

Abstract
We present a method for creating a network of gene co-occurrences from the literature and partitioning it into communities of related genes. The way in which our method identifies communities makes it likely that the component genes of each community will be related by their function. The method processes a large database of article abstracts, synthesizing information from many sources to shed light on groups of genes that have been shown to interact. It is a tool to be used by researchers in the biomedical sciences to swiftly search for known interactions and to provide insight into unexplored connections. The partitioning procedure is designed to be particularly applicable to large networks in which individual nodes may play a role in more than one community. In this paper, we explain the details of the method, in particular the partitioning process. We also apply the method to produce communities of genes related to colon cancer and show that the results are useful.
The automated analysis of biomedical text is useful in any form, because knowledge in the biomedical sciences is predominantly disseminated in the form of journal articles. However, when applied to the subject of human gene function, automated text analysis is critically important. There are ≈15,000 currently known human genes and >1 million related articles in the Medline databaseb alone. Moreover, genes act in a complex interrelated way, so information from many experiments is necessary to explain the function of a typical gene. A comprehensive study of even a simple cellular process involving several genes might require a researcher to be familiar with hundreds of articles. Merely locating all relevant articles in a database by using a simple search utility would be time consuming, not to mention inefficient and difficult, because of shortcomings of the human gene nomenclature system. In contrast, our method indexes gene symbol occurrences in all articles of large database such as Medline in <1 dayc and then can produce a list of communities of functionally related genes in another half day.d
In this article, we present a method to find communities of related genes. The method creates a network of gene symbol co-occurrences from Medline article abstracts and partitions this network into communities. The genes in each community are likely to be functionally related because of the way in which the communities are identified, and because most recent research on genes and proteins has been devoted to their function. This method can thus be a valuable tool that both summarizes available information and indicates possible directions of research. The format of the results is designed to make them easy to use. The results can easily include a list of the Medline PubMed identification numbers (PMID) for articles containing each gene and pair of genes to facilitate research. Varying the user-selected key words (see Method Overview) allows the method to be applied repeatedly and focused on particular topics of interest.
We apply our method to the Medline database to identify communities of genes related to colon cancer. We show that genes placed together in a community that are not explicitly connected in any Medline article or in the Online Mendelian Inheritance in Man (OMIM)e listing for either gene can nevertheless be related by their function. The communities thereby imply connections among genes that may otherwise be overlooked or that would require much time and effort to be found manually. We also show that our method separates genes that co-occur but are not functionally related into different communities. Finally, we demonstrate cases in which a node common to two communities indicates a link between two groups of related genes.
It is important to note that the gene communities in the results are not meant to perfectly reproduce biological reality. The communities are simply interesting artifacts within the network that provide a powerful method for organizing and presenting information from the literature.
Method Overview
Gene symbol mentions are first extracted from almost allf 12.5 million Medline article titles and abstracts. We then select sets of genes found to be statistically correlated to a set of user-selected (related) key words. These two steps are performed following the procedure of ref. 1. This procedure includes steps to account for alias symbols and to distinguish gene symbol abbreviations from identical abbreviations referring to other conceptsg (2). Selecting genes correlated to certain key words ensures continuity of biological function of the genes considered and reduces the number of genes considered so the results can be readable and useful.
Networks are then created from these sets of genes. In the networks, each node represents a gene, and an edge connects two genes if they co-occur in at least one article. The degree distribution of the networks follows a power law, as we show, so their clustering structure is scale-free and there is no typical community size. Therefore, to find communities, we partition the graph using a nonlocal process exploiting the concept of betweenness centrality (3).
The partitioning process may be applied to any network, but it is particularly applicable to networks of several hundred to 1,000 nodes in which nodes may play a role in more than one community. It is based on the process of Girvan and Newman (GN) (ref. 4; for a faster algorithm for finding communities, see ref. 5), which was shown to give very good results for a variety of small graphs. The general idea of our process is the same as that of GN, but the details are significantly different. Our modifications allow nodes to be placed in several communities if the structure of the network indicates that the nodes belong there, and they provide a quantitative estimate of how strongly each node belongs to each community. This is important when single nodes play a role in several communities or when the source information is incomplete or flawed. It can also indicate a link between two communities that have one or more nodes in common, and it “smooths” the process of partitioning, which for any large network is somewhat arbitrary. The modifications also allow communities to be identified as discrete units. Identifying discrete communities is particularly useful when community sizes are not known in advance and makes the results easier to use if the network is large (6).
Motivation and Previous Work
For the most part, biologists now understand the rules by which the system of genes, proteins, RNAs, and other cellular constituents operates; what remains is to determine the exact details of this system. A worldwide effort is underway in the biomedical community to identify and understand the cellular interactions at the root of human health. Given the enormous number of human genes and the complex interrelated nature of gene and protein interaction, this task is more than a little daunting, and accomplishing it will involve an unprecedented level of collaboration and information exchange. However, the current condition of knowledge organization in the field makes extensive collaboration and complete information exchange difficult.
As mentioned above, information pertinent to human gene function exists largely in the form of an astoundingly large number of journal articles. Medline yields 1.5 million hits when queried for “gene” or “protein” with “human,” ≈150,000 of which were published in 2002 alone. Our results, taking into account co-occurrences within the set of 682 genes we identified as correlated to colon cancer, were created from the 7,985 article abstracts from an astonishing 904h different journals. Given these numbers, it is easy to see that an expert, although familiar with many hundreds of articles, could nonetheless be unaware of developments related to his or her area of interest. And, whereas online biomedical databases provide easy access to abstracts, a manual literature survey would encounter difficulties beyond the large number of results, due to the nomenclature system for human genes. Both the existence of multiple alias symbols for many genes and the frequent occurrence of unrelated abbreviations equivalent to gene symbols interfere with any simple search utility.
Despite the impracticality of an exhaustive manual search, online databases of journal abstracts present a gold mine of available information. In fact, the ability to sift through millions of abstracts, extract pertinent information, and present it in a useful format is arguably essential to the understanding of human gene function. Accordingly, automated text analysis has been an area of focus in the field of bioinformatics.
One approach has been to extract detailed information by using natural language-processing techniques (7-17). Our method follows a different line of attack: only simple information, such as gene and protein names, is extracted from each article, and more detailed conclusions are then inferred from this information. Gene and protein term identification in particular has been simplified by the recent appearance of online libraries of gene and protein symbols (refs. 18-20 show this can otherwise be a major task). However, data obtained by simple term matching will be highly error-prone due to false positive identifications of human gene symbols, unless carefully treated.
A reasonable conclusion that can be drawn from gene occurrence data is that genes mentioned in the same article are related in some way. This has been shown to be true both on large (21) and small (22, 23) scales. It is also possible to connect genes to key words found in articles and thus to biological processes, as in refs. 1 and 24. These results have been applied in conjunction with natural language-processing techniques to find related groups of genes, from among a restricted set of genes mentioned in a restricted set of articles, in refs. 25 and 26. Our method, while similar, has a very different way of finding communities that requires neither the preprocessing step of selecting genes or articles nor natural language processing.
Obtaining Co-occurrence Data
As stated above, the first step of the method is to identify literature co-occurrences of genes relevant to a disease by using the procedure of ref. 1. This section is simply a brief summary of this procedure; for more detail, please see the referenced article.
Using a list of all official and alias symbols for human genes compiled from the Human Genome Organisation (HUGO) (www.gene.ucl.ac.uk/nomenclature), OMIM, and Locuslink (www.ncbi.nlm.nih.gov/LocusLink) web sites, we automatically extracted the gene name symbols and disease mentions from all Medline article titles and abstracts. Where possible, we replaced alias symbols with official ones. We also extracted key words related to a certain disease and used them to determine which genes were statistically correlated with this disease.
To test a gene for statistical relevance to a disease, we simply compared the observed number of gene-disease co-occurrences to the number we would expect given no correlation. Because the distribution of co-occurrences of two uncorrelated terms follows a binomial distribution, a value of observed gene-disease co-occurrences more than one SD greater than the binomial expected value indicates correlation. This statistical method is preferable to the “term frequency, inverse document frequency” metric, because it accurately handles infrequently mentioned genes, which are very common.
The final step in obtaining data was to remove false positives, which occur frequently because gene symbols generally coincide with other abbreviations having nothing to do with genes. For example, the symbol HDC, representing the gene histidine decarboxylase, was commonly used in the literature as an abbreviation for high dose chemotherapy. We disambiguated the data, using a method shown in ref. 2, which yielded unambiguous symbol identifications with a low error rate.
Gene Graph
The creation of gene graphs from the co-occurrence data was performed following a well known procedure (21, 23). Each vertex in the graph represents a gene, and an edge exists between two vertices if the genes they represent co-occur at least once. We did not use weighted edges. In creating the graph, we neglected articles published before 1990 and articles that listed more than five genes, as mentioned in the Introduction.
The resulting graph has a power law distribution in its degree. That is, the number of vertices of degree x is given by Ax-β, where β < 0. This is shown in Fig. 1, where we plot the data on a log-log scale for gene graphs corresponding to several diseases.
Fig. 1.

The number of vertices (y axis) is plotted against the degree of the vertex (x axis) for several diseases on a log-log scale. We followed the usual binning procedure in plotting the data. The deviation from the power law for low vertex degree is typical. AML, acute myelogenous leukemia.
The properties of such power law graphs have been extensively studied (27-29). It has been shown that random graphs with 2<β<3.5 consist of one giant connected component and other small components of size O(ln(N)) (28). Here N is the size of the graph, and β is the power law exponent. The component structure of the gene graphs agrees with the predictions of ref. 28 for random graphs, as shown in Table 1.
Table 1. Sizes of connected components in several gene graphs.
| Components
|
||
|---|---|---|
| Disease (no. of statistically relevant genes) | Size | No. |
| Acute myelogenous leukemia (488) | 460 | 1 |
| 4 | 1 | |
| 3 | 4 | |
| 2 | 6 | |
| Breast cancer (816) | 686 | 1 |
| 6 | 2 | |
| 5 | 1 | |
| 4 | 5 | |
| 3 | 9 | |
| 3 | 33 | |
| Colon cancer (682) | 561 | 1 |
| 4 | 4 | |
| 3 | 15 | |
| 2 | 30 | |
Because the smaller components contain few genes with few neighbors, they are of limited interest. They usually consist of little-known genes that have not been related to other genes. In what follows, we focus exclusively on identifying communities within the giant component.
Partitioning the Graph into Communities
There is no formal definition for a community of vertices within a graph. A graph can be said to have community structure if it consists of subsets of genes, with many edges connecting vertices of the same subset but few edges lying between subsets (4). Finding communities within a graph is an efficient way to identify groups of related vertices.
As mentioned in the Introduction, the community discovery process we use is based on that of GN (the GN process or method), which has been shown to identify communities in graphs with known community structure to a high degree of accuracy (4). Our modifications were necessary to make the method applicable to gene graphs, which are large and are created from source information that may by nature be incomplete or flawed. In particular, we identify many possible community structures and average them into a final list of communities. The statistical character of this step provides a more accurate picture of the complicated nature of community structure of a gene graph, without undermining the effectiveness of the basic principle of the algorithm.
A concept central to the community discovery process is the betweenness centrality (hereafter betweenness) of a vertex or edge. The betweenness of an edge AB (or a vertex A) is defined as the number of shortest paths between pairs of other vertices that contain AB (or A). As mentioned before, this concept was introduced (3) as a measure of influence of an individual, with respect to information flow, within a social network., However, it was noticed (4) that betweenness may also be used to identify communities within a graph, because intercommunity edges (those that lie between different communities) are much more likely to have a higher betweenness than intracommunity edges (edges that lie within one community).
To explain the community discovery process, we consider as a first example the small graph shown in Fig. 2. This graph consists of two well defined communities: the four vertices denoted by squares, including vertex A, and the nine denoted by circles, including vertex B.
Fig. 2.

A graph consisting of two communities.
In the graph of Fig. 2, edge AB has the highest betweenness. If we were to remove it, the graph would split into two connected components, the square and circle communities. This illustrates the idea behind the GN method of imposing community structure on a graph. One repeatedly identifies intercommunity edges by the criterion that they have higher betweenness than intracommunity edges and removes them. This procedure splits the giant component into many separate components, which coincide with the communities of the original graph.
It is important to note that the removal of an edge strongly affects the betweenness of many others, so that one must repeatedly recalculate the betweenness of all edges. To do this quickly, we used the fast algorithm of ref. 29 or 30.
At a certain point in our procedure, as opposed to the GN method, we stop removing edges from a component when we cannot further meaningfully subdivide it into communities; for example, as in Fig. 2, after removing edge AB. This allows us to obtain distinct communities of nodes, such as the circles and squares of Fig. 2. What criterion tells us when to stop?
Structurally, a component of five or fewer vertices cannot consist of two viable communities. The smallest possible such component is size 6, consisting of two triangles linked by one edge (Fig. 3).
Fig. 3.

The smallest possible graph consisting of two communities.
Components of size ≥6 can also be individual communities, like the group of nine in Fig. 2. The criterion we used to identify this type of component as a community was that the largest betweenness of any edge in the component did not exceed N - 1, where N is the number of vertices in the component.
This threshold is based on the betweenness of an edge connecting a leaf vertex, or vertex of degree one, to the rest of the graph. Consider the graph of Fig. 4 below. It is clear that it consists of just one community. Applying the Brandes algorithm, we find that edge XY has the highest betweenness, indicating that the size of the largest distinct community within the graph has size 1. That is, there are no distinct communities within the graph. In general, the single edge connecting a leaf vertex (such as X in Fig. 4) to the rest of a component of N vertices has a betweenness of N - 1, because it contains the shortest path from X to all N - 1 other vertices. If no edge's betweenness exceeds N - 1, therefore, we can identify the component as a community.i
Fig. 4.

A partitioning algorithm should not separate this graph into two communities.
We can now explain the need to neglect survey-type articles that list many genes in creating our graph. The genes listed in these articles will all be linked to one another, forming a complete subgraph Kn. Such a grouping is very tightly knit and will likely not be split into different communities. This situation, due only to the survey article, may not accurately reflect the interactions between the genes. It is possible that a few articles mention many genes that are in fact functionally related, but in this case it is likely that the genes will be linked by other articles that discuss them three or four at a time.
Communities Consist of Functionally Related Genes. The communities thus created consist of genes that were strongly interrelated in the literature. Most, but not all, gene co-occurrences imply a functional relation; genes may also co-occur in an article abstract because of physical proximity, similarity of nomenclature or structure, historical association, or other reasons. However, because such nonfunctional edges are a minority, they are highly likely to be intercommunity, because the neighbors of two nonfunctionally related genes are unlikely to be linked.
For example, genes S100A4 and S100A6 are members of the S100 family and co-occur twice in articles related to colon cancer, but they are not functionally related (Medline PMIDs 10389988 and 10952782). In our results, S100A4 and S100A6 do not occur in a community together. The neighbors of one are not linked to the neighbors of the other, which causes them to be placed in separate communities. Further examples are given in Results.
Multiple Community Structures. The process of assigning the nodes of a graph to communities may be called identifying a community structure on the graph. In the small examples given thus far in Figs. 2, 3, 4, there was only one reasonable community structure on each graph, because each node clearly belonged to only one community. In contrast, complex real-world graphs contain many “ambiguous” nodes that can be said to belong to two or more different communities due to their placement in the graph. An example, described in detail later, is the subgraph in Fig. 5, in which node B is ambiguous. Gene graphs include many ambiguous genes that belong to several communities, both in the context of the graph and in the context of biological function.
Fig. 5.

In this graph, it is unclear to which community node B belongs.
Therefore, if we identify only one community structure on a real-world graph, such as a gene graph, we could only hope to be somewhat accurate in classifying the nodes. A large amount of information concerning ambiguous genes and communities related through ambiguous genes would be lost.
Our resolution to this problem is to identify many plausible community structures on the graph and compare them. To do this, we make a modification to the GN process that introduces an element of randomness into which edges of very high betweenness are removed early in the process. Tightly knit communities are not affected by the order of edge removal and will eventually be identified no matter which high-betweenness edges are removed first. However, the eventual placement of ambiguous genes is strongly affected by which high-betweenness edges are removed first. By varying which high-betweenness edges are removed early in the process, we may therefore identify many community structures on a graph. By then comparing the structures, we can easily identify tightly knit communities, which do not vary from structure to structure, and ambiguous genes, which migrate from group to group.j
The subgraph of Fig. 5 illustrates why the order of edge removal affects the placement of ambiguous genes and the need for multiple community structures. This subgraph consists of two communities, one on the left including vertex A and another on the right including C. Among its edges, BC initially has the highest betweenness, and AB's betweenness is also high. Once we remove BC, however, AB becomes an intracommunity edge with low betweenness, and it will never be removed. Gene B will eventually be placed in a community with gene A. Had we removed AB first, BC would be rendered intracommunity, and gene B would end up in the community with C. Moreover, in considering Fig. 5, it is not clear where B should end up. B is ambiguous and could rightfully be considered to be a part of both communities. If we considered multiple community structures on this subgraph, we would see that B ended up in the community with A in some structures (the ones in which BC was removed early) and in the community with C in others (those where AB was removed). By comparing the structures, we could see that B really plays a role in both communities and (depending on the meaning of the links in the graph) possibly ties the communities together.
To describe in detail our method of identifying multiple community structures, we briefly describe how the Brandes algorithm (31) computes the betweenness for all edges in a graph, and how the GN process decides which edge to remove at each step. We then explain our modification, which allows for the identification of multiple community structures.
The first step of the Brandes algorithm is to find the shortest paths from all vertices to one “center” vertex using a breadth first search. In the second step, the contributions of these paths to the betweenness of each vertex or edge are added to a running total. The center is then switched and the above steps repeated. After every vertex in the component has been the center, we will have considered every shortest path twice, and the running totals for each vertex or edge will equal twice the betweenness of that vertex or edge. At this point in the GN procedure, one simply chooses the edge of highest betweenness and removes it. However, this choice is somewhat arbitrary, because there are likely to be many intercommunity edges in the graph.
Our modification is as follows. Instead of using every vertex as the “center” once in the Brandes algorithm, we cycle randomly through at least m centers (where m is some cutoffk) until the betweenness of at least one edge exceeds a threshold, again based on the betweenness of a “leaf” vertex.l We then remove the edge whose betweenness is highest at that point and repeat. Because the betweenness of the edge we remove exceeds the threshold, it is very likely to be intercommunity. We continue removing edges from each component in this way, until all of the components become small.m We then perform the full Brandes algorithm and remove the edge of highest betweenness from each component until it is resolved into communities.
The random nature of the modification allows us to change the order in which edges are removed, because the edge with highest betweenness after m centers have been considered will vary depending on which centers are considered. Our modified process can therefore be applied repeatedly to identify different plausible community structures on the graph.
This process may erroneously remove an intracommunity edges, which can happen if a large percentage of the centers considered lies in one community. In a large graph with many small communities, this probability is small, especially because we perform only the modified removal step in large components. Additionally, when we compare many different community structures, anomalous placements due to errors will be suppressed.
Applying this modified process n times, we obtain n community structures imposed on the graph. We can then compare the different structures and identify communities, as well as the strength of each gene within the community. For example, after imposing 45 structures on our graph, we might find: a community of genes A, B, C, and D in 20 of the 45 structures; a community of genes A, B, C, D, and E in another 20; and one of genes A, B, C, D, E, and F in the remaining 5. We report this result in the following way: A(45) B(45) C(45) D(45) E(25) F(5), which signifies that A, B, C, and D form a well defined community, E is related to this community but also to some other(s), and F is only slightly, possibly erroneously, related to it.
Aggregating Communities from Different Structures. To aggregate communities from different structures and obtain a final list of communities in the form {A(45) B(45) C(45) D(45) E(25) F(5)}, we use a procedure that is straightforward but rather tedious to explain because of the terminology. To summarize briefly, we create an initial “master list” M1 of communities by choosing one structure at random from among our set of N (in our experiments, n = 50). We then perform N - 1 steps, each consisting of comparing one of the remaining N - 1 structures S to the master list, and based on the results of the comparison, aggregating S into the list. The final master list, obtained by aggregating all N structures, is the final result of the entire algorithm.
Let us introduce the notation Mt to denote the state of the master list created from aggregating the first t structures (chosen in arbitrary order from the set of structures we found). At step t + 1, we select a structure S from among those we have not yet considered and compare its communities to the communities of Mt. S is aggregated into Mt based on the results of the comparison, creating an updated master list Mt+1.
Mt is a list of communities, and we will denote the kth community of Mt by 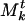 . The numbering system of communities in the list is arbitrary and serves only to distinguish them. Each community
. The numbering system of communities in the list is arbitrary and serves only to distinguish them. Each community 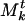 of Mt is a collection of genes and associated weights {(βj, ρj)}. The weight ρj associated with gene βj in a community
of Mt is a collection of genes and associated weights {(βj, ρj)}. The weight ρj associated with gene βj in a community 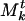 indicates how strongly it “belongs” to
indicates how strongly it “belongs” to 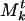 . To be precise, ρj is the number of structures, out of the t we have aggregated to form Mt, in which βj has been associated with the community that evolved (because structures were aggregated) into
. To be precise, ρj is the number of structures, out of the t we have aggregated to form Mt, in which βj has been associated with the community that evolved (because structures were aggregated) into 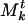 . This will be clearer when we explain the aggregation step. The communities evolve very little as the structures are aggregated, because the structures are on the whole quite similar. Thus the weight as defined is an accurate indication of how strongly a gene belonged to a community. One might expect that, because the communities in the master list evolve, the final result would depend on the order of aggregation. To the contrary, we found the order of aggregation had little effect on the final result due to the similarity of the different structures.
. This will be clearer when we explain the aggregation step. The communities evolve very little as the structures are aggregated, because the structures are on the whole quite similar. Thus the weight as defined is an accurate indication of how strongly a gene belonged to a community. One might expect that, because the communities in the master list evolve, the final result would depend on the order of aggregation. To the contrary, we found the order of aggregation had little effect on the final result due to the similarity of the different structures.
The details of the matching of communities S to the communities of Mt-1, and of the aggregation of S into Mt-1 to form Mt, are as follows. A basic metric to compare two communities A and B of genes α1, α2,..., αn, and β1, β2,..., βm, respectively, the traditional union/intersection metric
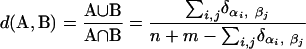 |
Here n and m are the number of genes in communities A and B, so the sums run over i = 1,..., n and j = 1,..., m. This notation is overcomplicated but useful for comparison to the weighted metric below. The sum over Δ functions just means we are counting how many of the genes in A and B are the same. The metric d(A, B) has a value between 0 and 1 and will be larger for a closer match. To compare a community A of S to a weighted community 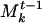 of Mt-1, we modify the traditional union/intersection metric to include the weight:
of Mt-1, we modify the traditional union/intersection metric to include the weight:
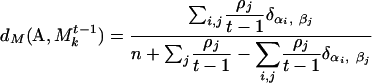 |
By comparing each community in one structure to all of the communities in the other using the weighted metric, we can find the closest match for each one.
Once a closest match for each community of S is found from among the Mt-1, the communities of S are aggregated into Mt-1. If community A of S is matched to community 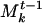 , we combine A and
, we combine A and 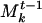 by incrementing the weights of the genes common to A and
by incrementing the weights of the genes common to A and 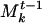 and appending the genes in A that were not in
and appending the genes in A that were not in 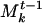 . For example, suppose that community {C, D, F} in S is matched to {B(5)C(5)D(3)E(5)} in Mt-1. We would update this community to become {B(5)C(6)D(4)E(5)F(1)} in Mt; that is, C and D would be incremented, and F would be appended.
. For example, suppose that community {C, D, F} in S is matched to {B(5)C(5)D(3)E(5)} in Mt-1. We would update this community to become {B(5)C(6)D(4)E(5)F(1)} in Mt; that is, C and D would be incremented, and F would be appended.
Occasionally, two or more communities in the structure were matched to one in M and vice versa. In this case, we assumed that the intracommunity edge had been erroneously removed to divide one community into two or more, either in the structure or the master list (in that case, it would have been in one of the previous structures aggregated into the master list). We thus melded the divided communities into one, altering M if need be, and then updated M as described above. This step could create a problem if one ended up with huge communities at the end, but we found that in general the largest communities in the final result had only 10 or 15 more genes than the largest communities in each individual structure, which incidentally indicates that our edge removal algorithm had a low error rate.
The entire process of determining community structure is displayed in Table 2.
Table 2. Algorithm for determining community structure.
| A. For n iterations, repeat { |
| 1. Break the graph into connected components. |
| 2. For each component, check to see whether component is a community. |
| a. If so, remove it from the graph and output it. |
| b. If not, remove edges of highest betweenness, using the modified Brandes algorithm for large components and the normal algorithm for small ones. Continue removing edges until the community splits in two. |
| 3. Repeat step 2 until all vertices have been removed from the graph in communities. |
| } |
| B. Aggregate the i structures into a final list of communities. |
Results
We applied the above technique using key words related to colon cancer. We considered articles that mentioned at least one of colon, colorectal, colonic, or gastrointestinal, and at least one of cancer or carcinoma. We identified 682 genes that were statistically correlated with colon cancer and that co-occurred in these articles with at least one other correlated gene. The graph of this co-occurrence network consisted of a giant component of 561 genes and other uninteresting smaller components (Table 1). The community discovery algorithm split the giant component into 79 different communities, with sizes ranging from 2 to 50 genes.
To present the usefulness of our results, we discuss features of these communities that demonstrate the utility of our method. Used in conjunction with the Medline and OMIM web sites, these communities allow us to suggest undocumented connections between genes of one community and between genes in different communities. They also demonstrate that our method tends to separate genes that co-occurred but were not functionally related into different communities, as discussed in Gene Graph. Genes that occur in two or more communities can indicate a link between the genes of each community.
We have published a full list of communities related to colon cancer and other diseases on our web site. Here we simply present one community to demonstrate the format of the results, discuss its features, and briefly mention similar features of other communities.
Table 3 shows one community of genes related to colon cancer from our results. Genes in this community are related to the overexpression of prostaglandin-endoperoxide synthase 2 (PTGS2), in colon cancer. Although PTGS2 is the official HUGO symbol, this gene is very commonly called cyclooxygenase 2 (COX-2), and we will use this term.
Table 3. A sample community of nine genes from our results for colon cancer.
| Gene symbol | Weight in community | Overall mentions with colon cancer | Neighbors with colon cancer |
|---|---|---|---|
| PTGS2 | 50 | 263 | PTGS1*DLD*MLH1*BCL2*PLA2G2A PLA2G4 APC*ERBB2*PGES ERBB3*PLA2 ACL4 WNT1*GRP*GRPR*LEF DLR*TCF4*TCF*MYB*VEGF*NOS2A TP53*MADH4*EGFR*S11*PDCD4 BRCA1*BRCA2*MSH2*ERBB4 |
| PLA2G2A | 50 | 12 | APC*PTGS2 PLA2G4 TP53*NF2*DCC*MLH1*SPLA2 |
| PLA2G4 | 50 | 1 | PLA2G2A PTGS2 |
| SPLA2 | 50 | 4 | PTGS2 PLA2G2A |
| FACL4 | 50 | 1 | PTGS2 |
| NOS2A | 50 | 7 | PTGS2 |
| PDCD4 | 50 | 1 | PTGS2 |
| PGES | 18 | 2 | ERBB2*PTGS2 ERBB3* |
| LEF1 | 5 | 18 | WNT1*TCF*PTGS2 TCF4*APC*FRA1*PLAUR*MYC*MMP7*TCF7* |
Here score in community denotes the number of community structures, out of 50, in which each gene was placed in this community (Partitioning the Graph into Communities). Genes with a score of 50 were members of this community only; genes with a lower score were members of this community and others.
Neighbor not in community.
The features of this community suggest the following possibilities: connections between some of the genes that co-occur with COX-2, but not each other; good reasons why many of the neighbors of COX-2 are not in this community; and possible connections to other communities via progesterone E synthase (PGES) and lymphoid enhancer-binding factor 1 (LEF1). We investigated these possibilities and present the results below.
Implied Connections. This community suggests a possible connection between the phospholipase A2 genes in this group and the gene FACL4. A Medline search for FACL4 or its alias ACS4 with each of PLA2, SPLA2, PLA2G4, and PLA2G2A turned up no result, and the OMIM entry for FACL4 has no mention of phospholipase A2. Nevertheless, by examining the abstracts of articles in which these genes were found, we see that these genes are related by their function, via COX-2 and arachidonic acid. COX enzymes convert arachidonic acid to prostaglandins (Medline PMID 11274413, for example). The three phospholipase A2 genes in the group {SPLA2, PLA2G4 [also known as cPLA (2)], PLA2GA2} are all sources of arachidonic acid (PMID 10706128, for example) and are thus related to COX-2. However, we found that the FACL4 enzyme also uses arachidonic acid, and that “the cellular level of unesterified arachidonic acid is a general mechanism by which apoptosis is regulated and that COX-2 and FACL4 promote carcinogenesis by lowering this level” (PMID 11005842). This indicates a clear link between the phospholipase A2 family of genes and FACL4 in carcinogenesis. It would have been time consuming for a researcher to ascertain this connection manually from Medline; even a search for arachidonic acid and colon cancer together produces 119 abstracts to sift through. Additionally, during this brief literature search, we discovered that nonsteroidal antiinflammatory drugs (NSAIDs) function by suppressing cPLA2 (PLA2G4) mRNA expression and thus depriving COX-2 of arachidonic acid.k Our method therefore suggests that these drugs may possibly affect FACL4 expression, although a Medline search of NSAID and FACL4 turned up no results.
Absent Neighbors. In examining neighbors of PTGS2 (COX-2) not present in this community, we noticed in particular the similarly named gene PTGS1 (also known as COX-1). These two genes are isoforms of cyclooxygenases (PMID 9099957, for example); they co-occurred in 70 articles related to colon cancer and 1,500 articles overall. However, they have been shown to regulate colon carcinoma-induced angiogenesis by two different mechanisms (Medline PMID 9630216). COX-2 has also been shown to be expressed much more frequently than COX-1 in tumors and less frequently in normal tissue (PMID 7780968, for example; note the use of the alias PGHS-1 or -2 for COX-1 or -2 in this article) The separation of COX-1 and -2 into different communities thus accurately reflects our current knowledge about how these genes function in relation to colon cancer. Although the enzymes they code for are structurally very similar, COX-2 plays a strong role in colon cancer, whereas COX-1's role is weaker and by a different mechanism.
Several other neighbors of PTGS2, such as MLH1, BRCA1, BRCA2, and MSH2, also proved to be weakly or nonfunctionally related. However, a few of PTGS2's noncommunity neighbors have been tentatively identified as functionally related, such as GRP and GRPR (GRP receptor; PMID 11292836) and EGFR (PMID 9012840). For this reason, we include a list of all neighbors of each gene in the results as a secondary list of possible connections to explore.
Links to Other Communities. We also looked for links to other communities through the genes PGES and LEF1, both of which show a weak connection to the COX-2 community and were often placed in other communities.
Both searches yielded good results. PGES co-occurs with other genes only once, in an abstract with COX-2, ERBB2, and ERBB3. Examining this abstract, we find a link between the COX-2 pathway and autocrine/panacrine activation of HER2/HER3 (also known as ERBB2 and ERBB3; 9927187). The ERBB genes are present in another community of 25 genes. In conjunction with the previous discussions about arachidonic acid, there is a possible link between not only COX-2 but all of the genes related to arachidonic acid (most of which never co-occur with ERBB2 or -3) to any gene related to the autocrine/panacrine activation of ERBB2/ERBB3. This conclusion depends on knowledge of many articles, in particular PMID 9927187, and could easily escape notice in a manual search.
LEF1 was found with COX-2 in only one article (PMID 10834941). It states that “NO (nitric oxide) may be involved in PGHS-2 (COX-2) overexpression in conditionally immortalized mouse colonic epithelial cells. Although the molecular mechanism of the link is still under investigation, this effect of NO appears directly or indirectly to be a result of the increase in free soluble β-catenin and the formation of nuclear β-catenin/LEF-1 DNA complex.” This article indicates a possible connection between COX-2, NOS2A (nitric oxide synthase, responsible for the production of NO) and the very important colon cancer gene β-catenin.
Importance of Alias Symbols. As a last note, this community demonstrates the crucial importance of considering alias symbols when extracting gene names. The aliases COX-2, PGHS-2, NOX2, and cPLA (2) were very commonly used in articles that tied this community together
Other Results. Here we present similar results from two other communities: A connection between PXR (pregnane X receptor) and GP170 (P-glycoprotein) is indicated because they are placed together in a community. PXR is implicated in the induction of the MDR1 gene (PMID 11297522), whereas MDR1 expression has been associated with the expression of functional P-glycoprotein (PMID 10334913). A Medline search turns up no results for GP170 or GP-170 with PXR or its aliases PAR, SXR, and NR1i2.
Another probable undocumented connection between GP200-MR6 and STAT6, via IL-4 and its receptor IL-4R is suggested by their placement together in a community. IL-4 induces STAT6, which is involved in mediating activation of IL-4R gene expression (PMID 8810328), whereas GP200-MR6 has been shown to be functionally associated with IL-4R (PMID 9178815). This example demonstrates the power of an automated method to bring together information from disparate, old sources (cited articles from J. Biol. Chem., Oct 11, 1996 and Int. J. Cancer, May 16, 1997).
Although large communities are more difficult to analyze for the nonexpert, we were nevertheless able to draw some conclusions. For example, we considered a 30-gene community largely concerned with apoptosis and genes related to BCL-2, containing in particular the gene TRAIL. TRAIL has been shown to induce procaspase-8 activation, triggering caspase-dependent apoptosis in colon cancer cells (PMID 11245478). It could thus be related to the function of genes such as BCLX, BCLXS, etc., which we find in this community but which do not co-occur with TRAIL via the genes BCL-2 and CASP8.
A good example of nonfunctionally related genes with similar names that are placed in different communities is MMP11 and MMP9 (PMID 8645587). Often nonfunctionally related neighboring genes do appear together in one community in a small number of structures (see Partitioning the Graph into Communities) but appear in different communities in the majority of structures. Examples of this include CYP3A4 or CYP3A5 and CYP1A2 (PMID 9202751) as well as SMAD3 and SMAD5 (PMID 10446110 and 11196171, for example; SMAD2 and SMAD4 are aliases for MADH2 and MADH4, respectively).
Conclusion
We have presented a data-mining technique for biological literature that produces detailed results while extracting only very simple data from each article abstract and title. The method produces a list of communities of functionally related genes that are designed to summarize available information and indicate genes that are likely to be complementary in their function. The genes within a community are weighted, indicating how strongly they belong to the community. We show that the communities produced in the case of colon cancer have interesting features that give one insight into the function of the component genes.
The identification of many similar community structures on each gene graph allows us to recognize those genes that belong in two or more different communities. In this sense, our method produces a richer result than previous methods that impose one rigid structure on the graph. This idea could be applied to social and other networks where individuals play a role in more than one community.
We introduce two statistical components into the process, which lessen the inevitable errors of text mining in the biological literature, particularly severe in our case because of the complex young nomenclature system for genes. However, our method retains the ability to detect relations among rarely mentioned genes, one of its strongest features.
To reiterate an important point from the Introduction, our results are not meant to perfectly model biological reality, only to function as a tool for biologists. It was not possible to compare our communities to a database or list of groups of related human genes, because such a list does not exist. The only justification we can provide that our communities were “accurate” is to cite ref. 4, in which the GN method was shown to be very effective in identifying communities. In fact, because genes within a community are linked by edges from a co-occurrence, it is almost certain that they are related somehow. A much more interesting measure of the effectiveness of the method is whether it separates genes that should be separated.
The factor that most limits our results is the absence of many gene symbols from HUGO and other online databases. Hopefully, these databases will soon be more complete. Related problems are the unorganized nomenclature system for human genes (see discussion in ref. 31) and small modifications to recognized symbols introduced by many authors, such as the addition of hyphens, parentheses, or spaces, which make the symbols difficult to detect. Efforts are being made to standardize the gene nomenclature system (33).
A less acute limiting factor was the placement of many genes in either large and very small communities in our results. Although still a step forward from raw co-occurrence data, such communities are of limited usefulness; they often did not provide much insight into the function of their component genes, other than that the genes were rarely related to others in the context of colon cancer. If such genes were more commonly mentioned in other contexts, a search using other diseases or key words would likely turn up more interesting communities with these genes. Large communities were difficult for us to analyze but nevertheless yielded some interesting results. These communities contained many of the most commonly mentioned genes in connection with colon cancer, such as APC and TP53. Strangely, a search for colon cancer genes is probably not the most efficient way to study these genes, which are simply too highly linked in this context. Instead, one could perform other searches with other key words, hoping to focus on particular aspects of these genes' function by confining them to smaller more informative communities.
We believe that large communities are a product of graph topology, not of the threshold we use to stop subdividing a community or of the aggregation process. To further subdivide large communities, one could consider a weighted graph, where the weight corresponds to the (normalized) number of times the two genes co-occur. This could increase the “distance” between, for example, two commonly studied distantly related co-occurring genes. They would then not end up in the same community and, more importantly, would not glue a false community together. The simplest such weighting would be to neglect all links below some (normalized) threshold weight. Another resolution to the problem of large communities would be to refine the step that aggregates the community structures into one result.
Acknowledgments
We thank Lada Adamic, Eytan Adar, and Melissa Wilkinson for many useful discussions.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Mapping Knowledge Domains,” held May 9-11, 2003, at the Arnold and Mabel Beckman Center of the National Academies of Sciences and Engineering in Irvine, CA.
Abbreviations: COX-2, cyclooxygenase 2; PTGS2, prostaglandin-endoperoxide synthase 2; GN, Girvan-Newman; PMID, PubMed identification number.
Footnotes
Medline is the foremost English-language database of biomedical articles. The search utility for Medline is PubMed (www.ncbi.nlm.nih.gov/entrez/query.fcgi).
The machine we used is a standard 1-GHz machine with an Intel Pentium 3 processor running red hat linux.
The time to perform this step increases as nm2, where n is the number of genes in the network and m is the number of pairs of genes that co-occur. As we explain later, genes are selected to create a network, and if the network is too large, this step could be very slow. We found that a size of <1,000 genes is generally tractable for our method.
http://www3.ncbi.nlm.nih.gov/omim. This web site provides detailed information about many genes, proteins, and other biological objects as well as references to related articles.
We omitted the small fraction of abstracts published before 1990, because they very rarely discuss gene function and tend to use outmoded nomenclature. In addition, we neglect abstracts that mention more than four genes, because they are typically abstracts of survey-type articles that impair the community identification process. These two types of articles form a very small fraction of the Medline database.
An example is DCC, which may be used to refer to the gene “deleted in colon cancer” or the cancer assay method “dextran-coated charcoal.” Such ambiguous symbols are very common because of the frequent use of abbreviations in biological texts.
This number was determined by comparing the International Standard Serial Numbers of the journals in the Medline listings of the 7,985 abstracts involved in creating the network of genes related to colon cancer.
It is not in general true that an intercommunity edge must have betweenness greater than N-1, although such a situation is extremely unlikely in a power law graph. For a community of size m within a graph of size N, there is a total betweenness of m(N-m) divided among the edges connecting the community to the graph. So, if there are more than m such edges, it is possible that none of them will have betweenness greater than N. However, remember that few of these edges, or the extracommunity vertices they connect, should be adjacent, because otherwise m would not be a community. Even in GN′s highly nonpower law college football graph, the criterion only occasionally fails when an intercommunity edge has a betweenness slightly less than N-1.
This process is essentially a form of soft clustering, although it differs significantly from existing methods of soft clustering. These methods (see ref. 34 for an example) are essentially restricted to clustering objects such as documents that comprise many individual elements (e.g., words). The words of one document are compared to the words of another, and a relative closeness can be established. The soft clustering presented here is affected only by a node's placement in the graph, not by a comparison of elements comprising neighboring nodes.
The cutoff we used was m(N) = 10log(N)-25, where N is the size of the component. This function has m(50) ≈ 15, and m(800) ≈ 41. We found that 15 was a reasonable number of centers to consider for a component of size 50, whereas 40 centers is more than enough for any component, however large. Basically, an intracommunity edge will be erroneously removed if we repeatedly choose centers from the same community. For a component of 50 vertices and 4 communities, the probability of choosing 8 of 15 centers from one community is ≈1%. For a large component with many communities, the probability of error is very low for a cutoff of 40 centers.
The value of the threshold in this case is (N + 1)/2-1, where N is the size of the component, and i is the number of centers that have been considered up to that point in the process.
We never attempted to precisely define “small.” We used values in the 35-50 node range and, as one might expect, it made little or no difference in the final result. An exact definition would depend on the community size, the graph size, and a desired probability of error (see discussion on this page). However, even when we used a number as large as 50, the randomness of the method was sufficient to produce a slightly different community structure every time.
References
- 1.Adamic, L., Wilkinson, D., Huberman, B. & Adar, E. (2002) in Proceedings of the IEEE Bioinformatics Conference (Institute of Electrical and Electronic Engineers, Los Alamitos, CA), pp. 109-117. [PubMed]
- 2.Adar, E. (2004) Bioinformatics, in press. [DOI] [PubMed]
- 3.Freeman, L. (1977) Sociometry 40, 35-41. [Google Scholar]
- 4.Girvan, M. & Newman, M. (2002) Proc. Natl. Acad. Sci. USA 99, 8271-8276.12060770 [Google Scholar]
- 5.Wu, F. & Huberman, B. A. (2004) Eur. J. Phys. B, in press.
- 6.Tyler, J. R., Wilkinson, D. M. & Huberman, B. A. (2003) Proceedings of the International Conference on Communities and Technologies (Kluwer, Amsterdam).
- 7.Blaschke, C., Andrade, M., Ouzounis, C. & Valencia, A. (1999) in Proceedings of the AAAI Conference on Intelligent Systems in Molecular Biology (American Association for Artificial Intelligence Press, Heidelberg), pp. 60-67. [PubMed]
- 8.Ng, S. K. & Wong, M. (1999) Genome Inf. 10, 104-112. [PubMed] [Google Scholar]
- 9.Humphreys, K., Demetriou, G. & Gaizauskas, R. (2000) Pac. Symp. Biocomput. 5, 502-513. [DOI] [PubMed] [Google Scholar]
- 10.Rindflesch, T. C., Tanabe, L., Weinstein, J. N. & Hunter, L. (2000) Pac. Symp. Biocomput. 5, 514-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas, J., Milward, D., Ouzounis, C., Pulman, S. & Carroll, M. (2000) Pac. Symp. Biocomput. 5, 538-549. [DOI] [PubMed] [Google Scholar]
- 12.Friedman, C., Kra, P., Yu, H., Krauthammer, M. & Rzhetsky, A. (2001) Bioinformatics 17, S74-S82. [DOI] [PubMed] [Google Scholar]
- 13.Pustejovsky, J., Castaño, J., Zhang, J., Cochran, B. & Kotecki, M. (2002) Pac. Symp. Biocomput. 7, 362-272. [DOI] [PubMed] [Google Scholar]
- 14.Tanabe, L., Scheft, U., Smith, L., Lee, J., Hunter, L. & Weinstein, J. (1999) BioTechniques 27, 1210-1217. [DOI] [PubMed] [Google Scholar]
- 15.Craven, M. & Kumlien, J. (1999) in Proceedings of the ISMB Conference (International Society for Computational Biology, Brisbane, Australia), pp. 77-86
- 16.Shatkay, H. & Wilbur, W. (2000) in Proceedings of the IEEE Conference on Advances in Digital Research (Institute of Electrical and Electronic Engineers, Los Alamitos, CA), pp. 183-192
- 17.Raychaudhuri, S., Chang, J., Sutphin, P. & Altman, R. (2002) Genome Res. 12, 203-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrade, M. & Valencia, A. (1997) in Intelligent Systems for Molecular Biology (American Association for Artificial Intelligence Press, Heidelberg), pp. 25-32.
- 19.Fukuda, K, Tsunoda, T., Tamura, A. & Takagi, T. (1998) Pac. Symp. Biocomput. 3, 705-716. [PubMed] [Google Scholar]
- 20.Proux, D., Rechenmann., F., Julliard, L., Pillet, V. & Jacq, B. (1998) in Genome Informatics Workshop (Universal Academic, Tokyo), pp. 72-80. [PubMed]
- 21.Jenssen, T.-K., Laegrid, A., Komorowski, J. & Hovig, E. (2001) Nat. Genet. 28, 21-28. [DOI] [PubMed] [Google Scholar]
- 22.Stephens, M., Palakal, M., Mukhopadhyay, S. & Raje, R. (2001) Pac. Symp. Biocomput. 6, 483-396. [DOI] [PubMed] [Google Scholar]
- 23.Stapley, B. & Benoit, G. (2000) Pac. Symp. Biocomput. 5, 529-540. [DOI] [PubMed] [Google Scholar]
- 24.Masys, D., Welsh, J., Fink, L., Gribskov, M., Klacansky, I. & Corbeil, J. (2001) Bioinformatics 17, 319-326. [DOI] [PubMed] [Google Scholar]
- 25.Shatkay, H., Edwards, S. & Boguski, M. (2002) in IEEE Intelligent Systems, Special Issue on Intelligent Systems in Biology (Institute of Electrical and Electronic Engineers, Los Alamitos, CA), pp. 45-53.
- 26.Raychaudhuri, S., Schutze, H. & Altman, R. (2002) Genome Res. 12, 1582-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huberman, B. A. & Adamic, L. (1999) Nature 401, 131.10490019 [Google Scholar]
- 28.Aiello, W., Chung, F. & Lu, L. (2000) in Proceedings of the 32nd Annual ACM Symposium on Theory of Computing (Association for Computing Machinery, New York), pp. 171-180.
- 29.Albert, R. & Barabasi, A.-L. (2002) Rev. Mod. Phys. 74, 47-97. [Google Scholar]
- 30.Newman, M. (2001) Phys. Rev. E 64, 026118-1-026118-17. [Google Scholar]
- 31.Brandes, U. (2001) J. Math. Soc. 25, 163-177. [Google Scholar]
- 32.Pearson, H., (2001) Nature 411, 631-632. [DOI] [PubMed] [Google Scholar]
- 33.Ashburner, M., Ball, C., Blake, J., Botstein, D., Butler, H., Cherry, J., Davis, A., Dolinski, K., Dwight, S. & Eppig, J. (2000) Nat. Genet. 25, 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tishby, N., Pereira, F. & Bialek, W. (1999) in Proceedings of the 37th Annual Allerton Conference on Communication, Control and Computing (UIUC Press, Champaign-Urbana, IL), pp. 368-377.