

Abstract
Prediction of HLA binding affinity is widely utilized to identify candidate T cell epitopes, and an affinity of 500 nM is routinely used as a threshold for peptide selection. However, the fraction (%) of peptides predicted to bind with affinities of 500 nM varies by allele. For example, of a large collection of about 30,000 dengue virus derived peptides only 0.3% were predicted to bind HLA A*0101, while nearly 5% were predicted for A*0201. This striking difference could not be ascribed to variation in accuracy of the algorithms utilized, as predicted values closely correlated with affinity measured in vitro with purified HLA molecules. These data raised the question whether different alleles would also vary in terms of epitope repertoire size, defined as the number of associated epitopes or, alternatively, whether alleles vary drastically in terms of the affinity threshold associated with immunogenicity. To address this issue, strains of HLA transgenic mice with wide (A*0201), intermediate (B*0702) or narrow (A*0101) repertoires were immunized with peptides of varying binding affinity and relative percentile ranking. The results show that absolute binding capacity is a better predictor of immunogenicity, and analysis of epitopes from the Immune Epitope Database (IEDB) revealed that predictive efficacy is increased using allele-specific affinity thresholds. Finally, we investigate the genetic and structural basis of the phenomenon. While no stringent correlate was defined, on average HLA B alleles are associated with significantly narrower repertoires than HLA A alleles.
Introduction
Molecular structures recognized by immune system receptors are called epitopes (1). Epitopes that bind, and are presented in the context of, class I and class II MHC molecules are typically recognized by CD8+ and CD4+ T cells, respectively. Binding of a peptide to the MHC molecule is one of the most selective steps in the classical MHC I pathway of antigen processing (2-4). The affinity with which an epitope binds to the MHC molecule plays an important role in determining its immunogenicity (5), and high affinity MHC-epitope interactions tend to be associated with higher immune responsiveness. However, while MHC binding is necessary for recognition by T cells, it is in itself not sufficient to define immunogenicity. Indeed, recognition appears to be influenced by several other factors, such as abundance of proteins, antigen processing, immunodominance and the presence of a suitable T-cell repertoire (2-4, 6-10). Previous studies indicated 500 nM as an MHC affinity threshold associated with potential immunogenicity for HLA class I restricted T cells (5).
Computational prediction of MHC class I binding capacity has been used in epitope identification and vaccine discovery studies for many years (11-19). Various bioinformatics tools and resources that allow prediction of the binding affinity of peptides to MHC class I and II molecules are provided at a number of publically accessible websites, including the Immune Epitope Database and Analysis Resource (20, 21), Bimas (22), SYFPEITHI (23), NetMHC (24), ProPred (25), ProPred1 (26), ABCpred (27), Multipred (28) and Rankpep (29). In general, MHC class I binding prediction tools scan a protein’s amino acid sequence to determine each subsequence’s ability to bind a specific MHC class I molecule. While the majority of MHC class I epitopes are 9 and 10 amino acids in length (20, 21, 23) it is known that shorter or longer peptides can also be antigenic targets of class I responses. However, the availability of predictive tools for non-canonical sizes (i.e., other than 9- and 10-mers) is more limited, and their performance is generally less robust, likely due to the fact that limited data is available to train and improve the corresponding algorithms.
Several different computational approaches towards prediction algorithms are available, including those based on Artificial Neural Networks (ANN) (30), the Average Relative Binding (ARB) method (31), Stabilized Matrices (SMM) (32, 33), scoring matrices derived from positional scanning combinatorial peptide libraries (Comblib) (34), the NetMHCpan method (35), Hidden Markov Models (HMM) (28) and Position Specific Scoring Matrices (PSSMs) (29). The output of the different methods is typically given either in units of predicted affinity (IC50 nM), or as a percentile score reflecting the relative affinity of a selected peptide compared to a universe of random sequences. The efficacy of different methodologies for predicting high affinity MHC binding peptides has been addressed in several studies by our group, from both the bioinformatics (21, 38) and T cell epitope identification perspectives (4, 13, 39-42).
However, a key question to be addressed is whether predicted binding affinity or percentile rank is the best predictor of potentially immunogenic peptides, especially across a diverse set of different HLA class I molecules. The present study set out to address this question by investigating the MHC binding and associated T cell epitope repertoires of a panel of different HLA class I molecules using bioinformatics analyses, experimental testing of in vitro binding with purified HLA molecules, and in vivo immunogenicity testing of selected peptides in HLA transgenic mice. The results unexpectedly revealed that different HLA class I molecules are associated with distinctively different repertoires of peptide binders and associated epitope affinities.
Materials and methods
Peptide sets
A set of Dengue virus (DENV) sequences recently utilized for epitope identification studies in endemic areas (43) was used in the initial analysis presented herein. Essentially, full-length DENV polyprotein sequences for each serotype were retrieved from the NCBI Protein database using the query, “txid11053 AND polyprotein AND 3000:5000[slen]” with the corresponding NCBI taxonomy ID being substituted for each serotype. To avoid geographical bias, the number of unique isolates (varying by at least 1 amino acid from all other isolates) from any one country was limited to 10. Polyproteins were then broken down into all possible 9-mer sequences for binding predictions, corresponding to a set of 38,845 unique peptides.
The binding affinities of all 9-mer peptides were predicted for 27 common HLA class I alleles (Table 1). The set of alleles was selected on the basis of allele frequencies in the general worldwide population, and together they are estimated to provide coverage of over 90% of individuals at the A and B loci (44-46). Binding predictions were performed using the command-line version of the SMM prediction tool available on the Immune Epitope Database website (http://www.iedb.org) (21, 32). The SMM tool was selected because it consistently performs as one of the best prediction tools across a wide array of alleles, and also provides predicted IC50 nM values for the complete set of 27 common alleles considered here. In addition to predicted affinity (IC50) the SMM algorithm also provides a percentile score expressing the relative capacity of each peptide to bind each specific allele, compared to a universe of potential sequences of the same size.
Table 1. Comparison of predicted and actual binding peptides in the top 1% percentile for common HLA A and B molecules.
| HLA allele |
# of peptides tested |
# of predicted binders |
# of actual binders |
Ratio of predicted/ actual binders |
|---|---|---|---|---|
|
| ||||
| A*0101 | 152 | 37 | 41 | 0.90 |
| A*0201 | 160 | 160 | 157 | 1.02 |
| A*0203 | 165 | 165 | 163 | 1.01 |
| A*0206 | 146 | 146 | 141 | 1.04 |
| A*0301 | 156 | 156 | 151 | 1.03 |
| A*1101 | 151 | 151 | 133 | 1.14 |
| A*2301 | 145 | 125 | 91 | 1.37 |
| A*2402 | 150 | 68 | 96 | 0.71 |
| A*2601 | 136 | 77 | 43 | 1.79 |
| A*3001 | 154 | 154 | 120 | 1.28 |
| A*3002 | 146 | 136 | 82 | 1.66 |
| A*3101 | 136 | 136 | 110 | 1.24 |
| A*3201 | 166 | 165 | 116 | 1.42 |
| A*3301 | 143 | 143 | 132 | 1.08 |
| A*6801 | 137 | 137 | 133 | 1.03 |
| A*6802 | 145 | 145 | 143 | 1.01 |
| B*0702 | 153 | 149 | 141 | 1.06 |
| B*0801 | 142 | 134 | 124 | 1.08 |
| B*1501 | 136 | 136 | 135 | 1.01 |
| B*3501 | 142 | 142 | 102 | 1.39 |
| B*4001 | 141 | 140 | 109 | 1.28 |
| B*4402 | 154 | 24 | 122 | 0.20 |
| B*4403 | 143 | 88 | 97 | 0.91 |
| B*5101 | 145 | 9 | 34 | 0.26 |
| B*5301 | 156 | 146 | 108 | 1.35 |
| B*5701 | 136 | 127 | 127 | 1.00 |
| B*5801 | 152 | 152 | 148 | 1.03 |
|
| ||||
| Average = 1.09 | ||||
| Std Dev = 0.33 | ||||
For the analysis of self sequences, protein sequences were randomly selected from the human genome and a set of 9-mer peptides equal in size to the DENV set was generated. Binding predictions were performed as described above. For the analysis of previously identified epitopes, all 9-mer epitopes with defined HLA class I restriction were retrieved from the IEDB (47). Binding predictions were then generated for alleles that had more than 15 epitopes.
Selection of peptides for immunogenicity testing
Selected dengue 9-mer peptides were tested for immunogenicity in A*0101, B*0702 and A*0201 transgenic mice, as described below. For each HLA transgenic mouse strain 4 sets of 30 peptides each were constructed to represent specific predicted percentile score ranges: (i) 0.0 – 0.30 (ii) 0.30 – 1.25 (iii) 1.25 – 5.0 (iv) 5.0 – 15.0. In total 360 9-mer peptides were selected for immunogenicity testing (3 alleles × 4 categories × 30 peptides). All peptides used in this study were synthesized as crude material by Mimotopes (Clayton, Victoria, Australia).
MHC-peptide binding
Purification of HLA class I molecules and quantitative competitive inhibition assays to measure the binding affinity of peptides to purified MHC were performed as described elsewhere (48). Briefly, 0.1-1 nM of a high affinity radiolabeled peptide is co-incubated at room temperature with 1 μM to 1 nM of purified MHC in the presence of a cocktail of protease inhibitors and 1 μM B2-microglobulin. Following a two-day incubation, MHC bound radioactivity is determined by capturing MHC/peptide complexes on W6/32 (anti-class I) antibody coated Lumitrac 600 plates (Greiner Bio-one, Frickenhausen, Germany), and measuring bound cpm using the TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter, and the concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide is calculated. Under the conditions utilized, where [label]<[MHC] and IC50 ≥ [MHC], the measured IC50 values are reasonable approximations of true Kd values (49, 50). Each competitor peptide is tested at six different concentrations covering a 100,000-fold dose range, and in three or more independent experiments. As a positive control, the unlabeled version of the radiolabeled probe is also tested in each experiment.
Mice and immunizations
HLA-A*0201/Kb, A*0101, and B*0702-transgenic mice were bred at the La Jolla Institute for Allergy and Immunology animal facility (La Jolla, CA) as previously described (43). All mouse experiments were performed following Institutional Animal Care and Use Committee approved animal protocols. Mice between 8 and 12 weeks of age were immunized s.c with a pool of 10 individual peptides [10 μg peptide] in 100 μl PBS emulsified in CFA. Two weeks post immunization the mice were sacrificed, and splenic CD8+ T cells were isolated.
IFNγ ELISPOT assay
CD8+ T cells were isolated by magnetic bead positive selection (Miltenyi Biotec, Bergisch Gladbach, Germany). A total of 2 ×105 CD8+ T cells were stimulated with 1 × 105 naive splenocytes as APCs and 10 μg/ml individual peptides in 96-well flat-bottom plates (Immobilon-P; Millipore, Bedford, MA) coated with anti-IFN-γ mAb (clone AN18; Mabtech, Stockholm, Sweden). Each peptide was tested in triplicates. Following a 20-h incubation at 37°C, the wells were washed with PBS/0.05% Tween 20 and then incubated with biotinylated IFN-γ mAb (clone R4-6A2; Mabtech) for 2 h. The spots were developed using Vectastain ABC peroxidase (Vector Laboratories, Burlingame, CA) and 3-amino-9-ethylcarbazole (Sigma-Aldrich, St. Louis, MO) and counted by computer-assisted image analysis (KS-ELISPOT reader; Zeiss, Munich, Germany).
Results
The predicted peptide binding repertoires of HLA class I alleles are widely variable in size
Previously, based on analyses undertaken in the context of HLA A*0201, it was noted that the majority of HLA class I restricted epitopes bound with an affinity of 500 nM (IC50 ≤ 500 nM) or better (5). In the present study we sought to examine whether the number of peptides predicted to bind at this affinity threshold was fairly uniform, or whether different alleles were associated with different repertoire sizes.
Accordingly, the binding affinity of all 9-mer peptides encoded in a database of sequences corresponding to a set of dengue virus (DENV1- 4) proteomes (43) was predicted for a panel of 27 common HLA class I alleles, chosen to provide global population coverage of over 90% (44-46). For each allele the percentage of peptides predicted by the SMM algorithm to have an affinity of 500 nM or better was compiled (Fig. 1a). It was found that the percentage of the predicted binders varied widely, ranging from 0.07% for HLA-B*5101 to 10.40% for HLA-A*0206. It was also noted that the binding affinity of predicted peptides for any given percentile range also varied significantly from allele to allele. As shown in Figure 1b, the geometric mean of predicted affinity of the top 1% predicted binders varied from 14 nM for A*6801 to 1110 nM for HLA-B*5101.
Figure 1. The predicted binding repertoire is highly variable among HLA alleles.

(A) The repertoire size (cumulative percentage) of predicted binders among the 27 alleles considered in the study is shown. The peptides were considered to be binders if the binding affinity (IC50) predicted by SMM algorithm was ≤ 500 nM. (B) The geometric mean of binding affinity (IC50 predicted by SMM algorithm) of the top 1% peptides based on SMM IC50 is shown.
The range of repertoire sizes is verified by in vitro binding assays with purified HLA molecules
The algorithms utilized in the predictions for the different HLA molecules have been trained with varying numbers of measured peptide binding affinities, and have been shown to vary in performance (13, 21, 36, 37, 51). To exclude the possibility that the observed variability in predicted repertoires was an artifact of the variable accuracy of the algorithms utilized, we next undertook a series of experiments to empirically determine corresponding repertoires with in vitro binding assays utilizing purified MHC molecules.
For each HLA allele, the top 1% predicted binders were synthesized and tested for binding capacity as described in the Materials and Methods (Table 1). The complete dataset is available in the IEDB (http://www.iedb.org/subId/1000490). The number of peptides binding each allele with an affinity of 500 nM or better was tabulated (Table 1) and compared with the fraction of peptides predicted to bind with an affinity of 500 nM or better. It was found that, for vast majority of alleles, the number of predicted and actual binders were very similar. The ratio of the predicted to measured binders was between 0.66-1.5 for 85% of the alleles (Table 1), with a general trend that larger predicted repertoires map to larger measured repertoires. This data confirms that different alleles have very different absolute affinities associated with the same relative percentile, and that differences in predicted repertoires tend to reflect differences in repertoires as measured in actual MHC-peptide binding assays. Accordingly, for the analyses that follow, predicted peptide class I binding affinities and corresponding allele repertoires have been utilized as metrics of actual binding affinities and repertoires.
Selection of peptide sets to experimentally test the correlation between potential immunogenicity and i) binding affinity or ii) percentile rank
In the next series of analyses we sought to determine whether absolute binding affinity or relative rank of affinity (i.e., percentile score) is most predictive of potential immunogenicity. For this analysis we focused on A*0101, B*0702 and A*0201 as representative of alleles associated with small, medium and large binding repertoires, respectively (see Fig. 1).
For each allele considered independently we randomly selected 30 9-mer peptides for each of 4 contiguous categories of percentile ranks: (i) 0 – 0.30 (ii) 0.30 – 1.25 (iii) 1.25 – 5.0 (iv) 5.0 – 15.0. This resulted in a set of 360 9-mer peptides (30 peptides × 4 categories × 3 alleles). As shown in Table 2, in case of A*0101 only the 30 peptides that belonged to the 1st category of cumulative percentages were predicted to be binders (SMM IC50 ≤ 500 nM), but none of the peptides in any of the three remaining categories were predicted to be binders. As expected, B*0702 had more predicted binders, with all peptides in both the first and second categories predicted to bind at the 500 nM level. Finally, in the case of A*0201 all peptides in the first, second and third categories were predicted to bind at the 500 nM level. For all three alleles, all peptides in the fourth category, corresponding to the 5 to 15% percentile range, were predicted to be non-binders (SMM IC50 ≥ 500 nM). Thus, as assembled, these panels allow correlation of both predicted percentile scores and predicted IC50s with immunogenicity propensity.
Table 2. Selected peptides for immunogenicity study.
| HLA allele |
Selected percentile |
# of peptides |
Predicted IC50 [nM] |
|---|---|---|---|
|
| |||
| A*0101 | 0 – 0.30 | 30 | 122-477 |
| 0.30 – 1.25 | 30 | 527-1850 | |
| 1.25 – 5.0 | 30 | 1985-5931 | |
| 5.0 – 15.0 | 30 | 6522-20859 | |
|
| |||
| A*0201 | 0 – 0.30 | 30 | 7-22 |
| 0.30 – 1.25 | 30 | 23-78 | |
| 1.25 – 5.0 | 30 | 82-489 | |
| 5.0 – 15.0 | 30 | 516-3790 | |
|
| |||
| B*0702 | 0 – 0.30 | 30 | 3-86 |
| 0.30 – 1.25 | 30 | 89-448 | |
| 1.25 – 5.0 | 30 | 510-2861 | |
| 5.0 – 15.0 | 30 | 3238-15320 | |
Absolute, rather than relative, HLA binding affinity is a better correlate of immunogenicity
To define if peptide immunogenicity is more reflective of relative (percentile) or absolute (IC50) binding affinity, we immunized A*0101, A*0201 and B*0702 transgenic mice with the corresponding peptide sets. For each allele, peptides were administered as 12 pools of 10 individual peptides, corresponding to 3 pools for each percentile range), as shown in Table 2. Two weeks after immunization CD8+ T cells were isolated and screened for reactivity (IFNγ) against the immunized peptide pools as shown in Figure 2.
Figure 2. Immunogenicity of predicted binders varies between HLA alleles.

A) For each HLA allele 120 peptides were predicted to represent four percentile ranges: (i) 0 – 0.30, (ii) 0.30 – 1.25 (iii) 1.25 – 5.0 (iv) 5.0 – 15.0. For each percentile range 30 randomly selected peptides were pooled into 3 pools of 10 individual peptides [10μg/peptide]. Groups of 3 HLA transgenic mice between 8 and 12 weeks of age were immunized s.c with each pool diluted in 100 μl PBS emulsified in CFA. Two weeks post immunization the mice were sacrificed, and splenic CD8+ T cells were isolated and screened for IFNγ production. Data are expressed as mean number of SFC/106 CD8+ T cells from three independent experiments. Error bars represent SEM. Responses against peptides were considered positive if the stimulation index (SI) exceeded double the mean negative control wells (CD8+ T cells plus APCs without peptide) and net spots were above the threshold of 20 SFCs/106 CD8+ T cells experiments. Asterisks indicate peptides able to elicit a significant IFNγ response in two out of three individual experiments, according to the criteria described above. (B) Pools eliciting a significant IFNγ response were subsequently deconvoluted to identify individual epitopes. For each allele the number of identified epitopes per percentile is shown.
In case of the A*0201 transgenic mice, six of the 12 pools representing peptides from the first three percentile categories ((i) 0 – 0.30, (ii) 0.30 – 1.25 (iii) 1.25 – 5.0) were determined to be immunogenic on the basis of their capacity to elicit an IFNγ response (Fig. 2a, upper panel). Four pools of B*0702 predicted peptides derived from the first two categories ((i) 0 – 0.30 and (ii) 0.30 – 1.25) were determined to be immunogenic (Fig. 2b, middle panel). Finally, A*0101 transgenic mice recognized only one peptide pool from the first category (0 – 0.30). Interestingly, for all alleles the majority of the responses in terms of magnitude were detected in the first percentile category. No positive responses were detected for peptides from the fourth category (5.0 – 15.0) in any of the HLA transgenic mouse strains (Fig. 2a).
Deconvolution of all peptide pools eliciting an IFNγ response revealed 12 A*0201, 7 B*0702 and 1 A*0101 restricted epitopes (Fig. 2b). A list of all epitopes identified, as well as their predicted binding affinities, predicted percentile scores, and magnitude of response is shown in Table 3. Analysis of the data revealed that all of the B*0702 and A*0101 epitopes had percentile scores less than 0.4 and 0.2, respectively, while the percentile scores of the A*0201 epitopes ranged between 0.1 and 3, with 5 of the 12 A*0201 epitopes having scores greater than the first percentile. That is, essentially, a score of 0.4 was sufficient to identify all B*0702 epitopes and the single A*0101 epitope, while a score of 3 percent was necessary to select the entirety of A*0201 epitopes. Thus, given this approximately 7-fold difference in effective selection percentiles, no universally applicable percentile threshold associated with immunogenicity could be defined.
Table 3. Epitopes identified in this study.
| HLA allele |
Selected percentile |
# of epitopes |
Sequence | SMM Rank |
SMM IC50 [nM] |
Average T cell response [SFC/106 CD8+] |
|---|---|---|---|---|---|---|
|
| ||||||
| A*0201 | 0 – 0.30 | 6/30 | KLAEAIFKL | 0.1 | 9 | 469 |
| TLLCLIPTV | 0.2 | 11 | 377 | |||
| TIMAVLFVV | 0.2 | 12 | 245 | |||
| VLNPYMPTV | 0.2 | 12 | 240 | |||
| LVISGLFPV | 0.3 | 17 | 185 | |||
| GLYPLAIPV | 0.3 | 17 | 122 | |||
|
| ||||||
| 0.3 – 1.25 | 4/30 | ILAKAIFKL | 0.5 | 31 | 61 | |
| IMAVGIVSI | 1.1 | 68 | 108 | |||
| VLLLVTHYA | 1.2 | 43 | 99 | |||
| ALCEVLTLA | 1.2 | 69 | 92 | |||
|
| ||||||
| 1.25 – 5.0 | 2/30 | SLLKNDVPL | 2.2 | 159 | 172 | |
| SGMLWMAEV | 3.0 | 274 | 83 | |||
|
| ||||||
| B*0702 | 0 – 0.30 | 6/30 | RPAKSGTVM | 0.1 | 3 | 103 |
| RPTPRGAVM | 0.1 | 7 | 74 | |||
| RPMPGTRKV | 0.1 | 29 | 300 | |||
| LPSIVREAL | 0.1 | 19 | 456 | |||
| RPRWLDARV | 0.2 | 49 | 659 | |||
| TPRSPSVEV | 0.3 | 66 | 681 | |||
|
| ||||||
| 0.30 – 1.25 | 1/30 | IPKIYGGPI | 0.4 | 91 | 119 | |
|
| ||||||
| A*0101 | 0 – 0.30 | 1/30 | VIDLEPIPY | 0.2 | 382 | 307 |
At the same time, it was noted that all 7 epitopes identified in the HLA B*0702 mice had predicted IC50s of about 100 nM, or better, while a threshold of 274 nM was needed to identify all of the A*0201 epitopes, and 382 nM the single A*0101 epitope. At the same time, however, in all cases, irrespective of the allele, the epitopes identified had predicted binding affinities of 500 nM, or better, suggesting that this previously defined threshold may be universally applicable.
Taken together, this data suggest that alleles associated with higher number of predicted binders, and corresponding higher predicted binding affinities (Figure 1a and b) are associated with higher numbers of epitopes and higher cumulative magnitude of responses. Furthermore, immunogenicity more closely corresponds to predicted affinity, rather than percentile scores.
Allele specific HLA repertoire and immunogenicity thresholds
The data presented above suggest that each HLA allele is associated with a different number/proportion of peptides that bind below the 500 nM affinity threshold and are immunogenic. We expected this finding to be reflected in the predicted binding affinity of HLA class I restricted epitopes described in the literature.
To address this issue we retrieved all epitopes with defined HLA restrictions from the IEDB (47). Next, we predicted the corresponding binding affinity of all epitopes restricted by alleles for which at least 15 data points (reported unique epitopes) were available. Figure 3 shows a cumulative plot of the predicted binding affinities of the corresponding epitopes for each allele, and the results are also summarized in Table 4. An important caveat to consider for the foregoing analysis is that HLA binding predictions using the 500 nM affinity threshold have been widely used in the studies reported in the literature, and thus some bias may be present in the data set.
Figure 3. IC50 threshold of epitopes derived from the IEDB.

The cumulative epitope distribution is compared with the binding affinity (SMM IC50 nM) for alleles for which at least 15 data points were available from the IEDB. While the affinity distribution for different alleles varied significantly, 500 nM was found to be a useful binding threshold. Overall, the median % fraction of epitopes identified by this threshold is 79%.
Table 4. IC50 threshold of epitopes derived from the IEDB.
| Alleles | No. of data points |
% of epitopes at 500 nM |
nM at 75% | nM at 90% |
|---|---|---|---|---|
|
| ||||
| A*0101 | 33 | 58 | 606 | 3,328 |
| A*0201 | 833 | 80 | 331 | 1,635 |
| A*0202 | 23 | 100 | 19 | 65 |
| A*0203 | 23 | 96 | 35 | 154 |
| A*0206 | 26 | 100 | 77 | 129 |
| A*0301 | 40 | 63 | 646 | 2,341 |
| A*1101 | 68 | 81 | 372 | 1,213 |
| A*2402 | 98 | 53 | 1,573 | 3,687 |
| A*2902 | 18 | 89 | 91 | 112 |
| A*6802 | 16 | 56 | 952 | 2,767 |
| B*0702 | 73 | 79 | 285 | 1,324 |
| B*0801 | 18 | 56 | 1,235 | 1,970 |
| B*1501 | 15 | 93 | 199 | 274 |
| B*2705 | 39 | 77 | 439 | 1,953 |
| B*3501 | 45 | 76 | 383 | 2,861 |
|
| ||||
| Median | 33 | 79 | 372 | 1,635 |
Nevertheless, is apparent that the affinity distribution of the reported epitopes for the different alleles varies significantly. The median affinity threshold necessary to identify 75% of the epitopes is 372, ranging from 19 nM for A*0201 to 1235 for B*0801. The median affinity threshold necessary to identify 90% of the epitopes is 1635 nM, ranging from 65 nM to 3687 for B*0801. Despite these large variations, 500 nM is still a useful binding threshold. Overall, the median % fraction of epitopes identified by this threshold is 79%.
Relation between HLA repertoire size and associated epitope binding affinity
The data presented above suggest that certain alleles, such as HLA A*0101, are relatively inefficient in terms of antigen presentation, binding fewer peptides with an overall lower affinity. As a result, these alleles are associated with a correspondingly smaller T cell epitope repertoire, and those epitopes are associated with lower binding affinity. Conversely, other alleles, such as A*0201, bind a larger repertoire of peptides, are associated with a higher number of epitopes, and those epitopes are associated with higher binding affinity on average.
To test the validity of this theory, we compared the percent of peptides predicted to bind in the set of DENV peptides (See Table 1), with the IC50 threshold associated with ≥75% of the epitopes retrieved from IEDB. This analysis revealed that epitopes restricted by alleles with larger repertoires were, in general, also associated with higher predicted binding affinity (IC50 <500 nM) (Pearson correlation coefficient, r = −0.77) (Fig. 4). Conversely, alleles with more limited predicted repertoires the 75% threshold for their corresponding epitopes were associated with lower affinities (IC50 >500 nM). Notably, the correlation in Figure 4 can be utilized to derive an allele specific affinity cutoff for any allele of interest, based on the breadth of their binding repertoires. Supplemental Table 1 shows revised thresholds selected according to this method for 38 most common HLA A and B alleles, representative of the nine major supertypes (52).
Figure 4.
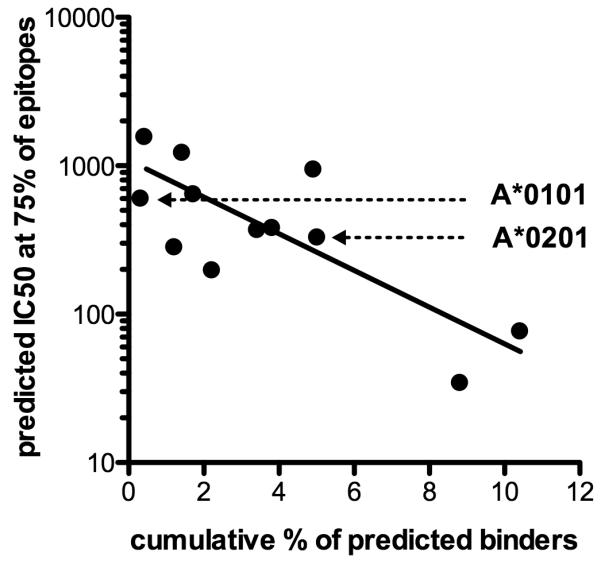
Correlation between cumulative % of predicted binders and binding affinity (IC50) of the epitopes retrieved from IEDB at IC50 500nM among different alleles. Alleles with higher number of predicted binders had epitopes with stronger binding affinity.
HLA binding prediction strategies based on allele-specific thresholds
Based on the data presented above we next sought to compare the practical implications of our findings in terms of epitope prediction strategies. First we calculated the number of peptides required by a prediction strategy utilizing a generalized 500 nM threshold, which is associated with prediction of 79% of the epitopes (Table 4). For this analysis we utilized the same allele list and set of DENV sequences form Table 1. We found that on average 1184 peptides/allele would be required (Fig 5).
Figure 5.
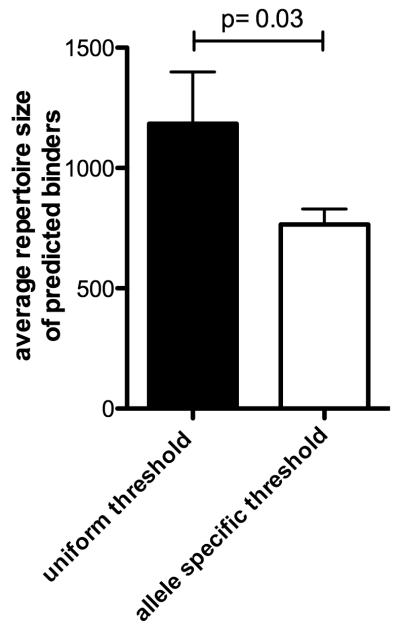
Average number of peptides/allele required by different prediction strategies using a uniform threshold of IC50 of 500 nM (black bar) or an allele specific IC50 threshold (white bar). The same set of alleles and DENV sequences from Figure 1 was utilized. Error bars represent SEM.
Next we calculated the number of peptides required by allele specific thresholds, as Tabulated in the Supplemental Table 1. In this case an average of 765 peptides/allele would be required (Fig.5). This is significantly less (p=0.03) than the 500n nM general threshold. In addition it would be expected that this approach, would be more accurate, as it would by definition accommodate effective prediction of alleles with larger repertoire sizes.
Genetic and structural basis of allele associated differences in repertoire and epitope affinity
These observations raised the question of whether a genetic or structural/molecular basis for these differences can be discerned. First, when the ranking of HLA A versus HLA B alleles in Figure 1a was compared, it became apparent that HLA A alleles are in general associated with broader repertoires (Student’s T-test, P = 0.01). Figure 6 illustrates the comparison of repertoire size for HLA A versus B alleles. To investigate the possible molecular basis of this phenomenon, within each subset of HLA A and B alleles we examined whether any significant correlation existed between certain structural features of associated motifs, high/low repertoire size, and epitope affinity. For this purpose, we sorted the alleles from Figure 1, according to their main supertypic specificities, as cataloged by Sidney et al. (52) (Table 5). It was found that repertoire size was only loosely correlated with supertype specificities. That is, in some cases alleles within a supertype were consistently associated with large or small repertoires. For example, the broadest repertoire was associated with the A2 supertype (A*0201, A*0203, A*0206 and A*6802), where an average repertoire of 7.26% was predicted, and little difference was noted between the individual alleles (± 2.77%), while the narrowest repertoires were associated with the A24 (A*2301 and *2401; 0.7% ± 0.36) and B44 supertypes (B*4001, *4402 and *4403; 0.83% ± 0.65). In other cases dramatic differences were detected between alleles in the same supertype. This is particularly exemplified by the A1 supertype, where A*0101 was associated with one of the narrowest repertoires (0.29%) and A*3201 was associated with one of the largest repertoires (7.46%), of the alleles analyzed herein. Similarly, in the case of the B7 supertype, repertoire sizes ranged widely, with a low of 0.07% for B*5101 to a high of 3.79% for B*3501.
Figure 6.
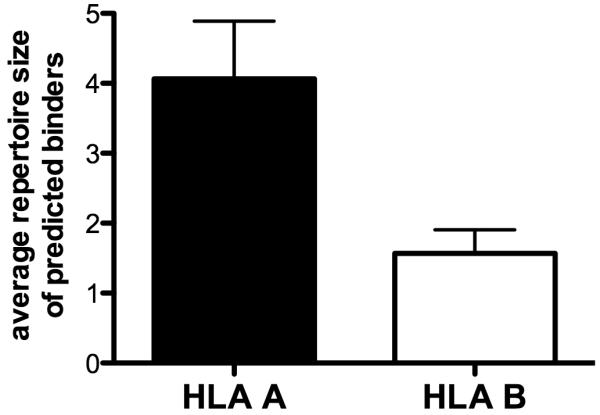
Average repertoire size of predicted binders restricted by HLA A and B alleles. HLA A alleles in general were found to be associated with broader predicted binder repertoires. Error bars represent SEM.
Table 5. Repertoire sizes of the HLA allele supertypes covering the 27 alleles included in the study.
| Supertype | Alleles | Repertoire size (%) |
Average repertoire size per supertype |
SD |
|---|---|---|---|---|
|
| ||||
| A01 | A*0101 | 0.29% | 2.42% | 3.39% |
| A*2601 | 0.60% | |||
| A*3002 | 1.31% | |||
| A*3201 | 7.46% | |||
|
| ||||
| A02 | A*0201 | 4.96% | 7.26% | 2.77% |
| A*0203 | 8.77% | |||
| A*0206 | 10.40% | |||
| A*6802 | 4.90% | |||
|
| ||||
| A03 | A*0301 | 1.74% | 4.15% | 2.50% |
| A*1101 | 3.44% | |||
| A*3001 | 8.13% | |||
| A*3101 | 4.00% | |||
| A*3301 | 1.71% | |||
| A*6801 | 5.92% | |||
|
| ||||
| A24 | A*2301 | 0.96% | 0.70% | 0.36% |
| A*2402 | 0.45% | |||
|
| ||||
| B07 | B*0702 | 1.24% | 1.81% | 1.57% |
| B*3501 | 3.79% | |||
| B*5101 | 0.07% | |||
| B*5301 | 2.16% | |||
|
| ||||
| B08 | B*0801 | 1.37% | 1.37% | |
|
| ||||
| B44 | B*4001 | 1.51% | 0.83% | 0.65% |
| B*4402 | 0.21% | |||
| B*4403 | 0.76% | |||
|
| ||||
| B58 | B*5701 | 1.08% | 1.97% | 1.26% |
| B*5801 | 2.86% | |||
|
| ||||
| B62 | B*1501 | 2.22% | 2.22% | |
It has been suggested that HLA B molecules in particular have evolved to maximize their recognition of viral sequences (53, 54). To test whether this concerted evolution might be related to the phenomena we observed, we examined whether the repertoire size differences we detected in the case of the DENV sequence set was still detected in a set of self-sequences of similar size. To this end, we randomly selected 38,845 9-mer sequences from the human genome. When binding predictions were done similarly to what described above in Figure 1, similar differences in repertoire size were detected (data not shown). These data underline the generality of this observation and thus argue against the suggestion that B evolved to maximize viral recognition.
Discussion
Herein, we show that different HLA class I alleles are associated with different peptide binding repertoire sizes, defined as the fraction of all possible peptides that are bound at a given affinity threshold. These repertoire differences translate into a correspondingly different number of epitopes being recognized, and with correspondingly different median affinities. This observation has implications both at the basic level, in terms of generation of epitope repertoires, and at the practical level, in terms of guiding optimal epitope predictions by bioinformatics means. The present study provides means to efficiently select peptides from pathogens, allergens or other antigens of immunological relevance, which may in turn facilitate studies probing the correlates of immunity and antigen recognition. It is also possible that these results may facilitate the design of peptide-based subunit vaccines and immunotherapeutics. However, in this context, it is important to recognize that the utility and effectiveness of such subunit vaccines is still an issue in need of further study and validation. And, indeed, the use of whole proteins for such constructs implicitly diminishes the issue of HLA allelic variation in the population.
One immediate question arising from these observations pertains to the structural and molecular basis of the phenomenon. Our analysis demonstrates that the breadth of repertoire and associated epitope affinities is likely influenced by the structure of the HLA molecule, as differences are apparent between the HLA A and B loci. It is generally assumed that pathogen escape is a major driver of evolution of HLA polymorphism. In this regard, it is reasonable to hypothesize that repertoire size and average epitope affinity should be considered as broad variables potentially influencing HLA evolution at the immunogenetic and immunochemical level.
It is currently unknown to what degree these observations might be generalizable. It is noteworthy that similar variation in repertoire size was recently noted in the case of the class I molecules expressed by the rhesus macaque (Macaca mulatta (55)). Therefore, it is reasonable to speculate that similar allele-related variation in repertoire size will be broadly observed. Future studies will address this point by combined immunological and bioinformatics analysis.
A theoretical point of potential interest is whether the presence within a species or a population of different MHC allelic variants associated with different repertoire sizes might confer some advantage and be optimal in terms of overall biological function of the MHC system. A wide repertoire will render pathogen escape by loss of MHC binding less likely, but on the other hand might dilute the focus of the immune response, be more likely to trigger autoimmunity, or even paradoxically result in a too narrow T cell repertoire, because of overzealous negative selection.
It is worth noting that in evolutionary terms, HLA B polymorphism is more recent, more diverse and more rapidly evolving (53, 54). Thus, in a broad sense, HLA A seems, on average, to be associated with a broader repertoire approach than HLA B, which seems to be enacting a strategy based on somewhat narrower but more flexible and diverse repertoires and binding motifs. Indeed, most examples of HLA class I molecules associated with disease resistance to viral infections, such as HIV or DENV, are HLA B.
Last but not least, our data has significant implications in terms of the practical use of bioinformatics predictions of HLA binding and T cell epitopes. First, our data indicates that if a single criterion for alleles has to be chosen, it is preferable to use absolute binding affinity, rather than a relative percentile. Second it indicates that 500 nM is a reasonably good “universal” threshold. Thirdly our data indicates that more effective allele-specific thresholds can be derived either experimentally or by linear regression equations as the one shown in Figure 4 and Supplemental Table 1. In this our data indicates that the number of peptides necessary to predict a similar fraction of epitopes will vary as a function of the particular allele considered and thus the optimal prediction strategy can be adapted to the specific experimental goal.
In conclusion, our data highlights how different HLA loci and alleles are associated with different repertoire sizes, and sheds new light on the mechanisms governing allelic polymorphism of MHC molecules. We predict that this enhanced understanding will further increase the accuracy and ease of prediction of MHC restricted T cell epitopes.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health (NIH) contracts HHSN272200900042C and HHSN272201200010C (to A.S.)
References
- 1.Murphy K. Janeway’s Immunobiology. 8th Ed. Garland Science; New York: 2011. [Google Scholar]
- 2.Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 3.Yewdell JW. Confronting Complexity: Real-World Immunodominance in Antiviral CD8+ T Cell Responses. Immunity. 2006;25:533–543. doi: 10.1016/j.immuni.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Assarsson E, Sidney J, Oseroff C, Pasquetto V, Bui H, Frahm N, Brander C, Peters B, Grey H, Sette A. A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. The Journal of Immunology. 2007;178:7890–7901. doi: 10.4049/jimmunol.178.12.7890. [DOI] [PubMed] [Google Scholar]
- 5.Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, Melief C, Oseroff C, Yuan L, Ruppert J. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. The Journal of Immunology. 1994;153:5586–5592. [PubMed] [Google Scholar]
- 6.Dick LR, Aldrich C, Jameson SC, Moomaw CR, Pramanik BC, Doyle CK, DeMartino GN, Bevan MJ, Forman JM, Slaughter CA. Proteolytic processing of ovalbumin and beta-galactosidase by the proteasome to yield antigenic peptides. The Journal of Immunology. 1994;152:3884–3894. [PMC free article] [PubMed] [Google Scholar]
- 7.Daly K, Nguyen P, Woodland DL, Blackman MA. Immunodominance of major histocompatibility complex class I-restricted influenza virus epitopes can be influenced by the T-cell receptor repertoire. J. Virol. 1995;69:7416–7422. doi: 10.1128/jvi.69.12.7416-7422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heemels M, Ploegh H. Generation, translocation, and presentation of MHC class I-restricted peptides. Annu. Rev. Biochem. 1995;64:463–491. doi: 10.1146/annurev.bi.64.070195.002335. [DOI] [PubMed] [Google Scholar]
- 9.Niedermann G, King G, Butz S, Birsner U, Grimm R, Shabanowitz J, Hunt DF, Eichmann K. The proteolytic fragments generated by vertebrate proteasomes: structural relationships to major histocompatibility complex class I binding peptides. Proceedings of the National Academy of Sciences. 1996;93:8572–8577. doi: 10.1073/pnas.93.16.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eggers M, Boes-Fabian B, Ruppert T, Kloetzel P, Koszinowski UH. The cleavage preference of the proteasome governs the yield of antigenic peptides. J. Exp. Med. 1995;182:1865–1870. doi: 10.1084/jem.182.6.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Groot AS, Bosma A, Chinai N, Frost J, Jesdale BM, Gonzalez MA, Martin W, Saint-Aubin C. From genome to vaccine: in silico predictions, ex vivo verification. Vaccine. 2001;19:4385–4395. doi: 10.1016/s0264-410x(01)00145-1. [DOI] [PubMed] [Google Scholar]
- 12.Sylvester-Hvid C, Nielsen M, Lamberth K, Røder G, Justesen S, Lundegaard C, Worning P, Thomadsen H, Lund O, Brunak S. SARS CTL Vaccine Candidates - HLA Supertype, Genome-Wide Scanning and Biochemical Validation. Scand. J. Immunol. 2004;59:632–632. doi: 10.1111/j.0001-2815.2004.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui H, Grey H, Sette A. A consensus epitope prediction approach identifies the breadth of murine TCD8 -cell responses to vaccinia virus. Nat. Biotechnol. 2006;24:817–819. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 14.Lin H, Zhang G, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics. 2008;9:S22. doi: 10.1186/1471-2105-9-S12-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moise L, McMurry JA, Buus S, Frey S, Martin WD, De Groot AS. In silico-accelerated identification of conserved and immunogenic variola/vaccinia T-cell epitopes. Vaccine. 2009;27:6471–6479. doi: 10.1016/j.vaccine.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen MV, Lelic A, Parsons R, Nielsen M, Hoof I, Lamberth K, Loeb MB, Buus S, Bramson J, Lund O. Identification of CD8 T cell epitopes in the West Nile virus polyprotein by reverse-immunology using NetCTL. PloS One. 2010;5:e12697. doi: 10.1371/journal.pone.0012697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundegaard C, Hoof I, Lund O, Nielsen M. State of the art and challenges in sequence based T-cell epitope prediction. Immunome Research. 2010;6:S3. doi: 10.1186/1745-7580-6-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sette A, Rappuoli R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity. 2010;33:530–541. doi: 10.1016/j.immuni.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lund O, Nascimento EJ, Maciel M, Jr, Nielsen M, Larsen MV, Lundegaard C, Harndahl M, Lamberth K, Buus S, Salmon J. Human leukocyte antigen (HLA) class I restricted epitope discovery in yellow fewer and dengue viruses: importance of HLA binding strength. PloS One. 2011;6:e26494. doi: 10.1371/journal.pone.0026494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salimi N, Fleri W, Peters B, Sette A. Design and utilization of epitope-based databases and predictive tools. Immunogenetics. 2010;62:185–196. doi: 10.1007/s00251-010-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40:W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. The Journal of Immunology. 1994;152:163–175. [PubMed] [Google Scholar]
- 23.Rammensee HG, Bachmann J, Emmerich NPN, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 24.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh H, Raghava G. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 26.Singh H, Raghava G. ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics. 2003;19:1009–1014. doi: 10.1093/bioinformatics/btg108. [DOI] [PubMed] [Google Scholar]
- 27.Saha S, Raghava G. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Structure, Function, and Bioinformatics. 2006;65:40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- 28.Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic Acids Res. 2005;33:W172–W179. doi: 10.1093/nar/gki452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reche PA, Reinherz EL. Immunoinformatics. Springer; 2007. Prediction of peptide-MHC binding using profiles; pp. 185–200. [DOI] [PubMed] [Google Scholar]
- 30.Buus S, Lauemøller S, Worning P, Kesmir C, Frimurer T, Corbet S, Fomsgaard A, Hilden J, Holm A, Brunak S. Sensitive quantitative predictions of peptide-MHC binding by a ‘Query by Committee’ artificial neural network approach. Tissue Antigens. 2003;62:378–384. doi: 10.1034/j.1399-0039.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 31.Bui HH, Sidney J, Peters B, Sathiamurthy M, Sinichi A, Purton KA, Mothé BR, Chisari FV, Watkins DI, Sette A. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics. 2005;57:304–314. doi: 10.1007/s00251-005-0798-y. [DOI] [PubMed] [Google Scholar]
- 32.Peters B, Sidney J, Bourne P, Bui H, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biology. 2005;3:e91. doi: 10.1371/journal.pbio.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394. doi: 10.1186/1471-2105-10-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Research. 2008;4:2. doi: 10.1186/1745-7580-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trost B, Bickis M, Kusalik A. Strength in numbers: achieving greater accuracy in MHC-I binding prediction by combining the results from multiple prediction tools. Immunome Research. 2007;3:5. doi: 10.1186/1745-7580-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64:177–186. doi: 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- 38.Peters B, Bui H, Frankild S, Nielsen M, Lundegaard C, Kostem E, Basch D, Lamberth K, Harndahl M, Fleri W. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Computational Biology. 2006;2:e65. doi: 10.1371/journal.pcbi.0020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotturi MF, Peters B, Buendia-Laysa F, Sidney J, Oseroff C, Botten J, Grey H, Buchmeier MJ, Sette A. The CD8 T-cell response to lymphocytic choriomeningitis virus involves the L antigen: uncovering new tricks for an old virus. J. Virol. 2007;81:4928–4940. doi: 10.1128/JVI.02632-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiskopf D, Angelo MA, de Azeredo EL, Sidney J, Greenbaum JA, Fernando AN, Broadwater A, Kolla RV, De Silva AD, de Silva AM. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8 T cells. Proceedings of the National Academy of Sciences. 2013;110:E2046–E2053. doi: 10.1073/pnas.1305227110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pasquetto V, Bui H, Giannino R, Mirza F, Sidney J, Oseroff C, Tscharke DC, Irvine K, Bennink JR, Peters B. HLA-A* 0201, HLA-A* 1101, and HLA-B* 0702 transgenic mice recognize numerous poxvirus determinants from a wide variety of viral gene products. The Journal of Immunology. 2005;175:5504–5515. doi: 10.4049/jimmunol.175.8.5504. [DOI] [PubMed] [Google Scholar]
- 42.Oseroff C, Peters B, Pasquetto V, Moutaftsi M, Sidney J, Panchanathan V, Tscharke DC, Maillere B, Grey H, Sette A. Dissociation between epitope hierarchy and immunoprevalence in CD8 responses to vaccinia virus western reserve. The Journal of Immunology. 2008;180:7193–7202. doi: 10.4049/jimmunol.180.11.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiskopf D, Yauch LE, Angelo MA, John DV, Greenbaum JA, Sidney J, Kolla RV, De Silva AD, de Silva AM, Grey H. Insights into HLA-restricted T cell responses in a novel mouse model of dengue virus infection point toward new implications for vaccine design. The Journal of Immunology. 2011;187:4268–4279. doi: 10.4049/jimmunol.1101970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sette A, Sidney J. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics. 1999;50:201–212. doi: 10.1007/s002510050594. [DOI] [PubMed] [Google Scholar]
- 45.Middleton D, Menchaca L, Rood H, Komerofsky R. New allele frequency database: www.allelefrequencies.net. Tissue Antigens. 2003;61:403. doi: 10.1034/j.1399-0039.2003.00062.x. www.allelefrequencies.net [DOI] [PubMed] [Google Scholar]
- 46.Meyer D, Singe R, Mack S, Lancaster A, Nelson M, Erlich H, Fernandez-Vina M, Thomson G. Single locus polymorphism of classical HLA genes. 2007;1:653–704. [Google Scholar]
- 47.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B. The immune epitope database 2.0. Nucleic Acids Res. 2010;38:D854. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidney J, Southwood S, Moore C, Oseroff C, Pinilla C, Grey HM, Sette A. Measurement of MHC/Peptide Interactions by Gel Filtration or Monoclonal Antibody Capture. Current Protocols in Immunology. 2013;100:18.3.1–18.3.36. doi: 10.1002/0471142735.im1803s100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 50.Gulukota K, Sidney J, Sette A, DeLisi C. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J. Mol. Biol. 1997;267:1258–1267. doi: 10.1006/jmbi.1997.0937. [DOI] [PubMed] [Google Scholar]
- 51.Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1471–2172. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, Rathnavalu P, Moore C, Pfafferott KJ, Hilton L. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 54.Goulder PJ, Walker BD. HIV and HLA class I: an evolving relationship. Immunity. 2012;37:426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mothé BR, Southwood S, Sidney J, English AM, Wriston A, Hoof I, Shabanowitz J, Hunt DF, Sette A. Peptide-binding motifs associated with MHC molecules common in Chinese rhesus macaques are analogous to those of human HLA supertypes and include HLA-B27-like alleles. Immunogenetics. 2013:1–16. doi: 10.1007/s00251-013-0686-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.