

Abstract
Introduction
Pompe’s disease is a metabolic myopathy caused by a deficiency of the enzyme alpha-glucosidase. Patients with late-onset Pompe’s disease have progressive muscle weakness, which also affects pulmonary function. Since the advent of specific treatment for Pompe’s disease, enzyme replacement therapy with alpha-glucosidase, the prognosis of the disease has changed.
Methods
We report the case of the first patient treated in Spain with home therapy, and the effects on her clinical status of a reduction in treatment frequency.
Results
A worsening was seen in the patient’s neuromuscular assessment on different scales, after two discontinuations during the patient’s usual administration frequency.
Conclusion
It is essential to keep an adequate administration schedule to maintain the clinical benefits of enzyme therapy.
Keywords: alpha-glucosidase replacement enzyme, maltase acid deficiency, neuromuscular disease, Pompe’s disease
Introduction
Lysosomal storage diseases (LSD) are caused by congenital metabolic errors and are characterized by a deficiency of a given enzyme within the lysosome, resulting in progressive accumulation of substances causing a storage disease. The combined prevalence of these diseases is one per 7700 live births, although there is variability depending on the type of disease.1
Pompe’s disease (PD) is an LSD, and results from the deficiency of acid alpha-glucosidase (GAA), the enzyme responsible for degradation of glycogen to glucose. It was first described in 1932 by the Dutch pathologist J.C. Pompe.2 It is also known as maltase acid deficiency, glycogen storage disease type II, or glycogenosis type II. It is inherited in an autosomal recessive manner.3 It is a disease with a continuous and variable spectrum of symptoms. The data on the combined incidence of all clinical subtypes is one per 40,000 births.4
The accumulation of glycogen primarily affects muscle tissue, causing progressive musculoskeletal degeneration, respiratory impairment, and involvement of the heart muscle in children in particular.5,6 It presents with a variable clinical spectrum in terms of age of onset, progression, and organ involvement. Two forms of onset are distinguished: early (infantile) and late onset. When onset is prior to 12 months of age, the first symptoms usually occur in the first 3 months. Death usually occurs before 1 year of age.7 In the late onset forms, the clinical picture resembles that of muscular dystrophy. Motor delay occurs due to weakness in proximal limb muscles and the involvement of respiratory muscles.8 In adults, symptoms may start from the 2nd to the 6th decade of life as progressive myopathy predominantly in the proximal pelvic muscles.9,10 Disease progression leads to the use of assisted ventilation, and a wheelchair due to muscle weakness.
In 2006, both the Food and Drug Administration and the European Medicines Agency approved the marketing of recombinant human GAA (rh-GAA) as specific treatment for PD. Administration of treatment is by intravenous infusion every 2 weeks, and is lifelong. As it is a chronic disease, the patient must go to a center where the drug can be administered, thus becoming dependent on the health system.
Strict adherence to the treatment schedule and adequate dosing becomes essential in these patients to optimize the results, however, there is no evidence as to how inadequate dosing may affect the clinical symptoms of the patient. This article describes the case of a female patient with PD who experienced worsening of her clinical condition after suppression of two doses of her treatment. This is also the first case in Spain of a patient who received home therapy with this drug.
Report
The case was a 39-year-old woman, with no relevant family history, who had been operated on three times in childhood for bilateral clubfoot. Informed consent was obtained. From 14 years of age, the patient experienced progressive muscle weakness in the four extremities, predominately proximal, and more significant in the legs, making it difficult for her to climb stairs, rise from the floor, and run. At 18 years of age, weakness manifested in the arms and she started to experience dyspnea on exertion.
At 20 years of age, a biopsy of the biceps muscle was performed, revealing a vacuolar myopathy with glycogen accumulation, increased phosphatase acid activity, and breakdown of muscle fibers; changes consistent with myopathy due to maltase acid deficiency or PD. Examination revealed tetraparesia of a predominantly proximal nature with scapular winging. An electromyogram showed a myopathic pattern. Transaminases and creatine phosphokinase levels were elevated. Cardiac function appeared normal, and pulmonary function tests showed a vital capacity of 32% with a muscular inspiratory pressure of 16 cm H2O and an O2 saturation of 96%.
At 23 years of age, she required the use of a wheelchair to move due to her inability to stand or walk. At 26 years of age, treatment was started with noninvasive assisted ventilation via a nasal mask during sleep. At this time, the vital capacity was 600 cc (20%) and blood gases showed pO2 67 mmHg and paCO2 58 mmHg. At 31 years of age, a gastrostomy was performed, given her difficulties for oral nutrition. At this point, her dependence on the ventilator was virtually total.
In January 2005, she started replacement therapy with rh-GAA with a dosing regimen of 20 mg/kg every 15 days (2 weeks), with no adverse reactions. The neuromotor and functional assessment before starting replacement therapy were the most significant findings (using the Muscle Strength Scale [Medical Research Council Scale]):11,12 neck flexors-extensors - 3/5, trunk muscles - 0 (does not control trunk), right arm: proximal muscles - 0/5, elbow flexors - 1/5, carpal bone extensors - 3/5, finger flexors - 3/5, thumb approximator - 2/5. Rest of hand muscles - 0/5. Left arm: overall proximal muscles - 0/5, elbow flexors-extensors - 0/5, carpal bone extensors - 3/5, finger flexors - 2/5. Rest of hand muscles - 0/5. Right leg: hip flexors - 1/5, hip approximators - 1/5, knee extensors - 0/5, knee flexors - 1/5, finger flexors - 2/5. Rest of muscles - 0. Left leg: hip flexors - 1/5, hip approximators - 2/5, knee extensors - 0/5, knee flexors - 1/5, finger flexors - 2/5. Rest of muscles - 0. The patient had a Functional Independence Level (Functional Independence Measure [FIM] scale) score of 53 out of 126. She had a Walton & Gardnerd-Medwin Scale13 grade of 9 (out of 10). The patient was dependant on a ventilator for virtually 24 hours a day.
Four months after the start of treatment, new assessments were performed on the patient, showing the changes from baseline (described in Table 1). The most notable were the ability to remain upright for over 30 minutes, the improved functionality of the right hand, and particularly the ventilator-free time (over 7 hours), and recovery of oral feeding (Images 1 and 2).
Table 1.
Comparison of efficacy between dosing schedules
| Baseline | 20 mg/2 weeks | 20 mg/month | |
|---|---|---|---|
| Neck flexors-extensors | 2/5 | 3/5 | 2/5 |
| Trunk muscles | 0 | Stay upright (30 mins) | 0 |
| Proximal arm muscles | 0/5 | 0/5 | 0/5 |
| Right elbow extensors | 1/5 | 2/5 | 1/5 |
| Right hand | 2/5 | 4/5 | 3/5 |
| Proximal leg muscles | 0/5 | 0/5 | 0/5 |
| Hours without assisted ventilation | 0 | 7 | 3 |
| FIM Scale | |||
| Feeding | 1 | 3 oral and enteral | 1 only enteral |
| Personal care | 1 | 3 | 2 |
| Total (FIM scale) | 52/126 | 56/126 | 53/126 |
FIM=Functional Independence measure scale.
Image 1 and 2.
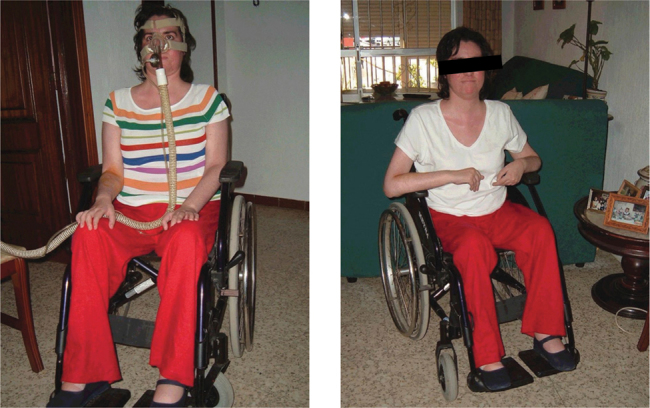
Patient affected by Pompe’s disease after 6 months of treatment with enzyme replacement therapy.
These results remained unchanged until March 2009 when, after decreasing the administration schedule to 20 mg/kg every 30 days for 2 months because of a supply problem, a clear regression was noted in the previously observed positive clinical findings (Table 1). There was no intercurrent infection or any other situations that could explain this worsening.
After reinitiation of treatment with a 2-weekly dosing schedule, the results of tests were in the range of the values prior to treatment interruption, with the patient recovering the ventilator-free time of up to 7 hours. Clinical and functional conditions of this patient have continuously been stable since then, up to the present day.
Discussion
The importance of adequate treatment is reflected in the case reported here. The efficacy of rh-GAA treatment has been shown in different trials with the drug. In the phase 2 trial, it was shown that children treated with rh-GAA had a survival rate of 73% versus 37% in the control group. This trial also showed improvement in weight gain and height. Early treatment improved results.14 In a phase 3 trial in patients aged 5 to 15 years, increases were seen in respiratory function and distance walked.15,16 There is very recent data on the efficacy of replacement therapy,17 however, there are still many unknowns on the importance of the dose and dosing intervals of the enzyme. It is also important to emphasize the fact that patients can obtain great benefits if they are treated at home.
The main limitation of the study is that it shows the experience of only one patient who is much more severely affected by the disease (Images 1 and 2) than the patients included in many other reported studies.
In the case of this patient, it is clear that the organic damage caused until initiation of treatment made it difficult to obtain a very marked improvement, but the recovery of functional control of the trunk, substantial improvement in ventilator-free time, and increased strength and functionality of the right arm has allowed her a certain degree of independence, making it possible for her to partially feed herself orally.
Preliminary data from the study carried out in adult patients to establish efficacy and safety showed a clinical benefit in the test of distance walked in 6 minutes and stabilization of lung function.17 These preliminary data showed the importance of treatment, compared with the historical cohort without treatment.
Conclusion
What is relevant in this case is not only the efficacy of treatment with the enzyme and the benefits for the patient and family to be treated at home, but that despite being a patient with a substantially worse baseline status than the patients included in the different reported studies, the results were relevant for the autonomy of the patient (Images 1 and 2).
It is also essential that dosing is adequate, as was shown in this patient, who recovered the levels of autonomy lost after reducing the frequency of treatment. Therefore, it can be concluded that maintaining dosing frequency is important for achieving optimum results, even in patients in whom the disease is severe.
Acknowledgments
The authors declare that they have no personal, commercial, academic, or financial interests and there was no funding/sponsorship related to this paper. Emilia Barrot is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Open Access. This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
To view enhanced content go to www.biologicstherapy-open.com
This article is published with open access at Springerlink.com
References
- 1.Meikle P.J., Hopwood J.J., Clague A.E. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 2.Pompe J.C. Over idiopatische hypertrophie van het hart. Ned Tijdshr Geneeskd. 1932;76:304. [Google Scholar]
- 3.Hers H.G. α-Glucosidase deficiency in generalized glycogen storage disease (Pompe’s disease) Biochem J. 1963;86:11–16. doi: 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kishnani P.S., Steiner R.D., Bali D. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirschhorn R., Reuser A.J. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver C.R., Beaudet A., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3389–420. [Google Scholar]
- 6.Kishnani P.S., Howell R.R. Pompe disease in infants and children. J Pediatr. 2004;144:S35–S43. doi: 10.1016/j.jpeds.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 7.Van den Hout H.M., Hop W., van Diggelen O.P. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 8.Slonim A.E., Bulone L., Ritz S., Goldberg T., Chen A., Martiniuk F. Identification of two subtypes of infantile acid maltase deficiency. J Pediatr. 2000;137:283–285. doi: 10.1067/mpd.2000.107112. [DOI] [PubMed] [Google Scholar]
- 9.Hagemans M.L., Winkel C.L.P.F., Van Doorn P.A. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain. 2005;128:671–677. doi: 10.1093/brain/awh384. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Felber W., Horvath R., Gempel K. Late onset Pompe disease: Clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Dis. 2007;17:698–706. doi: 10.1016/j.nmd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Medical Research Council . Aids to the examination of the peripheral nervous system. London: Her Majesty’s Stationery Office; 1981. [Google Scholar]
- 12.Hahn A.F., Bolton C.F., Pillay N. Plasma exchange therapy in chronic inflammatory demyelinating polyneuropathy. A double-blind, sham controlled, cross-over study. Brain. 1996;119:1055–1066. doi: 10.1093/brain/119.4.1055. [DOI] [PubMed] [Google Scholar]
- 13.Hamilton B.B., Laughlin J.A., Fiedler R.C., Granger C.V. Interrater reliability of the 7-level functional independence measure (FIM) Scand J Rehabil Med. 1994;26:115–119. [PubMed] [Google Scholar]
- 14.Amalfitano A., Bengur A.R., Morse R.P. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. doi: 10.1097/00125817-200103000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Kishnani P.S., Corzo D., Nicolino M. Recombinant human acid-glucosidase. Major clinical benefits in infantile-onset Pompe disease. Neurol. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 16.Van der Ploeg A., Marsden D.L. Response to enzyme replacement therapy in 18 juvenile and adult patients with severe Pompe disease. Mol Genet Metabol. 2007;90:227–265. doi: 10.1016/j.ymgme.2006.12.009. [DOI] [Google Scholar]
- 17.Van der Ploeg A., Clemens P.R., Corzo D. A randomized study of Alglucosidase alfa in late-onset Pompe’s disease. N Eng J Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]