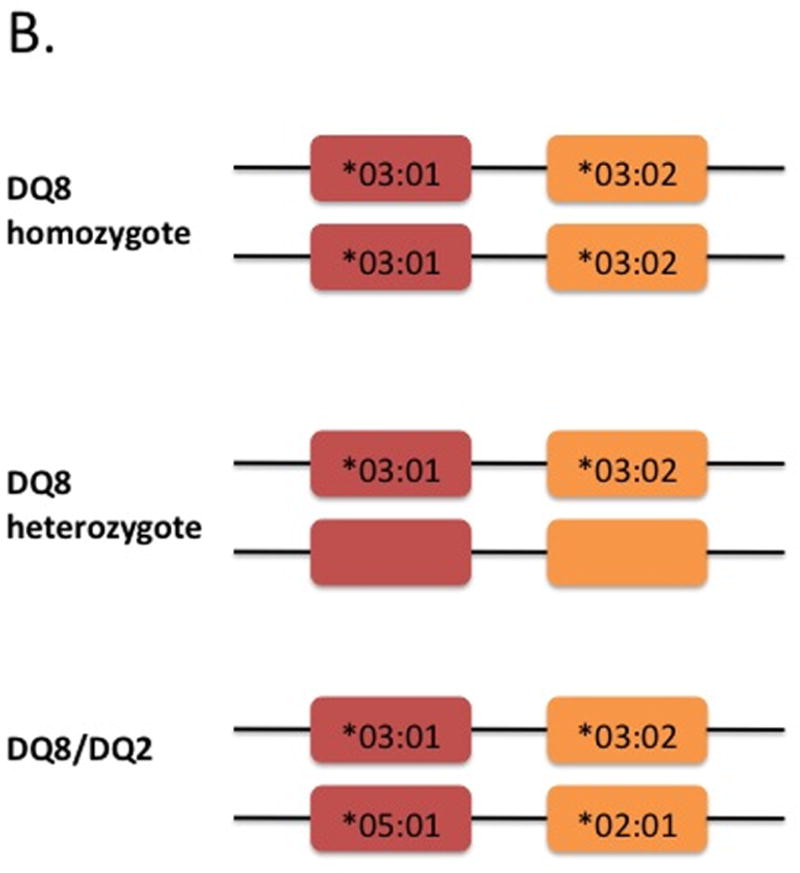
Red boxes denote the DQA1 gene encoding the alpha-chain and orange boxes denote the DQB1 gene encoding the beta-chain (see figure 2). Shown in the boxes are the specific alleles for each gene. Current WHO nomenclatures uses an asterix followed by the allele group (e.g., 05), a colon then the protein group (e.g., 01). An empty box refers to other HLA alleles not associated with celiac disease. Shown below the genes are the isoforms. †cis acting (i.e. on the same chromosome); ††trans acting (i.e. on opposite chromosomes); ††† risk of celiac disease for DQ2 half heterodimers is lower than the general population especially individuals carrying only DQA1*05 (ref 15)
A small minority of celiac patients carry only one of the alleles of the risk HLA-DQ2 heterodimer: HLA-DQA1*05 (05:01 or 05:05) or HLA-DQB1*02 (02:01 or 02:02). This is called the “half-heterodimer”. The European Genetic Cluster on Celiac Disease typed over 1000 celiac patients and found that 6% carried neither HLA-DQ2 nor –DQ8. Of these patients, 93% (57/61) carried the DQ2.5 half heterodimer with almost three-quarters carrying only the DQB1*02 allele. 15 The prevalence of individuals carrying only one copy of DQB1*02 was increased in celiac patients compared with controls, while those carrying only one DQA1*05 was higher in controls compared to patients indicating a negative association for the DQA1*05 half heterodimer.9
DQ8 is a heterodimer composed of α-chains encoded by DQA1*03:01 and β-chains encoded by DQB1*03:02. When they are inherited on the same chromosome, they are found on a haplotype with DRB1*04 notated as DR4-DQ8. The prevalence of HLA-DQ8 in the general population varies geographically with higher rates in individuals from the Middle East and South America.16 In celiac disease overall, HLA-DQ8 is found in 5–10% of patients.9,15 As with DQ2, risk of disease with HLA-DQ8 follows a gradient. The highest risk appears to be in individuals who inherit DQ8 and DQ2; though, the overall prevalence of carrying both DQ8 and DQ2 is low at 2.5%.9 In individuals with HLA-DQ8 and DQ2.2 or DQ2.5, risk is estimated at 1:24, while those with HLA-DQ8 but not DQ2.2 or DQ2.5, risk is estimated at 1:89.9 DQ8 homozygosity confers increased risk compared to DQ8 heterozygotes.17
Development of celiac disease in individuals who are HLA-DQ2 and -DQ8 negative is extremely rare. In a large European collaborative study, only 4 of 1008 patients (0.4%) fulfilled criteria for celiac disease but did not carry DQ2 (including half heterodimer) nor DQ8.15 No other class I or II associations were identified in this small group. In support of these findings, two additional studies in the US and Italy found the prevalence of DQ2/8 negativity in celiac disease to range from 0.16–0.9%. 9,17 Thus, in a very small group of patients, if clinical suspicion is high with supporting serological and histological findings, celiac disease can be diagnosed in the absence HLA-DQ2 or -DQ8. However, the overall risk of celiac disease in individuals who do not carry DQ2 or DQ8 is very low. These findings support the use of HLA testing for its high negative predictive value (Figure 4).
Figure 4. Clinical application of HLA testing.

HLA testing should be considered for screening, disease exclusion or to support a diagnosis. Testing is unaffected by a gluten-free diet. Providers should ensure that both DQ2 alpha and beta chains are tested. If a patient carries HLA-DQ2 or –DQ8, they carry a risk factor (or varying magnitude) for celiac disease and additional work-up should be considered. Individuals carrying HLA-DQ2 half-heterodimers, are also at risk for celiac disease (albeit substantially lower than other HLA-DQ2 and –DQ8 positive patients). If HLA-DQ2 and –DQ8 are not present, then celiac disease risk is highly unlikely and antibody screening is not necessary.
HLA peptide binding
HLA-DQ2 and –DQ8 play a key role in celiac disease due to their physiochemical properties and binding of specific peptides deamidated by tissue transglutaminase 2 (tTG2). Both HLA-DQ2 and –DQ8 contain positively charged pockets with a preference for binding negatively charged particles. Specifically, in DQ2, the lysine position at β71 has a preference for binding negatively charged residues at positions P4, P6 and P7 (Figure 5). 18 The DQ8 β57 polymorphism creates a basic environment with a preference for binding the negatively charged residue at P9 (Figure 5).19
Figure 5. MHC class II-gluten peptide complexes.
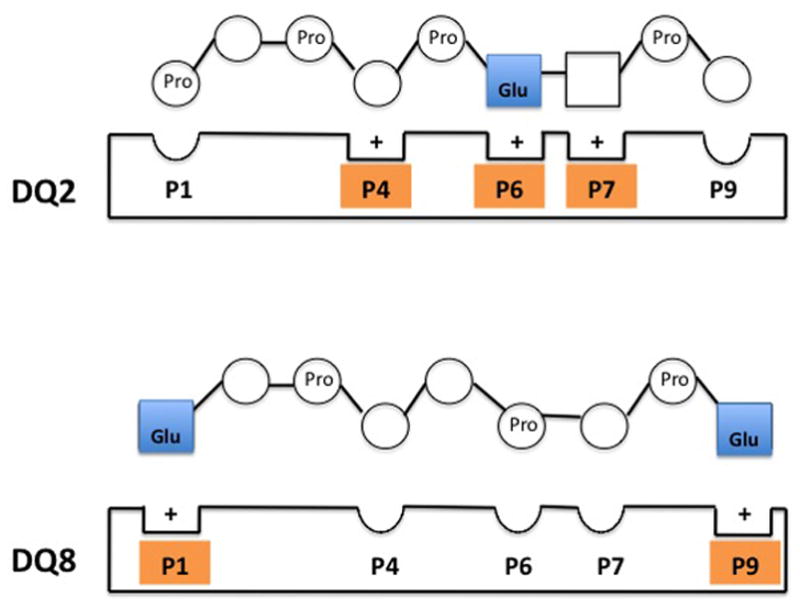
MHC class II molecules HLA-DQ2 and –DQ8 preferentially bind a glutamate residue of the gluten peptide at position 6 and position 1/9 respectively. This binding is enhanced by a negatively charged glutamate and positively charged pocket of the HLA molecule.
In celiac disease, these HLA molecules on APCs present gluten peptides to CD4+ T cells thereby activating them.20,21 The size of the peptide fragment defines stimulatory activity with larger fragments showing increased CD4+ T cell stimulation compared with smaller fragments.22–26 While deamidation favors binding to HLA-DQ2 or –DQ814, studies have suggested that it is not absolutely required for stimulation of CD4+ T cells especially in the case of HLA-DQ8.19,27 The mechanism for recognition of native peptides is that the polymorphism at position β57 allows DQ8 to switch from interaction with a negatively charged residue in TCR to one in the peptide.19
Non-HLA genetic susceptibility factors and role in disease pathogenesis
HLA is the best-characterized genetic susceptibility factor in celiac disease, but does not account for all disease heritability suggesting that additional genetic factors play a role. Genome-wide association studies (GWAS) have identified a number of candidate genetic susceptibility factors in celiac disease. The results of GWAS shed light on new genes and genetic pathways involved in disease pathogenesis. The immediate challenge is to identify variants within these regions that are functionally important in order to elucidate their role in celiac disease pathogenesis. To date, non-HLA genetic loci harboring 115 genes have been associated with celiac disease using GWAS.28–31 Of these genes, 28 are immune-related which can be broadly grouped into categories based on function and pathways. (Reviewed in 32,33). Enrichment analysis indicates that these genes are broadly involved in adaptive and innate immune response among others (Figure 6). Taken as a whole, these results underscore the importance of immune dysregulation in celiac disease by confirming the role of the adaptive immune response as well as highlighting pathways involved in innate immune response. Post-GWAS studies will need to focus on elucidating the functional basis of these genetic variants; in particular, the role of regulatory variation.
Figure 6. Enrichment analysis of non-HLA genes associated with celiac disease.

We used GeneTrail to test for enrichment of functional annotations among non-HLA genes associated with celiac disease from genome-wide association studies published through 2012. In this graph is shown the fold enrichment (y-axis) and significantly enriched biological functions (x-axis). Background expectations were based on all human genes. P-values were calculated using a hypergeometric distribution using the approach by Benjamini & Hochberg to control the false discovery rate. P-values for enrichment shown here ranged from 4.8 × 10−2 to 3.2 × 10−11.
An intriguing finding to emerge from GWAS is the overlap of variants identified in a number of diseases and traits including several immune-related diseases. Common loci have been identified with type 1 diabetes, rheumatoid arthritis and Crohn’s disease suggesting common genetic backgrounds for these immune-related diseases. However, non-HLA loci in celiac disease are estimated to account for a small portion of overall genetic risk. The reason for “missing heritability” is still under investigation and could be explained by the contribution of highly penetrant genetic variants with lower allele frequencies than those studied in GWAS. These rare variants may have greater impact on disease susceptibility than common variants discovered to date and, as large-scale sequencing studies are completed, it will become clear what role rare genetic variants play in celiac disease pathogenesis. Moreover, the role of gene-gene and gene-environment interactions need to be explored further in celiac disease.
Environment
Environmental factors clearly play an important role in celiac disease pathogenesis. The primary trigger in the disease is gluten, and, over the past decade, many studies have contributed to our understanding of gluten biochemistry and antigenic epitopes, transport through the small intestinal epithelium, modification by tTG, and binding to antigen presenting cells in the lamina propria with subsequent activation of adaptive immunity. Moreover, it has become clear that gluten is associated with innate immune responses in the gut epithelium and that cytotoxic intraepithelial lymphocytes appear to play a central role. In addition, emerging data implicates microbiota (both commensal and pathogenic) in disease pathogenesis, while epidemiological studies have suggested that early (and possibly late) gluten introduction to children, ceasarean section delivery as well as lack of breast-feeding are important risk factors for development of celiac disease.
“Gluten” and epithelial transport of peptide fragments
Wheat, rye and barley belong to the same tribe called triticeae that diverged from oats belonging to the aveneae tribe. (Figure 7) While “gluten” is used as the general term to describe the trigger of celiac disease, gluten technically refers to the disease-activating peptides found only in wheat. Gluten comprises two different protein types, gliadins and glutenins, capable of triggering disease.34–36 The peptides in barely and rye, hordeins and secalins respectively, are also capable of activating disease.37 In contrast, oats, comprised of more distantly related peptides called avenins, rarely trigger celiac disease.38 Gliadins, glutenins, hordein and secalins contain high contents of prolines and glutamines which makes them resistant to degradation by gastric acid, pancreatic and brush border enzymes because these are lacking in prolyl endopeptidase activity.39,40 There is ongoing interest in leveraging certain bacterial or fungi endopeptidase activities as a therapeutic strategy.39–42
Figure 7. Divergence of oats from wheat, rye and barley.
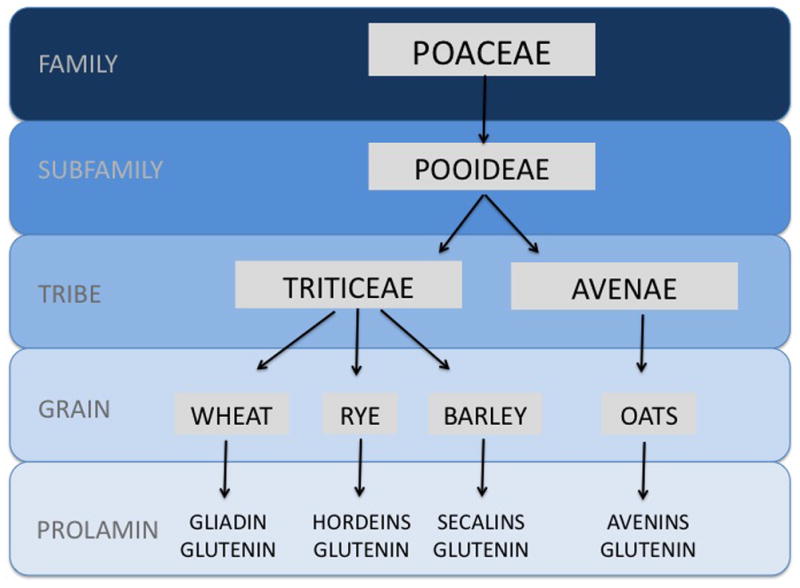
Wheat, rye, barley and oats belong to the same grain family (Poaceae) and subfamily (Pooideae). However, they belong to distinct tribes: wheat, rye and barley (Triticeae) and oats (Avenae). The prolamins from the triticeae tribe are immunogenic and contribute to celiac disease, while avenins from pure, uncontaminated oats are safe for the vast majority of celiac patients.
Transport of peptide fragments across the small intestinal epithelium and intestinal permeability have been areas of intense research in celiac disease, though their primary role in disease pathogenesis remains incompletely understood. Peptide fragments that have been resistant to degradation can be transported across the epithelium primarily by transcellular pathways (reviewed in 43). Tight junctions play a role in peptide transport and genome-wide association studies in celiac disease have found susceptibility SNPs in tight junction-associated genes.29,44,45
However, it is unclear whether altered intestinal permeability is a primary cause or a consequence of intestinal inflammation. Moreover, the role of tight junction blockade as a therapeutic strategy has been studied using pre-haptoglobin-2, an analogue of the zonnula occludens toxin.46–48 However, this study did not directly measure intestinal permeability and, therefore, the mechanism of action remains unclear. An alternate mechanism of transcellular transport of gliadin involves abnormal retro-transport of IgA-gliadin by the CD71 receptor.49 CD71, a transferrin receptor, was shown to be upregulated and apically expressed in active celiac disease leading to escape of gliadin degradation and translocation to the lamina propria known as the so-called “Trojan Horse” phenomenon.49 Further study is required to determine the role of peptide fragment transport and intestinal permeability in pathogenesis.
Microbiota
An emerging field of investigation is the role of the human microbiome in human health and disease.50 The human intestine harbors a vast number and variety of commensal microorganisms that are complex and dynamic (reviewed in 51). In the past 5 years, there have been important technological developments in high-throughput sequencing that have enable investigators to characterize the human microbiome using culture-free methods known as metagenomics.52 While an individual’s microbiome is unique, there is evidence of sharing among family members.53 The microbiome is influenced by diet54, and the interplay between diet and the microbiome affects metabolic function.55 Importantly, there are important interactions between the gut microbiome, diet and the immune system that appear to contribute to phenotypes such as obesity53, inflammatory bowel disease56 and celiac disease.
Studies of the gut microbiome in celiac disease are still in their early stages and have yielded conflicting results likely due to different experimental approaches on fecal or biopsy samples in various patient populations from different countries. All of these factors can bias microbiome results. In 2004, a study identified rod-shaped bacteria in intestinal biopsies of celiac patients suggesting a role for the microbiome in celiac disease.57 Further studies analyzed samples for metabolic readouts of the gut microbiome (e.g., short chain fatty acid and volatile compounds) in celiac patients58,59 as well as first-degree relatives of celiac patients60 and found significant differences compared to controls. Additional studies using various methodologies found differences in fecal and/or mucosal-associated composition primarily of Bacteroides, Clostridium, Bifidobacteria, Lactobacillus, Escheheria coli and Staphylococcus59,61–65 between celiac patients (both untreated and treated) and controls. Differences in microbial composition were also found between adult and children with celiac disease.66 However, other studies have failed to find differences in the microbiome among cases and controls.67 A recent paper hypothesized that the intestinal microbiome as a whole determines the switch from tolerance to immune response in genetically susceptible infants and found a lack of Bacteroidetes and increased abundance of Firmicutes in a longitudinal study of at-risk infants followed from birth to 24 months.68 Further studies using combined genomic approaches are needed to clarify the role of the microbiome in celiac disease.
Consistent with the role of diet in modulating the gut microbiome, the gluten free diet alone in healthy individuals led to decreases in Bifidobacterium and Lactobacillus.69 Moreover, animal and human studies suggest possible interactions between commensal bacteria and immune responses in celiac disease.70,71 Animal studies have suggested that the microbiome in celiac disease might alter intestinal permeability thereby contributing to disease pathogenesis.72 To date, probiotic studies in celiac disease investigated the proteolytic activity of VSL#3 or sourdough lactobacilli42,73, but none has studied the role in modulating commensal flora, although there is data of therapeutic effect of probiotics in irritable bowel syndrome.74 Animal and human studies in this area are ongoing.
Despite technological advances in studying the human intestinal microbiome, many questions remain to be answered about the role of commensal bacteria in immune-mediated gastrointestinal diseases such as celiac disease or inflammatory bowel diseases (reviewed in 75). First, and perhaps most important, among these is whether the intestinal microbiota is a cause or a consequence of intestinal inflammation. There is evidence to support both sides and additional studies are needed to elucidate cause and effect. Moreover, there is interest in how microbial alterations could be used for therapeutic interventions, though clinical trials are lacking in celiac disease. There are questions about how diet impacts and alters intestinal microbiota as well as the effect of different microbes on immune function. Finally, the role of commensal fungi and viruses has not been studied in celiac disease.
Other environmental risk factors
Besides the commensal microbiome, a number of other factors including childhood infections notably rotavirus, mode of delivery, gluten introduction to infants, and breast-feeding have been studied in celiac disease. The data on these factors stems primarily from epidemiological and ecological studies, and their role in disease pathogenesis remains to be fully elucidated.
The role of pathogenic organisms in celiac disease had been suggested in the 1980s when Kagnoff et al described a 12 amino acid sequence homology between A-gliadin and the E1b protein from human adenovirus type 1276 and that celiac patients had a significantly higher rate of previous adenovirus type 12 infection compared to controls.77 It was hypothesized that there may be immunological cross-reactivity between antigenic elements shared by viruses and α-gliadin.78 However, follow-up studies are inconsistent in their findings regarding adenovirus type 12 and celiac disease.79–81 The finding of a seasonal pattern of higher rates of summer births in children with celiac disease also suggests a role for infectious agents.82 More recent studies implicate rotavirus in celiac disease pathogenesis. Stene et al prospectively followed children with HLA risk and determined that frequent rotavirus infections (as measured by rotavirus antibody titers) showed a moderate, but statistically significant increased risk of celiac disease.83 Zanoni et al used a peptide library approach using sera of active celiac patients and found an autoantigen peptide recognize rotavirus serotype 1 major neutralizing protein VP7 as well as HSP60, desmoglein 1 and toll-like receptor4 (TLR4).84 Anti-peptide antibodies altered intestinal permeability and activate monocytes via TLR4 signaling suggesting a role of innate immunity and viral infection in disease pathogenesis.
Mode of delivery has also been studied as a possible risk factor for celiac disease perhaps due to altered exposure to commensal bacteria in the perinatal period. While not confirmed in all studies85, cesaerean section, particularly performed electively, is associated with a modest increased risk of later celiac disease.86,87 Intriguingly, a recent study found that children born vaginally have microbiota in various tissues that resemble their mother’s vaginal flora including Lactobacillus, Prevotella and Sneathia spp, while children born by cesearean section harbored flora resembling skin bacterial communities such as Staphylococcus, Corynebacterium and Propionibacterium spp.88 Additional work is needed to correlate neonatal bacterial colonization with future risk of celiac disease.
The effect of timing of gluten introduction on risk of celiac disease came to the forefront with the Swedish celiac “epidemic” in the 1980–90s. Prospective, population-based data noted that, in 1985, there was a four-fold increase in celiac disease incidence in children under age 2 that precipitously dropped to pre-1985 rates a decade later.89,90 Ten years later, the prevalence of celiac disease in Swedish children born during the epidemic remains three times higher than the population prevalence.91 This rapid rise and decline in disease incidence correlated with changes in infant feeding practices including younger age of gluten introduction, increase amount of gluten in diet and reduced breast-feeding.89,92 A prospective, ten year observational study in children at risk for celiac disease noted a five-fold increased risk of celiac disease autoimmunity when gluten was introduced in the first 3 months compared to 4–6 months of life further evidence that early gluten introduction is a risk factor.93 Despite these ecological and epidemiological studies, reasons why early gluten introduction causes higher risk of celiac disease remains unexplained.
Breast-feeding has also been shown in some studies to be protective against celiac disease. A meta-analysis pooled five case-control studies and found a 52% reduction in celiac disease correlating to duration of breast-feeding.94 Hypotheses for the protective effect of breast-feeding on celiac disease include avoidance of early gluten introduction, protection against infections, decreased immune response due to IgA antibodies in breast milk and T cell-specific suppressive effects. Mothers with at-risk infants are therefore counseled to continue breast-feeding as long as possible and introduce gluten between 4–6 months.95
Immune Dysregulation
Introduction
While celiac disease requires genetic susceptibility (primarily HLA-DQ2 or –DQ8) as well as environmental exposures (foremost gluten ingestion), these alone are insufficient to trigger the disease and do not explain ongoing small intestinal inflammation. Immune dysregulation, therefore, is a core feature of celiac disease pathogenesis and has been the subject of intense research over the last few decades. The role of tTG in the deamidation of specific toxic epitopes as well as the initiation of gluten-specific T cell adaptive immune responses have been elucidated. Moreover, the role of innate immune responses in disease pathogenesis has recently received attention especially in small intestinal epithelial damage via CD8+CD4- intraepithelial lymphocytes.
Toxic epitopes and tissue transglutaminase
Once undigested peptide fragments from wheat, rye and barley are transported to the lamina propria, they are subject to deamidation by tTG2 which converts glutamine to glutamate thereby introducing negative charges that have stronger binding affinity for HLA-DQ2 and -DQ8 on APCs. tTG2 belongs to a family of calcium-dependent transamidating enzymes that catalyze covalent and irreversible cross-linking of proteins expressed in all cell types. In an inactive, closed form, tTG2 is located intracellularly and is enzymatically inactive.96 For reasons that are incompletely understood, tTG2 is transported extracellularly, where, in the presence of calcium, tTG2 is in an open reduced form and is enzymatically active.97 Under normal physiological conditions, tTG2 is rapidly inactivated via oxidation. While in a reducing environment such as ongoing inflammation, tTG2 remains active extracellularly which might facilitate ongoing tTG2 activity (Figure 8).98
Figure 8. Active and inactive states of tissue transglutaminase (tTG2).
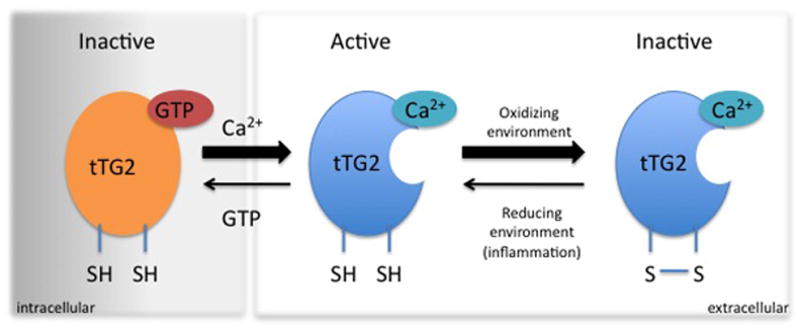
tTG2 is active in an open conformation in a reduced state. In presence of GTP and in the absence of Ca2+ (i.e. intracellular environment), tTG2 is in a reduced, closed state and the enzyme is inactive. Upon release to the extracellular environment with low GTP and high Ca2+, tTG2 takes on an open conformation and is active. Usually oxidizing conditions in the extracellular environment render tTG2 inactivated in its open conformation by the formation of a disulphide bond between two vicinal cysteine residues in the enzyme. Upon creation of reducing conditions (i.e. inflammation), the disulphide bond is reduced and the enzyme can again take an active open conformation.
Certain glutamine residues, so-called toxic epitopes, have higher specificity for tTG2 deamidation in the small intestine. Peptides derived from wheat, rye and barley are heterogeneous populations. Gliadin peptides are sub-divided intro α-, γ-, and ω-gliadins, while glutenins are characterized as high molecular weight or low molecular weight. Among gliadin, glutenin, hordein and secalin peptides (as well as a few avenin peptides derived from oats), toxic epitopes composed of a nine amino acid core sequence elicit gluten-specific T-cell responses in celiac disease dependent on HLA type. A nomenclature system has been proposed recently for celiac disease-relevant gluten epitopes based on specific criteria.99
A hallmark of celiac disease is the presence of anti-tTG2 antibodies that can be detected in the serum by ELISA. Anti-tTG2 antibodies (especially IgA) are highly sensitive and specific for the disease.100 However, the mechanism of auto-antibody formation remains incompletely understood (reviewed in 101). Furthermore, there is controversy about the role of anti-tTG2 antibodies in disease pathogenesis (reviewed in 102,103). A recent study104 provided evidence favoring a T cell-dependent model of antibody formation in celiac disease suggesting that tTG-specific B cells act as APCs for the gluten-specific T cell immune response. Additional studies suggest that auto-antibodies could modulate small intestinal biology by enhancing passage of gliadin peptides49, inhibiting angiogenesis105,106, or alter tTG2 activity;104–110 although there is conflicting data as to whether tTG2 activity is inhibited or enhanced. Support for a role of auto-antibodies in disease pathogenesis is provided by extra-intestinal manifestations of celiac disease notably dermatitis herpetiformis. In this dermatological condition associated with celiac disease, anti-tTG3 antibodies are expressed in the dermal papillae and are thought to mediate lesion formation.111
Adaptive immune response
The role of the adaptive immune system in the gut is to distinguish between harmful and beneficial antigens derived from microorganisms (commensal and pathogenic) as well as ingested food peptides. As a result, the intestinal mucosa holds a large proportion of all immune cells in the body that reside in gut-associated lymphoid tissue (GALT) where naïve T and B cells are found (reviewed in 112). Immune cells residing in the lamina propria and epithelial layer, in contrast, have effector and memory function. APCs patrol areas of naïve B or T cells and give costimulatory signals that induce T- or B-cell differentiation that, in turn, leads to elimination of harmful antigens or tolerance of harmless antigens. Maintenance of an adaptive tolerogenic T-cell response to a soluble protein antigen is termed oral tolerance. Under normal physiological conditions, oral tolerance is maintained in an environment of retinoic acid along with the cytokine TGF-β that together induce development of regulatory T cells to suppress pro-inflammatory effector T cells. 113,114 However, in celiac disease, it appears that retinoic acid, in the context of high IL-15, promotes destructive immune responses to gluten rather than oral tolerance.115 These findings also underscore the close association between adaptive and innate immunity in celiac disease pathogenesis (see section on innate immunity below). An integrative model of immune dysregulation in celiac disease is shown in Figure 9.
Figure 9. Immune dysregulation in celiac disease.

a) In health, gluten is tolerated in the presence of anti-gluten Foxp3+ regulatory T cells. Moreover, intraepithelial lymphocytes (IELs) express inhibitory natural killer (NK) receptors that prevent uncontrolled T cell activation.
b) With inflammation (e.g., celiac disease shown here) or infection, HLA-DQ2 or –DQ8 bind gluten on antigen presenting cells and present to T cells leading to an anti-gluten T cell response which release IFN-γ and possibly IL-21 leading to epithelial damage. The upregulation of IL-15 and IFN-α in the lamina propria induce dendritic cells to acquire a pro-inflammatory phenotype. The innate immune system is also dyregulated in celiac disease in that IELs undergo reprogramming to acquire a natural killer phenotype characterized by upregulation of NKG2D and CD94/NKG2C receptors that recognize MICA, MICB and HLA-E on epithelial cells mediating tissue damage. IL-15 upregulates NK receptors and promotes T-cell receptor independent killing as well as blocking Foxp3+ regulatory T cell action on IELs. Finally, the humoral immune system produces gluten-specific antibodies that mediate systemic manifestations notably dermatitis herpetiformis.
The role of adaptive immunity in celiac disease pathogenesis was first described in the 1970s when Ferguson and MacDonald116,117 reported an association of celiac disease with a lymphocyte-mediated immunity to gluten in the small intestine and that T cell-mediated immunity led to characteristic pathological changes such as villous atrophy in an allograft rejection model. Further studies found that T cells recognize gluten peptides presented by HLA-DQ2 or –DQ8 molecules on APCs in the lamina propria.118,119 Gluten-specific T cells from small intestines of celiac patients reveal high levels of interferon-γ (IFN-γ)120 and messenger RNA for IFN-γ was high in biopsies from celiac patients treated with short-term gluten in vitro.121 In celiac disease, IFN-γ is produced by TH1 cells induced by IL-15, IFN-α and possibly IL-18.115,122,123 IFN-α, in particular, is highly expressed in small bowel from celiac patients, and it likely plays an important role in differentiation of proinflammatory dendritic cells. In support of this hypothesis, clinical observations have been made of celiac disease development after IFN-α treatment for hepatitis C124 and higher risk of celiac disease in patients with Downs syndrome in whom IFN-α receptor expression and type I IFN response are increased as chromosome 21 harbors the IFN-α receptor. 125,126
Innate immune response
While gluten-specific CD4+ T cells play a central role in celiac disease, they are not sufficient to produce characteristic epithelial damage and villous atrophy. This is mediated by innate immune signals with intraepithelial lymphocytes (IELs) playing a primary role (reviewed in127). IELs are a prominent histological feature in the spectrum of celiac disease and aberrant IEL populations underlies refractory sprue (polyclonal in type I and monoclonal in type II) as well as enteropathy-associated lymphoma (EATL).128 Intestinal IELs are a heterogeneous population composed primarily of TCRαβ+ CD8+ cells but also TCRγδ+ and few natural killer(NK)-like cells.129
Epithelial stress can be triggered by inflammation, infection and gluten peptides leading to expression of stress signals on enterocytes primarily MHC class I-related chain A and B (MICA and MICB) molecules and HLA-E.130 (Figure 9) In healthy intestine, IELs typically express inhibitory CD94/NKG2A receptors. In celiac disease, on the other hand, IELs express NK receptors NKG2D131 and CD94/NKG2C132 that recognize MICA and MICB133 and HLA-E on epithelial cells134 which mediate epithelial destruction. IL-15 plays a key role here by upregulating NK receptors on cytotoxic IELs and enables T-cell receptor independent killing.131,135,136 Cytokine secretion (e.g. IFN-γ) and proliferation is mediated by CD94-NKG2C.132 Activation of cytotoxic IELs might also be induced by gluten-specific CD4+ T cells through IL-21123,137 and IFN-γ.121,138
In refractory sprue, IELs acquire a highly activated NK-like phenotype.128 In this condition, the inflammatory state in the small intestine persists despite avoidance of wheat, rye and barley. There are two types (RCD I and II) characterized by their IEL phenotypes(reviewed in 139). In RCD type I, IELs express CD3 and CD8 as well as TCR-β similar to that found in celiac disease. In these cases, prognosis is good with immunsuppressive therapy.128,140 RCD type II, on the other hand, lack CD8, CD4 and TCR-αβ, have intracellular CD3ε, have a clonal TCR gene rearrangement and carry a dismal prognosis.140 The NK-like phenotype of IELs in refractory sprue is promoted and maintained by elevated IL-15 expression in the small intestinal epithelium. 141,142
Unanswered questions and future directions
We have come a long way in our understanding of celiac disease pathogenesis since Dicke’s first clinical observations in the 1950s. However, several questions remain unanswered in all three domains of genetics, environment and immmunology. In celiac disease genetics, there has been an explosion in the number of susceptibility variants identified due to technological improvements in genotyping. The next phase of study will need to elucidate the functional consequences of these variants and their contribution to disease pathogenesis. The full impact of rare variants in celiac disease has not yet been studied and could explain some of the missing heritability. In addition, the role of epigenetics (e.g., methylation) has not been investigated in celiac disease and could play an important role in disease susceptibility. Finally, the application of genetics discoveries in clinical practice remains undetermined. Presently, HLA genetic testing is used primarily for its negative predictive value, and it is not clear if additional, low or moderately penetrant susceptibility variants will alter clinical diagnosis and management.
Regarding environmental factors, it remains unclear how microorganisms (both commensal and pathogenic) contribute to disease. To date, investigators have been unable to tease apart cause versus consequence in microbiome studies in celiac disease. Moreover, it remains to be studied how modulation of the microbiome through use of probiotics, for example, could alter disease onset or course. Importantly, the role of viruses and fungi has been understudied in celiac disease to date. While epidemiological studies suggest certain protective factors such as breast-feeding and timing of gluten introduction, mechanistic underpinnings of these observations remain incompletely understood.
Our immunological understanding of celiac disease now encompasses both adaptive and innate immunity. However, questions remain about transport of gluten peptides across the epithelium into the lamina propria. Moreover, the pathogenic role of anti-TG antibodies continues to be debated. In addition, the role of TCRγδ+ IELs in disease pathogenesis remains unexplored. Improved understanding of celiac disease pathogenesis is crucial to development of novel and effective treatment strategies.
Key Points.
Celiac disease results from the interplay of genetic, environmental and immunological factors.
HLA-DQ2 and –DQ8 are the strongest and best-characterized genetic susceptibility factors in celiac disease, although recent genome-wide association studies have identified additional susceptibility variants – many involved in the immune system and overlapping with other immune-mediated disease.
Environmental factors implicated in disease pathogenesis include gluten, commensal and pathogenic microorganisms, timing of gluten introduction, mode of delivery and length of breast-feeding; however, the mechanisms underlying these associations are incompletely understood.
Both the adaptive and innate immune systems are dysregulated in celiac disease pathophysiology.
Improved understanding of celiac disease pathophysiology will help uncover new potential therapeutic targets and provide insight into disease mechanisms relevant to other immune-mediated disease such as type I diabetes.
References
- 1.Troncone R, Jabri B. Coeliac disease and gluten sensitivity. J Intern Med. 2011;269:582–590. doi: 10.1111/j.1365-2796.2011.02385.x. [DOI] [PubMed] [Google Scholar]
- 2.Losowsky MS. A history of coeliac disease. Dig Dis. 2008;26:112–120. doi: 10.1159/000116768. [DOI] [PubMed] [Google Scholar]
- 3.DICKE WK, WEIJERS HA, VAN DE KAMER JH. Coeliac disease. II. The presence in wheat of a factor having a deleterious effect in cases of coeliac disease. Acta Paediatr. 1953;42:34–42. doi: 10.1111/j.1651-2227.1953.tb05563.x. [DOI] [PubMed] [Google Scholar]
- 4.Janeway CA, Travers P. Anonymous Immunobiology: The Immune System in Health and Disease. 3. Vol. 1. London and New York: Current Biology Ltd/Garland Publishing Inc; 1997. pp. 24–1.pp. 25 [Google Scholar]
- 5.Marsh SG for the WHO Nomenclature Committee for Factors of the HLA System. Nomenclature for factors of the HLA system, update January 2012. Tissue Antigens. 2012;79:393–397. doi: 10.1111/j.1399-0039.2012.01856.x. [DOI] [PubMed] [Google Scholar]
- 6.Ploski R, Ek J, Thorsby E, Sollid LM. On the HLA-DQ(alpha 1*0501, beta 1*0201)-associated susceptibility in celiac disease: a possible gene dosage effect of DQB1*0201. Tissue Antigens. 1993;41:173–177. doi: 10.1111/j.1399-0039.1993.tb01998.x. [DOI] [PubMed] [Google Scholar]
- 7.van Belzen MJ, Koeleman BP, Crusius JB, et al. Defining the contribution of the HLA region to cis DQ2-positive coeliac disease patients. Genes Immun. 2004;5:215–220. doi: 10.1038/sj.gene.6364061. [DOI] [PubMed] [Google Scholar]
- 8.Mearin ML, Biemond I, Pena AS, et al. HLA-DR phenotypes in Spanish coeliac children: their contribution to the understanding of the genetics of the disease. Gut. 1983;24:532–537. doi: 10.1136/gut.24.6.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Megiorni F, Mora B, Bonamico M, et al. HLA-DQ and risk gradient for celiac disease. Hum Immunol. 2009;70:55–59. doi: 10.1016/j.humimm.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Vader W, Stepniak D, Kooy Y, et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc Natl Acad Sci U S A. 2003;100:12390–12395. doi: 10.1073/pnas.2135229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zubillaga P, Vidales MC, Zubillaga I, Ormaechea V, Garcia-Urkia N, Vitoria JC. HLA-DQA1 and HLA-DQB1 genetic markers and clinical presentation in celiac disease. J Pediatr Gastroenterol Nutr. 2002;34:548–554. doi: 10.1097/00005176-200205000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Congia M, Cucca F, Frau F, et al. A gene dosage effect of the DQA1*0501/DQB1*0201 allelic combination influences the clinical heterogeneity of celiac disease. Hum Immunol. 1994;40:138–142. doi: 10.1016/0198-8859(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 13.Al-Toma A, Goerres MS, Meijer JW, Pena AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol. 2006;4:315–319. doi: 10.1016/j.cgh.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 14.Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2. 2 are related to sustained gluten antigen presentation. Nat Immunol. 2009;10:1096–1101. doi: 10.1038/ni.1780. [DOI] [PubMed] [Google Scholar]
- 15.Karell K, Louka AS, Moodie SJ, et al. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol. 2003;64:469–477. doi: 10.1016/s0198-8859(03)00027-2. [DOI] [PubMed] [Google Scholar]
- 16.The Allele Frequency Net Database. 2012;2012 Available at: www.allelefrequencies.net. [Google Scholar]
- 17.Pietzak MM, Schofield TC, McGinniss MJ, Nakamura RM. Stratifying risk for celiac disease in a large at-risk United States population by using HLA alleles. Clin Gastroenterol Hepatol. 2009;7:966–971. doi: 10.1016/j.cgh.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 18.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hovhannisyan Z, Weiss A, Martin A, et al. The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature. 2008;456:534–538. doi: 10.1038/nature07524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molberg O, Kett K, Scott H, Thorsby E, Sollid LM, Lundin KE. Gliadin specific, HLA DQ2-restricted T cells are commonly found in small intestinal biopsies from coeliac disease patients, but not from controls. Scand J Immunol. 1997;46:103–109. doi: 10.1046/j.1365-3083.1997.d01-93.x. [DOI] [PubMed] [Google Scholar]
- 21.Molberg O, Mcadam SN, Korner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 22.Arentz-Hansen H, Korner R, Molberg O, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. 2000;191:603–612. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arentz-Hansen H, McAdam SN, Molberg O, et al. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology. 2002;123:803–809. doi: 10.1053/gast.2002.35381. [DOI] [PubMed] [Google Scholar]
- 24.Qiao SW, Bergseng E, Molberg O, et al. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J Immunol. 2004;173:1757–1762. doi: 10.4049/jimmunol.173.3.1757. [DOI] [PubMed] [Google Scholar]
- 25.van de Wal Y, Kooy YM, van Veelen PA, et al. Small intestinal T cells of celiac disease patients recognize a natural pepsin fragment of gliadin. Proc Natl Acad Sci U S A. 1998;95:10050–10054. doi: 10.1073/pnas.95.17.10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shan L, Qiao SW, Arentz-Hansen H, et al. Identification and analysis of multivalent proteolytically resistant peptides from gluten: implications for celiac sprue. J Proteome Res. 2005;4:1732–1741. doi: 10.1021/pr050173t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henderson KN, Tye-Din JA, Reid HH, et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity. 2007;27:23–34. doi: 10.1016/j.immuni.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 28.van Heel DA, Franke L, Hunt KA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunt KA, Zhernakova A, Turner G, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trynka G, Hunt KA, Bockett NA, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trynka G, Wijmenga C, van Heel DA. A genetic perspective on coeliac disease. Trends Mol Med. 2010;16:537–550. doi: 10.1016/j.molmed.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol. 2011;29:493–525. doi: 10.1146/annurev-immunol-040210-092915. [DOI] [PubMed] [Google Scholar]
- 34.van de Wal Y, Kooy YM, van Veelen P, et al. Glutenin is involved in the gluten-driven mucosal T cell response. Eur J Immunol. 1999;29:3133–3139. doi: 10.1002/(SICI)1521-4141(199910)29:10<3133::AID-IMMU3133>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 35.Molberg O, Solheim Flaete N, Jensen T, et al. Intestinal T-cell responses to high-molecular-weight glutenins in celiac disease. Gastroenterology. 2003;125:337–344. doi: 10.1016/s0016-5085(03)00890-4. [DOI] [PubMed] [Google Scholar]
- 36.Dewar DH, Amato M, Ellis HJ, et al. The toxicity of high molecular weight glutenin subunits of wheat to patients with coeliac disease. Eur J Gastroenterol Hepatol. 2006;18:483–491. doi: 10.1097/00042737-200605000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Vader LW, Stepniak DT, Bunnik EM, et al. Characterization of cereal toxicity for celiac disease patients based on protein homology in grains. Gastroenterology. 2003;125:1105–1113. doi: 10.1016/s0016-5085(03)01204-6. [DOI] [PubMed] [Google Scholar]
- 38.Arentz-Hansen H, Fleckenstein B, Molberg O, et al. The molecular basis for oat intolerance in patients with celiac disease. PLoS Med. 2004;1:e1. doi: 10.1371/journal.pmed.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan L, Molberg O, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 40.Hausch F, Shan L, Santiago NA, Gray GM, Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol. 2002;283:G996–G1003. doi: 10.1152/ajpgi.00136.2002. [DOI] [PubMed] [Google Scholar]
- 41.Stepniak D, Spaenij-Dekking L, Mitea C, et al. Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am J Physiol Gastrointest Liver Physiol. 2006;291:G621–9. doi: 10.1152/ajpgi.00034.2006. [DOI] [PubMed] [Google Scholar]
- 42.De Angelis M, Rizzello CG, Fasano A, et al. VSL#3 probiotic preparation has the capacity to hydrolyze gliadin polypeptides responsible for Celiac Sprue. Biochim Biophys Acta. 2006;1762:80–93. doi: 10.1016/j.bbadis.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Heyman M, Abed J, Lebreton C, Cerf-Bensussan N. Intestinal permeability in coeliac disease: insight into mechanisms and relevance to pathogenesis. Gut. 2011 doi: 10.1136/gutjnl-2011-300327. [DOI] [PubMed] [Google Scholar]
- 44.Wapenaar MC, Monsuur AJ, van Bodegraven AA, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–467. doi: 10.1136/gut.2007.133132. [DOI] [PubMed] [Google Scholar]
- 45.Monsuur AJ, de Bakker PI, Alizadeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet. 2005;37:1341–1344. doi: 10.1038/ng1680. [DOI] [PubMed] [Google Scholar]
- 46.Fasano A, Baudry B, Pumplin DW, et al. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci U S A. 1991;88:5242–5246. doi: 10.1073/pnas.88.12.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 48.Tripathi A, Lammers KM, Goldblum S, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci U S A. 2009;106:16799–16804. doi: 10.1073/pnas.0906773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. 2008;205:143–154. doi: 10.1084/jem.20071204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55:856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muegge BD, Kuczynski J, Knights D, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wikoff WR, Anfora AT, Liu J, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy SF, Kwon JH, Boone DL. Novel players in inflammatory bowel disease pathogenesis. Curr Gastroenterol Rep. 2012;14:146–152. doi: 10.1007/s11894-012-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forsberg G, Fahlgren A, Horstedt P, Hammarstrom S, Hernell O, Hammarstrom ML. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol. 2004;99:894–904. doi: 10.1111/j.1572-0241.2004.04157.x. [DOI] [PubMed] [Google Scholar]
- 58.Tjellstrom B, Stenhammar L, Hogberg L, et al. Gut microflora associated characteristics in children with celiac disease. Am J Gastroenterol. 2005;100:2784–2788. doi: 10.1111/j.1572-0241.2005.00313.x. [DOI] [PubMed] [Google Scholar]
- 59.Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol. 2011;11:219. doi: 10.1186/1471-2180-11-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tjellstrom B, Stenhammar L, Hogberg L, et al. Gut microflora associated characteristics in first-degree relatives of children with celiac disease. Scand J Gastroenterol. 2007;42:1204–1208. doi: 10.1080/00365520701320687. [DOI] [PubMed] [Google Scholar]
- 61.Calvert VS, Collantes R, Elariny H, et al. A systems biology approach to the pathogenesis of obesity-related nonalcoholic fatty liver disease using reverse phase protein microarrays for multiplexed cell signaling analysis. Hepatology. 2007;46:166–172. doi: 10.1002/hep.21688. [DOI] [PubMed] [Google Scholar]
- 62.Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol. 2009;62:264–269. doi: 10.1136/jcp.2008.061366. [DOI] [PubMed] [Google Scholar]
- 63.Sanz Y, Sanchez E, Marzotto M, Calabuig M, Torriani S, Dellaglio F. Differences in faecal bacterial communities in coeliac and healthy children as detected by PCR and denaturing gradient gel electrophoresis. FEMS Immunol Med Microbiol. 2007;51:562–568. doi: 10.1111/j.1574-695X.2007.00337.x. [DOI] [PubMed] [Google Scholar]
- 64.Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol. 2010;10:175. doi: 10.1186/1471-2180-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanchez E, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Intestinal Bacteroides species associated with coeliac disease. J Clin Pathol. 2010;63:1105–1111. doi: 10.1136/jcp.2010.076950. [DOI] [PubMed] [Google Scholar]
- 66.Nistal E, Caminero A, Herran AR, et al. Differences of small intestinal bacteria populations in adults and children with/without celiac disease: Effect of age, gluten diet, and disease. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21830. [DOI] [PubMed] [Google Scholar]
- 67.Kalliomaki M, Satokari R, Lahteenoja H, et al. Expression of Microbiota, Toll-Like Receptors And Their Regulators In The Small Intestinal Mucosa In Celiac Disease. J Pediatr Gastroenterol Nutr. 2011 doi: 10.1097/MPG.0b013e318241cfa8. [DOI] [PubMed] [Google Scholar]
- 68.Sellitto M, Bai G, Serena G, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS One. 2012;7:e33387. doi: 10.1371/journal.pone.0033387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Palma G, Nadal I, Collado MC, Sanz Y. Effects of a gluten-free diet on gut microbiota and immune function in healthy adult human subjects. Br J Nutr. 2009;102:1154–1160. doi: 10.1017/S0007114509371767. [DOI] [PubMed] [Google Scholar]
- 70.De Palma G, Cinova J, Stepankova R, Tuckova L, Sanz Y. Pivotal Advance: Bifidobacteria and Gram-negative bacteria differentially influence immune responses in the proinflammatory milieu of celiac disease. J Leukoc Biol. 2010;87:765–778. doi: 10.1189/jlb.0709471. [DOI] [PubMed] [Google Scholar]
- 71.D’Arienzo R, Maurano F, Lavermicocca P, Ricca E, Rossi M. Modulation of the immune response by probiotic strains in a mouse model of gluten sensitivity. Cytokine. 2009;48:254–259. doi: 10.1016/j.cyto.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 72.Cinova J, De Palma G, Stepankova R, et al. Role of intestinal bacteria in gliadin-induced changes in intestinal mucosa: study in germ-free rats. PLoS One. 2011;6:e16169. doi: 10.1371/journal.pone.0016169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Angelis M, Rizzello CG, Scala E, et al. Probiotic preparation has the capacity to hydrolyze proteins responsible for wheat allergy. J Food Prot. 2007;70:135–144. doi: 10.4315/0362-028x-70.1.135. [DOI] [PubMed] [Google Scholar]
- 74.Ringel Y, Ringel-Kulka T. The rationale and clinical effectiveness of probiotics in irritable bowel syndrome. J Clin Gastroenterol. 2011;45 (Suppl):S145–8. doi: 10.1097/MCG.0b013e31822d32d3. [DOI] [PubMed] [Google Scholar]
- 75.Sartor RB. Key questions to guide a better understanding of host-commensal microbiota interactions in intestinal inflammation. Mucosal Immunol. 2011;4:127–132. doi: 10.1038/mi.2010.87. [DOI] [PubMed] [Google Scholar]
- 76.Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med. 1984;160:1544–1557. doi: 10.1084/jem.160.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kagnoff MF, Paterson YJ, Kumar PJ, et al. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut. 1987;28:995–1001. doi: 10.1136/gut.28.8.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kagnoff MF. Celiac disease: adenovirus and alpha gliadin. Curr Top Microbiol Immunol. 1989;145:67–78. doi: 10.1007/978-3-642-74594-2_6. [DOI] [PubMed] [Google Scholar]
- 79.Mahon J, Blair GE, Wood GM, Scott BB, Losowsky MS, Howdle PD. Is persistent adenovirus 12 infection involved in coeliac disease? A search for viral DNA using the polymerase chain reaction. Gut. 1991;32:1114–1116. doi: 10.1136/gut.32.10.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lahdeaho ML, Lehtinen M, Rissa HR, Hyoty H, Reunala T, Maki M. Antipeptide antibodies to adenovirus E1b protein indicate enhanced risk of celiac disease and dermatitis herpetiformis. Int Arch Allergy Immunol. 1993;101:272–276. doi: 10.1159/000236457. [DOI] [PubMed] [Google Scholar]
- 81.Vesy CJ, Greenson JK, Papp AC, Snyder PJ, Qualman SJ, Prior TW. Evaluation of celiac disease biopsies for adenovirus 12 DNA using a multiplex polymerase chain reaction. Mod Pathol. 1993;6:61–64. [PubMed] [Google Scholar]
- 82.Ivarsson A, Hernell O, Nystrom L, Persson LA. Children born in the summer have increased risk for coeliac disease. J Epidemiol Community Health. 2003;57:36–39. doi: 10.1136/jech.57.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol. 2006;101:2333–2340. doi: 10.1111/j.1572-0241.2006.00741.x. [DOI] [PubMed] [Google Scholar]
- 84.Zanoni G, Navone R, Lunardi C, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. 2006;3:e358. doi: 10.1371/journal.pmed.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts SE, Williams JG, Meddings D, Davidson R, Goldacre MJ. Perinatal risk factors and coeliac disease in children and young adults: a record linkage study. Aliment Pharmacol Ther. 2009;29:222–231. doi: 10.1111/j.1365-2036.2008.03871.x. [DOI] [PubMed] [Google Scholar]
- 86.Decker E, Engelmann G, Findeisen A, et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics. 2010;125:e1433–40. doi: 10.1542/peds.2009-2260. [DOI] [PubMed] [Google Scholar]
- 87.Marild K, Stephansson O, Montgomery S, Murray JA, Ludvigsson JF. Pregnancy outcome and risk of celiac disease in offspring: a nationwide case-control study. Gastroenterology. 2012;142:39–45. e3. doi: 10.1053/j.gastro.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ivarsson A, Persson LA, Nystrom L, et al. Epidemic of coeliac disease in Swedish children. Acta Paediatr. 2000;89:165–171. doi: 10.1080/080352500750028771. [DOI] [PubMed] [Google Scholar]
- 90.Ivarsson A, Persson LA, Nystrom L, Hernell O. The Swedish coeliac disease epidemic with a prevailing twofold higher risk in girls compared to boys may reflect gender specific risk factors. Eur J Epidemiol. 2003;18:677–684. doi: 10.1023/a:1024873630588. [DOI] [PubMed] [Google Scholar]
- 91.Myleus A, Ivarsson A, Webb C, et al. Celiac disease revealed in 3% of Swedish 12-year-olds born during an epidemic. J Pediatr Gastroenterol Nutr. 2009;49:170–176. doi: 10.1097/MPG.0b013e31818c52cc. [DOI] [PubMed] [Google Scholar]
- 92.Ivarsson A. The Swedish epidemic of coeliac disease explored using an epidemiological approach--some lessons to be learnt. Best Pract Res Clin Gastroenterol. 2005;19:425–440. doi: 10.1016/j.bpg.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 93.Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA. 2005;293:2343–2351. doi: 10.1001/jama.293.19.2343. [DOI] [PubMed] [Google Scholar]
- 94.Akobeng AK, Ramanan AV, Buchan I, Heller RF. Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies. Arch Dis Child. 2006;91:39–43. doi: 10.1136/adc.2005.082016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guandalini S. The influence of gluten: weaning recommendations for healthy children and children at risk for celiac disease. Nestle Nutr Workshop Ser Pediatr Program. 2007;60:139–51. doi: 10.1159/000106366. discussion 151–5. [DOI] [PubMed] [Google Scholar]
- 96.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci U S A. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sollid LM, Jabri B. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr Opin Immunol. 2011;23:732–738. doi: 10.1016/j.coi.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics. 2012 doi: 10.1007/s00251-012-0599-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rostom A, Dube C, Cranney A, et al. The diagnostic accuracy of serologic tests for celiac disease: a systematic review. Gastroenterology. 2005;128:S38–46. doi: 10.1053/j.gastro.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 101.Sollid LM, Jabri B. Is celiac disease an autoimmune disorder? Curr Opin Immunol. 2005;17:595–600. doi: 10.1016/j.coi.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 102.Lindfors K, Maki M, Kaukinen K. Transglutaminase 2-targeted autoantibodies in celiac disease: Pathogenetic players in addition to diagnostic tools? Autoimmun Rev. 2010;9:744–749. doi: 10.1016/j.autrev.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 103.Di Sabatino A, Vanoli A, Giuffrida P, Luinetti O, Solcia E, Corazza GR. The function of tissue transglutaminase in celiac disease. Autoimmun Rev. 2012 doi: 10.1016/j.autrev.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 104.Di Niro R, Mesin L, Zheng NY, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012;18:441–445. doi: 10.1038/nm.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caja S, Myrsky E, Korponay-Szabo IR, et al. Inhibition of transglutaminase 2 enzymatic activity ameliorates the anti-angiogenic effects of coeliac disease autoantibodies. Scand J Gastroenterol. 2010;45:421–427. doi: 10.3109/00365520903540822. [DOI] [PubMed] [Google Scholar]
- 106.Myrsky E, Kaukinen K, Syrjanen M, Korponay-Szabo IR, Maki M, Lindfors K. Coeliac disease-specific autoantibodies targeted against transglutaminase 2 disturb angiogenesis. Clin Exp Immunol. 2008;152:111–119. doi: 10.1111/j.1365-2249.2008.03600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barone MV, Caputo I, Ribecco MT, et al. Humoral immune response to tissue transglutaminase is related to epithelial cell proliferation in celiac disease. Gastroenterology. 2007;132:1245–1253. doi: 10.1053/j.gastro.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 108.Esposito C, Paparo F, Caputo I, et al. Expression and enzymatic activity of small intestinal tissue transglutaminase in celiac disease. Am J Gastroenterol. 2003;98:1813–1820. doi: 10.1111/j.1572-0241.2003.07582.x. [DOI] [PubMed] [Google Scholar]
- 109.Dieterich W, Trapp D, Esslinger B, et al. Autoantibodies of patients with coeliac disease are insufficient to block tissue transglutaminase activity. Gut. 2003;52:1562–1566. doi: 10.1136/gut.52.11.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kiraly R, Vecsei Z, Demenyi T, Korponay-Szabo IR, Fesus L. Coeliac autoantibodies can enhance transamidating and inhibit GTPase activity of tissue transglutaminase: dependence on reaction environment and enzyme fitness. J Autoimmun. 2006;26:278–287. doi: 10.1016/j.jaut.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 111.Sardy M, Karpati S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med. 2002;195:747–757. doi: 10.1084/jem.20011299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.du Pre MF, Samsom JN. Adaptive T-cell responses regulating oral tolerance to protein antigen. Allergy. 2011;66:478–490. doi: 10.1111/j.1398-9995.2010.02519.x. [DOI] [PubMed] [Google Scholar]
- 113.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun CM, Hall JA, Blank RB, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.DePaolo RW, Abadie V, Tang F, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011;471:220–224. doi: 10.1038/nature09849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.MacDonald TT, Ferguson A. Hypersensitivity reactions in the small intestine. 2. Effects of allograft rejection on mucosal architecture and lymphoid cell infiltrate. Gut. 1976;17:81–91. doi: 10.1136/gut.17.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ferguson A, MacDonald TT, McClure JP, Holden RJ. Cell-mediated immunity to gliadin within the small-intestinal mucosa in coeliac disease. Lancet. 1975;1:895–897. doi: 10.1016/s0140-6736(75)91689-x. [DOI] [PubMed] [Google Scholar]
- 118.Lundin KE, Scott H, Hansen T, et al. Gliadin-specific, HLA-DQ(alpha 1*0501, beta 1*0201) restricted T cells isolated from the small intestinal mucosa of celiac disease patients. J Exp Med. 1993;178:187–196. doi: 10.1084/jem.178.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lundin KE, Gjertsen HA, Scott H, Sollid LM, Thorsby E. Function of DQ2 and DQ8 as HLA susceptibility molecules in celiac disease. Hum Immunol. 1994;41:24–27. doi: 10.1016/0198-8859(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 120.Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. 1995;37:766–776. doi: 10.1136/gut.37.6.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nilsen EM, Jahnsen FL, Lundin KE, et al. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology. 1998;115:551–563. doi: 10.1016/s0016-5085(98)70134-9. [DOI] [PubMed] [Google Scholar]
- 122.Salvati VM, MacDonald TT, Bajaj-Elliott M, et al. Interleukin 18 and associated markers of T helper cell type 1 activity in coeliac disease. Gut. 2002;50:186–190. doi: 10.1136/gut.50.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Monteleone G, Pender SL, Alstead E, et al. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in coeliac disease. Gut. 2001;48:425–429. doi: 10.1136/gut.48.3.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cammarota G, Cuoco L, Cianci R, Pandolfi F, Gasbarrini G. Onset of coeliac disease during treatment with interferon for chronic hepatitis C. Lancet. 2000;356:1494–1495. doi: 10.1016/S0140-6736(00)02880-4. [DOI] [PubMed] [Google Scholar]
- 125.George EK, Mearin ML, Bouquet J, et al. High frequency of celiac disease in Down syndrome. J Pediatr. 1996;128:555–557. doi: 10.1016/s0022-3476(96)70369-4. [DOI] [PubMed] [Google Scholar]
- 126.Gerdes AM, Horder M, Bonnevie-Nielsen V. Increased IFN-alpha-induced sensitivity but reduced reactivity of 2′,5′-oligoadenylate synthetase (2,5AS) in trisomy 21 blood lymphocytes. Clin Exp Immunol. 1993;93:93–96. doi: 10.1111/j.1365-2249.1993.tb06502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jabri B, Sollid LM. Mechanisms of disease: immunopathogenesis of celiac disease. Nat Clin Pract Gastroenterol Hepatol. 2006;3:516–525. doi: 10.1038/ncpgasthep0582. [DOI] [PubMed] [Google Scholar]
- 128.Cellier C, Delabesse E, Helmer C, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet. 2000;356:203–208. doi: 10.1016/s0140-6736(00)02481-8. [DOI] [PubMed] [Google Scholar]
- 129.Jabri B, Ebert E. Human CD8+ intraepithelial lymphocytes: a unique model to study the regulation of effector cytotoxic T lymphocytes in tissue. Immunol Rev. 2007;215:202–214. doi: 10.1111/j.1600-065X.2006.00481.x. [DOI] [PubMed] [Google Scholar]
- 130.Hue S, Mention JJ, Monteiro RC, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 131.Meresse B, Chen Z, Ciszewski C, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21:357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 132.Meresse B, Curran SA, Ciszewski C, et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med. 2006;203:1343–1355. doi: 10.1084/jem.20060028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bauer S, Groh V, Wu J, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. [PubMed] [Google Scholar]
- 134.Braud VM, Allan DS, O’Callaghan CA, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 135.Roberts AI, Lee L, Schwarz E, et al. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol. 2001;167:5527–5530. doi: 10.4049/jimmunol.167.10.5527. [DOI] [PubMed] [Google Scholar]
- 136.Tang F, Chen Z, Ciszewski C, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J Exp Med. 2009;206:707–719. doi: 10.1084/jem.20071887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kasaian MT, Whitters MJ, Carter LL, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. 2002;16:559–569. doi: 10.1016/s1074-7613(02)00295-9. [DOI] [PubMed] [Google Scholar]
- 138.Perera L, Shao L, Patel A, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis. 2007;13:298–307. doi: 10.1002/ibd.20026. [DOI] [PubMed] [Google Scholar]
- 139.Rubio-Tapia A, Murray JA. Classification and management of refractory coeliac disease. Gut. 2010;59:547–557. doi: 10.1136/gut.2009.195131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rubio-Tapia A, Kelly DG, Lahr BD, Dogan A, Wu TT, Murray JA. Clinical staging and survival in refractory celiac disease: a single center experience. Gastroenterology. 2009;136:99–107. doi: 10.1053/j.gastro.2008.10.013. quiz 352–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Malamut G, El Machhour R, Montcuquet N, et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. J Clin Invest. 2010;120:2131–2143. doi: 10.1172/JCI41344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mention JJ, Ben Ahmed M, Begue B, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. 2003;125:730–745. doi: 10.1016/s0016-5085(03)01047-3. [DOI] [PubMed] [Google Scholar]