

Abstract
Obesity is a complex multifaceted disease resulting from interactions between genetics and lifestyle. The proportion of phenotypic variance ascribed to genetic variance is 0.4 to 0.7 for obesity and recent years have seen considerable success in identifying disease-susceptibility variants. Although with the advent of genome-wide association studies the list of genetic variants predisposing to obesity has significantly increased the identified variants only explain a fraction of disease heritability. Studies of gene–environment interactions can provide more insight into the biological mechanisms involved in obesity despite the challenges associated with such designs. Epigenetic changes that affect gene function without DNA sequence modifications may be a key factor explaining interindividual differences in obesity, with both genetic and environmental factors influencing the epigenome. Disentangling the relative contributions of genetic, environmental and epigenetic marks to the establishment of obesity is a major challenge given the complex interplay between these determinants.
Keywords: Gene–environment interaction, Lifestyle, Genetics, Environment, Epigenetics, Obesity, Epigenome
Introduction
Obesity was once considered a problem of economically developed countries, but the number of overweight and obese people is now dramatically increasing in low- and middle-income countries at a rate never seen before [1]. If recent trends continue unabated, by 2030, the absolute numbers could rise to a total of 2.16 billion overweight and 1.12 billion obese individuals, or 38 % and 20 % of the world’s adult population, respectively [2].
As the fundamental cause of obesity and overweight (defined by anthropometric measures: body mass index [BMI], waist circumference [WC] and/or waist-to-hip ratio [WHR]) is an energy imbalance between calories consumed on the one hand and calories expended on the other hand, increases in rates of obesity must reflect a state of positive energy balance, which is very likely a result of the profound changes in society and in behavioral patterns of populations over recent decades. Indeed, it is widely accepted that multiple factors contribute to this epidemic, including economic growth, modernization, urbanization and, most importantly, changes in our lifestyle, as eating habits have shifted to greater consumption of energy-dense foods that are high in fats and sugars, while at the same time, physical activity has decreased [1, 3]. Although a healthy lifestyle could be the apparent remedy for obesity, its implementation in the general population has proven difficult so far. Given the fact that people respond differently to the “obesogenic” environment owing to genetic predisposition, understanding the causes and pathophysiology of obesity is very important for prevention and therapy. Even in the presence of a strong “obesogenic” environment, hereditary factors remain key contributors to the disease etiology. Ethnic/racial differences in obesity even in comparable environments [4, 5] indicate that obesity is most likely the result of a complex interplay between multiple genetic, behavioral, social and environmental factors that affect energy balance and, thus, body weight regulation [6–9].
Over the past two decades, numerous strategies have been employed for the identification of genetic determinants of obesity, including studies of severe forms of obesity, genome-wide linkage studies, candidate gene analyses and genome-wide association studies (GWAS). Since 2005, the novel GWAS approach has led to breakthrough progress in our understanding of the genetic determinants of common obesity. Almost 50 loci have been identified and are collectively reported in the National Human Genome Research Institute GWAS catalogue (http://www.genome.gov/gwastudies/) [10]. Among those GWAS findings, the first obesity susceptibility locus identified was the FTO gene, which has the largest effect on obesity risk to date; each additional risk allele in FTO was shown to be associated with a 1- to 1.5-kg increase in body weight and a 20 % to 30 % increase in obesity risk [11, 12]. Since it is widely assumed that gene–environment interaction (GEI) must have an effect on adiposity, several epidemiologic studies have explored the relationship between lifestyle and obesity susceptibility genes, reporting significant interactive effects. Despite discrepancies in the reported results, some new insights into the role of gene–lifestyle interaction in obesity have been obtained. In the current review, we evaluate the recent successes in the examination of GEI in obesity and describe the main findings. We then examine the machinery that underlies GEI in obesity, focusing on epigenetics and particularly DNA methylation as a mechanism for these interactions. Finally, we discuss the challenges of the existing and emerging approaches in studying GEI.
Genetic Determinants of Obesity
Until recently, progress in finding obesity-susceptibility genes was rather slow. Numerous groups have been involved in research related to the genetics of common obesity, with a major focus on candidate gene studies. Those genes were selected based on their known functional role in physiologic pathways (e.g. regulating body weight or energy metabolism) [13]. Between 1996 and 2005, the Obesity Gene Map (http://obesitygene.pbrc.edu/) extensively evaluated all published results, including monogenic forms of obesity, transgenic and knockout animal models, quantitative trait loci from animal cross-breeding experiments, linkages from genome scans and candidate gene association studies [14]. In the last update of the Obesity Gene Map published in 2006, 127 candidate genes were reported, of which less than 20 % were replicated by 5 or more studies. Such a high level of non-replication was the result of many limitations of the candidate gene study approach, such as small sample size and, thus, insufficient statistical power to detect small effects, as well as lack of type 1 error control, among others [15•].
In the past few years, a novel approach (GWAS) that involves scanning of many thousands of samples using the latest advances in genotyping technology (i.e. high-density, genome-wide arrays to assay hundreds of thousands of single nucleotide polymorphisms [SNPs] that capture the majority of common variation in the human genome) have led to breakthrough progress in the identification of obesity-susceptibility genes. To date, large-scale meta-analyses of GWAS for overall and abdominal obesity along with a recent GWAS meta-analysis for percent body fat (%BF) have reported 32 genetic loci associated with BMI, 14 loci related to WHR and 2 loci for %BF (see Day and Loos [15•] for a recent overview of GWAS findings of obesity-related traits). The effect sizes of the 32 established BMI-associated loci ranged from 0.06 to 0.39 kg/m2 (or ~0.17 to ~1.13 kg for an adult of ~170 cm in height) per risk allele, with the FTO gene having the largest effect size [16]. For the 14 novel WHR loci, the effect sizes varied from 0.019 to 0.042 units per risk allele [17], while the risk alleles for the new %BF loci were associated with an increase ranging from 0.14 to 0.33 % in body fat [18]. Remarkably, the combined effect of all obesity-associated variants is very modest and explains less then 2 % of the BMI heritability [16]. Since the heritability of BMI is estimated to be relatively high (between 40 % and 70 % [15•]), the major question is: what accounts for the missing heritability? Among the suggested explanations is that the modifying effects of environmental factors on genetic predisposition to obesity might partially account for the unexplained interindividual variation in BMI [19••].
Lifestyle Risk Factors for Obesity
Many specialists and scientists in the obesity research field agree that the dramatic increase in the prevalence of overweight and obesity over the past few decades is mainly attributable to the modern (Western) lifestyle, which is characterized by an excessive caloric intake and a sedentary lifestyle [1]. On the basis of many observational and epidemiologic studies, we currently know that the major environmental risk factors for obesity are unhealthy dietary habits (e.g. low in vegetables and fruits high in fat), decreased physical activity and alcohol consumption [20–25]. Overall and abdominal obesity show a negative association with such modifiable lifestyle habits as a Mediterranean-type diet, moderate alcohol consumption and daily physical activity [26]. However, all these well-established risk factors for obesity cannot explain a large proportion of the obesity cases, as there is a high interindividual susceptibility to weight gain in a common “obesogenic” environment. Thus, the most accepted point of view is that the modern obesity epidemic occurs due to a complex interplay between multiple genetic, behavioral and environmental factors. Recently, Speakman and colleagues [27•] suggested a new interesting model for body weight regulation to explain the mechanism underlying the current obesity epidemic. Briefly, this model suggests the presence of upper and lower boundaries defining the set points at which physiologic regulation of body weight and/or fatness becomes activated. While the distance between these intervention points is genetically determined, the changes in body weight depend on the prevailing direction of the environmental pressure (e.g. in the presence of an “obesogenic” environment with increased food supply driving up food intake, only some people become obese). The hypothesis provides a compelling explanation for the observed complexity of the obesity problem and integrates data and research from both the behavioral–nutritional and the molecular genetic–physiologic fields [27•].
Studies on GEI in Obesity
It is well recognized that the investigation of GEI in obesity etiology has not been given sufficient attention in genetic studies [19••]. The majority of GWAS, in particular, have not examined GEI, mainly due to lack of data on environmental measurements [28]. GEI refers to the situation in which genotypes only have their effect in the context of an environment and environments have modifying effects that are dependent on genotypes. In other words, in the presence of the “obesogenic” environment, some individuals with a genetic predisposition to develop obesity will be more prone to gain weight compared with individuals with genetic “resistance” to obesity [29]. A growing body of recent evidence supports a significant role of GEI in obesity and related metabolic diseases [30–32, 33••]. To provide an overview on the most current publications in relationship to GEI in obesity, we searched PubMed (February 15, 2012) using a combination of keywords for genetic studies (i.e. gene, genetic variant, polymorphism, SNP), different obesity-related phenotypes (i.e. overweight, obesity, BMI, waist, hip, WHR, fat, adiposity) and environmental factors (i.e. feeding, diet, physical activity, alcohol, smoking, stress). This retrieved 522 papers published since January 1, 2011, of which 29 were selected as the most relevant to the present review.
The selected papers examined 1) candidate genes for obesity known to play a role in the functional pathways related to metabolic regulation and 2) novel obesity-susceptibility loci identified in recent GWAS. Two approaches were used to investigate the relationship between those genes and different lifestyle factors, such as dietary components, eating habits, physical activity, sedentary behavior and psychological stress: observational and intervention studies. The observational studies are relatively easy to perform. As soon as environmental exposures and genotyping information are collected, the GEIs are examined using cross-sectional or case–control designs. However, the major limitation of these designs is their inability to identify the individual and combined effects of the genetic and lifestyle risk factors or, in other words, to answer the question of how genetic predisposition and behavior combine to determine the risk of obesity [34]. Moreover, the observational studies are susceptible to multiple sources of bias (e.g. selection or recall bias) because environmental exposure and the outcome of interest are assessed simultaneously. In contrast, intervention study designs allow minimization of bias and provide direct control of the environmental factors by defining the experimental conditions a priori (e.g. a specific diet or level of physical activity). However, because these studies are usually small and short term, they have low statistical power and are not appropriate for investigating long-term effects [32].
GEI Studies for Candidate Genes
Overall, the investigation of GEI for biological candidate genes has not been very successful and only a few findings for GEI in obesity were replicated in independent studies. This is the result of small effect sizes and very modest levels of significance for the majority of candidate genes proven to be associated with obesity [34]. Furthermore, as interaction effect sizes are likely to be of even smaller magnitude, many published small-scale reports of GEI for obesity were underpowered and, thus, are probably false positive [33••].
Since January 1, 2011, a few studies have reported GEI consistent with those from previous studies (Table 1 provides a summary of the most relevant GEI studies in obesity published during the past year) [30–32]. For example, variants in the β2-adrenergic receptor (ADRB2)- rs1042714 (Gln27Glu) and rs1042713 (Arg16Gly)- were associated with higher risk of obesity among the individuals with unhealthy lifestyles (i.e. smoking and reduced physically activity) [35, 36] and were shown to have a moderating effect on diet-induced changes on body weight and body composition [37]. Significant genotype–dietary fat interactions for obesity traits have also been reported for the apolipoprotein genes that regulate lipid metabolism (APOA1, APOA2, APOA5, APOB) [38–40], confirming previously observed GEI: the APOA2–saturated fat interaction on body weight and the protective effect of the APO5-1131 C minor allele on obesity in individuals on high-fat diets [39–41]. In addition, an association between APOE genotypes and increased BMI and WC dependent on psychological stress was reported in Danish men [42]. Also, the peroxisome proliferator-activated receptor-γ (PPARγ) gene, which has been extensively studied for GEI related to obesity and type 2 diabetes [32, 43], was reported to have a diet-related effect on risk to obesity with the Pro12 allele being associated with increased adiposity in a high-fat diet group [44]. In addition, the lactase (LCT) gene was shown to be associated with risk to obesity only in individuals who had high milk consumption [45]. Three additional observational studies investigated multiple candidate genes from metabolically relevant pathways: a few positive associations between genes, dietary components and obesity were observed (Table 1) [46–48].
Table 1.
Selected gene–environment interaction studies on obesity for candidate genes
| Gene (SNP) | Obesity phenotype | Lifestyle factor | Type of study | Population | Sample size | Major findings | Reference |
|---|---|---|---|---|---|---|---|
| ADRB2 (Gln27Glu Arg16Gly) | BMI, body fat mass and lean mass | Energy-restricted diet | Intervention study | Spanish obese women | 78 | In response to a 12-wk energy-restricted diet, women carrying the Glu allele had a greater reduction in body weight and lost more lean mass than the non-Glu allele carriers | Ruiz et al. [37] |
| ADRB2 (R16G) | BMI | Overeating, smoking, parent’s obesity | Observational, family-based study | Korean population | 163 adolescents with parents | Smoking parents who overate and carried the Arg allele had an increased risk of obesity compared with nonsmoking parents who had none of these factors | Lee et al. [35] |
| ADRB2, APOB, NOS3 | BMI, WC, BF%, VAT, SAT | PA, diet | Observational study | EA and AA adolescents (13–19 years) | 621 | Significant interactions were revealed between the ADRB2 Arg16Gly SNP and vigorous PA on VAT, SAT and WC, suggesting that Gly16 homozygotes may benefit less from increased PA to reduce their weight | Lagou et al. [36] |
| APOA1/C3/A4/A5 (12 SNPs) | BMI, WC | Dietary fat intake | Observational study | The Boston Puerto Rican Health Study | 821 | Significant interactions were observed between dietary fat intake and APOA1-75 in association with WC. Homozygotes for the common allele of APOA1-75, APOA4 N147S and APOA5 S19W had lower WC when consuming <31 % of total fat from energy than participants with the minor allele | Mattei et al. [39] |
| APOA1 (rs670), APOB (rs512535) | BMI, WC | Dietary fat intake | Observational, case–control study | The LIPGENE-SU.VI.MAX study | 1,754 | Risk of metabolic syndrome was modified by dietary fat intake, whereby the deleterious effects conferred by GG homozygosity for APOB and APOA1 were exacerbated among individuals consuming a high-fat diet, particularly high in MUFA. The GG homozygotes had greater BMI compared with A allele carriers and with the GG homozygotes with the lowest fat intake | Phillips [38] |
| APOA2 (−265 T > C) | BMI | Saturated fat intake | Observational study | Mediterranean and multiethnic Asian populations | 4,602 | In Mediterranean individuals, the CC genotype was associated with a 6.8 % greater BMI in those consuming a high saturated fat diet; also, the CC genotype was significantly associated with higher obesity prevalence in Chinese and Asian Indians only with a high saturated fat intake | Corella et al. [41] |
| APOA5 (−1131 T > C) | BMI | Dietary fat intake | Observational study | Spanish overweight and obese adults | 1,465 | In homozygotes for the -1131 T major allele, fat intake was associated obesity, whereas in those carrying the APOA5-1131 C minor allele, higher fat intakes were not associated with higher BMI. | Sanchez-Moreno et al. [40] |
| APOE (4 SNPs) | BMI, WC | Psychological stress | Observational study | Danish men | Obese (n = 475), controls (n = 709) | The APOE rs439401 TT-genotype was associated with an adverse metabolic profile in a population of psychologically stressed Danish men. The TT genotype was associated positively with BMI and WC in stressed men compared with those not similarly stressed | Iqbal Kring et al. [42] |
| IRS1 (rs2943641) | Weight loss | Weight loss diets | Clinical trial | POUNDS LOST trial: overweight adults | 738 | Individuals with the CC genotype might obtain more benefits in weight loss than those without this genotype by choosing a high-carbohydrate and low-fat diet | Qi et al. [62•] |
| LCT (−13910 C > T rs4988235) | BMI, WC | Milk and dairy product intake | Observational study | Spanish individuals at high CVD risk | 940 | The LCT variant was strongly associated with BMI and obesity and its effect was modulated by lactose. CC individuals had lower weight, BMI and WC than T-allele carriers. These associations were found to be significant only among those consuming moderate or high lactose intakes | Corella et al. [45] |
| LEP (5 SNPs) | BMI, weight loss | Low-fat, Mediterranean and low-carbohydrate diets | Intervention study | The 2-y Dietary Intervention Randomized Controlled Trial (DIRECT) | 322 | Dynamics in leptin concentrations combined with genetic variability in the LEP gene may be a predictor of a long-term weight regain following a dietary intervention | Erez et al. [49] |
| PLIN1 (11482 G > A, 13041A > G) | BMI, WC, body fat mass, lean mass | Energy-restricted diet | Intervention study | Spanish obese women | 78 | In response to a 12-wk energy-restricted diet, women carrying the 11482A allele had a lower reduction in WC than non–A allele carriers, suggesting that the PLIN1 11482 G > A variant plays a modulating role on diet-induced changes in body fat and energy metabolism in obese women | Ruiz et al. [50] |
| PPARγ (Pro12Ala rs1801282) | BMI, WC, hip, skinfolds | Fat intake, PA | Observational study | Greek children | n = 2,102 (1–6 y), n = 794 (10–12 y) | The data suggested an age-dependent gene–diet (SFA, TF) interaction: when taking into account the dietary fat intake, the Pro allele homozygotes are at higher risk of increased adiposity | Dedoussis et al. [44] |
| 38 candidate genes (1,444 SNPs) | BMI | Smoking, PA, alcohol consumption, dietary energy intake | Observational study | The Southern Community Cohort Study | 1,173 (AA) and 1,165 (Caucasians) | In AAs, significant interactions were observed between smoking and an SNP in ADIPOR1, alcohol consumption, and an SNP in PPARGC1A and dietary energy intake, and an SNP in CYP19A1. In AA, significant interactions were observed between PA and a SNP in ADIPOR1 | Edwards et al. [46] |
| 15 candidate genes (123 SNPs): the hypothalamic genes | BMI, weight change | Protein intake, dietary GI | Observational case-cohort study | European individuals (Italy, UK, The Netherlands, Germany, Denmark) | 5,584 | The data suggested that individuals carrying the NMB minor allele (G allele) for rs7180849 are more vulnerable to the deleterious effects of a high GI diet in terms of weight gain | Du et al. [48] |
| 21 candidate genes (187 SNPs): cytokines, adipokines, neurotransmitters and transcription factors | BMI, WC, hip | Polyunsaturated fatty acids | Observational study | The second Bavarian Food Consumption Survey | 568 | SNPs in IL-2, IL-6, IL-10, IL-18, TNF-α, TNFRSF1A, TNFRSF1B, TNFRSF21, NPY, NPY1R, NPY5R, MC4R, POMC, PPY, PPARγ, PPARγC1A, LEP, LEPR, ADIPOQ and RETN were genotyped. The obesity risk of minor allele carriers significantly decreased with increasing fatty acid content. A reduced obesity risk for minor allele carriers of most variants with high PUFA content in erythrocyte membranes correlated with dietary PUFA intake, except for the SNP of TNFRSF21, ADIPOQ, rs2069762 (IL-2), rs4833248 (IL-2 region 5′), and rs10242595 (IL-6 region 3′). With the latter genes, subjects homozygous for the major allele benefited from an increased PUFA content | Jourdan et al. [47] |
AA African American; BF% body fat percentage; BMI body mass index; CVD cardiovascular disease; EA European American; GEI gene–environment interaction; GI glycemic index; PA physical activity; PUFA polyunsaturated fatty acid; SAT subcutaneous fat; SFA saturated fatty acid; SNP single-nucleotide polymorphism; TF total fat; VAT visceral fat; WC waist circumference
In the past year, several intervention studies reported GEIs for variants in the leptin (LEP) [49] and the perilipin (PLIN1) genes [50] being associated with difference in weight loss in responses to calorie-restricted diets. In contrast, no evidence was found for the effect of variation in the melanocortin-3 receptor (MC3R) gene on weight loss after a 10-week dietary intervention with hypo-energetic diets in obese Europeans (n = 760) [51].
GEI Studies for GWAS Genes
The investigation of GEI for obesity-susceptibility loci identified in recent GWAS is thought to be a more useful strategy than the candidate gene approach. The power to detect GEIs for GWAS loci proven to be robustly associated with obesity is very likely to be higher because of the gene’s strengthened causal inference for an interaction [33••]. Among the recent publications examining GEIs of GWAS loci (Table 2), the majority of observational studies evaluated whether dietary components and physical activity interact with variation in the FTO gene for their effect on obesity [52–56]. While three studies observed that the effect of the FTO risk alleles for obesity was modulated by energy intake or physical activity [52–54], one study with a sample size of more than 6,000 individuals found no evidence for this GEI [55]. The issue was clarified by an impressively large meta-analysis that included data from 45 studies involving 218,166 adults and 9 studies comprising 19,268 children and adolescents [57••]. This meta-analysis confirmed that the minor allele of the FTO rs9939609 variant increases the risk of obesity in adults and showed that this risk was reduced among physically active individuals by 27 %. This interaction was more pronounced in North American than in European individuals. The investigators highlighted that their finding is highly relevant for public health implications at the population level (i.e. the individuals with a high genetic susceptibility to obesity can reduce their risk by living a physically healthy lifestyle) [57••]. In addition, a novel finding of the breastfeeding protective effect on the relationship between FTO variants and adiposity indices in Greek children from the ages of three upward has been published [58]. A further three papers reported the effect of lifestyle modifications on the relationship between several GWAS genes and obesity-related traits in observational [59] and intervention studies (Table 2) [60, 61].
Table 2.
Selected gene–environment interaction studies on obesity for GWAS genes
| Gene (SNP) | Obesity phenotype | Lifestyle factor | Type of study | Population | Sample size | Major findings | Reference |
|---|---|---|---|---|---|---|---|
| FTO (rs8050136) | BMI | PA, caloric intake | Observational study | Healthy Caucasian women | 21,675 | The effect of the A-risk allele on BMI was larger among inactive or higher intake women, with additive effects of inactivity and high intake on the associated genetic risk | Achmad et al. [52] |
| FTO (rs9939609, rs1121980) | BMI | PA, fat and carbohydrate intake | Observational study | GOLDN and the BPRHS studies | GOLDIN (n = 1,069), BPRHS (n = 1,094) | The SFA intake modulated the association between FTO and BMI. Homozygotes for the FTO-risk allele had higher BMI compared with those with other genotypes only with a high SFA intake. Also, a significant interaction between PA and FTO on BMI was found | Corella et al. [53] |
| FTO (rs9939609) | BMI | PA | Meta-analysis | 45 studies of adults, 9 studies of children and adolescents | 218,166 adults, 19,268 children and adolescents | The association of the FTO risk allele with the odds of obesity is attenuated by 27 % in physically active adults, highlighting the importance of PA, in particular in those genetically predisposed to obesity | Kilpelainen et al. [57••] |
| FTO (rs9939609) | Childhood obesity | Dietary fatty acid intake | Observational study | Spanish children and adolescents (6–18 y) | 354 | Consumption of >12.6 % SFA (of total energy) and an intake ratio <0.43 PUFA:SFA were associated with higher obesity risk in A-risk allele carriers than TT subjects | Moleres et al. [56] |
| FTO (rs9939609, rs17817449) | BMI, WC, skinfolds | Breastfeeding | Observational study | Greek children from the GENDAI and GENESIS studies; British children from the ALSPAC study | 1,138 Greek peri-adolescent, 2,374 Greek children 1–6 y, ALSPAC (n = 4,325) | A short period of at least 1 month of breastfeeding was associated with reduced obesity indices (WHR, BMI and skinfolds triceps) for the Greek children of different ages homozygous for the rare allele, indicating the breastfeeding protective effect under an obesogenic environment | Dedoussis et al. [58] |
| 16 obesity-susceptibility SNPs | Weight reduction | Weight loss–inducing interventions | Randomized controlled trial | The Diabetes Prevention Program: overweight/obese adults with IGT | 3,234 | GEI for short-term (6 month) and long-term (2 years) weight loss and weight regain (6 mo to study end). Gene–lifestyle interactions were observed for short-term (LYPLAL1; GNPDA2; MTCH2) and long-term (NEGR1; FTO) weight loss | Delahanty et al. [63•] |
| 8 obesity-susceptibility SNPs | BMI, SAT | Resistance training program | Intervention study | Young individuals | 796 | Men carrying the A allele for rs9939609 (FTO) lost a significant amount of subcutaneous fat with exercise. Women with a copy of the G allele for rs7498665 (SH2B1) showed less change of subcutaneous fat volume after exercise | Orkunoglu-Suer et al. [61] |
| 6 obesity-susceptibility genes (6 SNPs) | Childhood obesity | Sedentary behavior, PA | Observational study | Chinese children (6 − 18 y) | 2,848 (1,229 obese cases/1,619 controls) | A higher obesity risk was observed in children who carried the high-risk alleles of the 6 SNPs (in FAIM2, NPC1, FTO, MC4R, BDNF, GNPDA2) and engaged in sedentary behavior outside of school or participated in low or moderate PA. The association between 5 genes (FAIM2, NPC1, FTO, MC4R and BDNF) and obesity risk was only observed in children who had moderate to low PA or engaged in sedentary behavior, regardless of which risk alleles they carried | Xi et al. [59] |
AA, African American; BF% body fat percentage; BMI body mass index; BPRHS The Boston Puerto Rican Health Study; GOLDN The Genetics of Lipid Lowering Drugs and Diet Network study; CVD cardiovascular disease; EA European American; GEI gene–environment interaction; GWAS genome-wide association study; IGT impaired glucose tolerance; PA physical activity; PUFA polyunsaturated fatty acid; SAT subcutaneous fat; SFA saturated fatty acid; SNP single nucleotide polymorphism; TF total fat; VAT visceral fat; WC waist circumference
Among the recently published papers, two clinical trials reported GEI in response to weight loss interventions. Qi and colleagues [62•] found a novel association between the variant in the insulin receptor substrate 1 (IRS1) gene and response to a weight loss diet: 738 overweight adults (61 % were women) were randomly assigned to 1 of 4 diets varying in macronutrient contents for 2 years. Participants with the IRS1 rs2943641 CC genotype had greater weight loss and improvement of insulin resistance than those without this genotype in response to a high-carbohydrate/low-fat diet. Interestingly, the variant rs2943650 (r 2 = 1.00 with rs2943641) near IRS1 has been reported in a recent GWAS for percentage body fat with the fat percentage–decreasing allele being associated with (counterintuitively) higher levels of insulin resistance [18]. Another study, a randomized controlled trial in overweight and obese adults (n = 3,234), investigated the effect of 16 novel GWAS obesity-susceptibility variants on weight loss during a 2-year intervention program. The researchers reported gene–lifestyle interactions for short-term and long-term weight loss [63•]. Altogether, these novel findings provide supportive information for the development of effective dietary intervention strategies based on genetic background.
So far, only one study has examined whether the genetic predisposition to obesity risk assessed by a genetic risk score (GRS) was modified by lifestyle factors. A large-scale population-based study (n = 20,430) investigated the effect of a GRS calculated by summing 12 BMI-increasing alleles across the 12 genetic variants and its interaction with physical activity on obesity risk. The researchers found that the genetic risk of obesity was attenuated by 40 % in physically active individuals compared with physically inactive individuals [64••]. These results provide further evidence that particular individuals who are genetically predisposed to obesity would benefit more from elevated physical activity levels than individuals who are genetically protected. Importantly, these findings also indicate that GEIs might contribute to the unexplained variance in obesity traits and suggest that future GWAS of obesity-related traits studying, for example, physically inactive individuals may discover new obesity-susceptibility loci because the effect sizes of genetic variants may be more pronounced and, thus, more easily identified. To our knowledge, numerous consortia-based meta-analyses are ongoing in which this innovative genome–environment-wide association approach is deployed, but so far, no results of these studies have been published.
Machinery That Underlies GEI
Environment has inarguably a large impact on human physiologic functions and health. Despite the recent successes in identifying genetic determinants accounting for obesity, the definition and quantification of GEIs has proven difficult. Environmental exposure to nutritional and other stimuli can alter the expression of a subset of genes through changes in the epigenome [65]. Although little is known about the exact role of the epigenome in the pathophysiology of obesity, epigenetic regulation of gene expression may be a key factor explaining interindividual differences in adiposity-related phenotypes [66] and the study of the epigenome offers hope in understanding the machinery that underlies complex GEIs.
Epigenetics
Epigenetics is loosely defined as the study of heritable changes in gene function without modifications in DNA sequences [67]. Epigenetic changes include DNA methylation, chromatin folding and binding, packaging of DNA around nucleosomes and covalent modifications of the histone proteins that make up the nucleosomes around which the DNA double helix is coiled [68]. The epigenome varies across different cell types and undergoes precise, coordinated changes during a lifetime [69••, 70].
DNA methylation is a well-studied epigenetic modification that involves the addition of a methyl (CH3) group to a cytosine located next to a guanine nucleotide (CpG) in CpG dinucleotide–rich regions [70]. Methylation in promoters-associated CpG islands is associated with a transcriptionally repressed state established by two main mechanisms: inability of transcriptional factors to bind to their cognate sequence due to the presence of a methyl group within the binding site or the attraction of methyl-CpG-binding proteins with repressive properties [71, 72].
Environmental and Genetic Effects on the Epigenome in the Context of Obesity
There is increasing evidence of epigenetic regulation of metabolic diseases further supporting a link between genes and environment through their influences on the epigenome [73]. Periconceptional and gestational periods are particularly sensitive to epigenetic perturbation, with the environment exerting different effects on the placenta and embryo [69••]. In particular, nutrition at different developmental stages can influence the epigenome, potentially contributing to an increased susceptibility to chronic diseases, such as obesity [65, 66]. In mammals, early nutrition and in particular dietary components, such as folate, vitamin B6, vitamin B12, betaine, methionine and choline have been associated with changes in DNA methylation patterns by affecting the one-carbon metabolism that ultimately provides the methyl groups for DNA and histone methylation [69••]. Furthermore, maternal food supplementation with bisphenol A—a DNA hypomethylated compound that can leach from polycarbonate plastics into their contents—has been associated with decreased methylation at the Avy allele (the viable yellow agouti allele is a murine metastable epiallele that is variably expressed due to epigenetic marks established during early development) in the offspring and with obesity in early and later life in mammals [68, 74], whereas supplementation of maternal diet with folic acid or genistein negated the hypomethylating effects of BPA [68]. Maternal exposure to several other chemicals (the so-called obesogens) has been associated with increased BMI in offspring, further suggesting that obesity is being programmed prenatally or in early childhood and disruption of normal epigenetic regulation that alters the expression of key genes in adipogenic pathways is likely to be involved [75]. Nevertheless, our understanding of how environmental influence on epigenetic marks can lead to obesity remains rather rudimentary. The potential interaction of environment with the epigenome mediating the expression of genes associated with increased adiposity has also been suggested [76]. For example, the FTO gene encodes for an enzyme that is able to remove methyl groups from DNA [77], long-term exposure to high-fat diet can decrease the melanocortin-4 receptor (MC4R) gene methylation [78] and high-fat diet–induced obesity can modify leptin methylation patterns [79]. The expression of the PPARγ gene, a key regulator of adipocyte differentiation, was found to be reduced due to DNA methylation of its promoter in adipocytes of visceral adipose tissue in mammals [80]. Several other genes involved in adiposity have promoters that seem to be epigenetic targets in relation to obesity (epi-obesogenic genes) [66]. One of the first genome-wide methylation studies revealed increased methylation levels at one CpG site (UBASH3A gene) and decreased methylation levels at one CpG site (TRIM3 gene) in obese subjects compared with lean controls, providing evidence that obesity is associated with epigenetic changes [81•]. Although collectively, these studies could indicate that epigenetic marks lead to obesity, it is not really clear whether they predict or precede obesity [82••]. The causal relationship between epigenetic marks and obesity has yet to be elucidated and other factors, such as nutrition or physical activity, that correlate with both DNA methylation and increased adiposity should be considered to this end [82••]. Genetic differences between individuals can also influence epigenetic regulation [69••] and genetic variants could account for the locus-specific variance in epigenetic states. In humans, it has been shown that 10 % of common SNPs are located in regions with differences in the propensity for local DNA methylation between the two alleles [83]. With this in mind, it is possible that the interplay of genetics and epigenetics could underlie the establishment of diseases, such as obesity. However, the extent to which DNA sequence determines epigenetic changes at specific loci and subsequently leads to obesity is poorly understood. Evaluating the relative contribution of genetic and environmental factors to the establishment of the epigenome and elucidating the causal relationship between epigenetics and obesity constitute major challenges given the complex interrelationship of those determinants (Fig. 1).
Fig. 1.
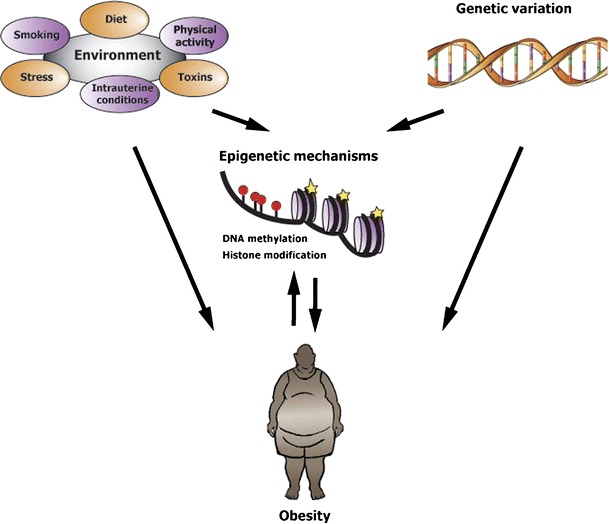
A model of the interplay between environmental/genetic factors and epigenetic changes in the establishment of obesity. Genes, environment, and epigenetic marks can directly lead to increased adiposity. Genes and environment can interact through their influence on the epigenome. Although epigenetic changes may cause obesity, it is often not really clear if they precede obesity, or vice versa
Transgenerational Epigenetic Inheritance
Currently, there is increased evidence that environmentally induced epigenetic changes can also be passed to the next generation via gametes and not only through the placenta in the developing embryo (maternal diet) or through breastfeeding in the infant [84]. Transgenerational epigenetic inheritance is also supported by the fact that some epigenetic marks escape reprogramming—that is, erasure and resetting of the gametic epigenome between generations [73]. This reprogramming escape, in combination with the observation that parental exposure to challenging environments, results in maladaptive responses that can be passed to the next generation renders trasngenerational epigenetic inheritance a mechanism of great interest, especially for obesity. A recent study in mice has examined the effect of a maternal exposure to a high-fat diet on body size, not only in the second generation (F2), but also in the third one (F3) in order to test whether the phenotype is transmitted by a germline-based epigenetic mark. Interestingly, the study has shown that the increased body size and length phenotypes were transmitted to F3 females through the F2 paternal lineage, suggesting that maternal high-fat diet programs a germline-based transgenerational phenotype in male gametes [85]. With this in mind, it is possible that environmentally induced epigenetic changes could in theory explain a significant fraction of the missing heritability for adiposity-related phenotypes by affecting both disease penetrance and heritability [86].
Challenges of GEI Studies
GEI studies can be very helpful in unraveling the biological pathways important for predicting disease risk and possibly in explaining some of the missing heritability through the identification of obesity-susceptibility genes that exert their effects through interaction with environment. Furthermore, GEIs could potentially be used for the identification of environmental factors that affect individuals with specific genotypes [19••]. However, the investigation of such interactions in complex diseases such as obesity remains a challenging task, with the major limitations including sample size/power, measurement of environmental factors, heterogeneity and lack of replication. Typically, thousands of samples are needed in candidate gene–based studies or even more in GWAS, in which very stringent cutoffs of significance are used. Not all reviewed studies have had a sufficient sample size to detect interactions and the lack of control for type 1 error continues to be a concern. In addition to that, accurate measurement of exposures that vary over time or are modifiable by other factors, such as time of exposure, has proven difficult and can create biases in the analysis. Another important issue is the observed heterogeneity in the study design that arises due to differences in the way that examined environmental exposures are assessed across studies and due to the possible study-specific characteristics of exposure [19••]. Recent efforts in the establishment of prospective cohorts (e.g. the National Children’s Study [http://www.nationalchildrensstudy.gov] and the Avon Longitudinal Study of Parents and Children [http://www.bristol.ac.uk/alspac/]) with robust and repeated measurements over time of environmental exposures can help in assessing the role of critical windows of susceptibility that likely correspond to the expression of specific genes. The challenges related to GEI studies were the topic of discussion in a recent workshop at which more than 150 researchers representing a wide range of scientific areas participated. Interesting questions were raised and useful recommendations were given regarding GEI study design for overcoming the aforementioned limitations. The need for integration of environment, genetics and epigenetics in the same study was also emphasized, as this could provide insight into their complex interactive role in the establishment of disease [87••]. In the post-GWAS era, the careful design of epidemiologic studies, accurate measurement of exposures and use of standardized methods across studies should facilitate collaborations, which will increase statistical power for assessing GEIs. With the completion of the Human Epigenome Project (http://www.epigenome.org/) [88], a more comprehensive picture of the genetic factors and epigenetic marks underlying cellular homeostasis will be achieved. Determination of disease-specific epigenetic changes and integration of this information with genetic and known environmental risks to obesity will provide more insights and will be proven valuable in predicting the onset and progress of obesity.
Conclusions
Obesity is a complex disease with multiple environmental and genetic causes. Over recent years, the GWAS experimental design has led to the identification of a number of obesity-susceptibility genes that, however, only explain a small portion of the interindividual variation in adiposity. Identifying the genes that predispose to obesity in combination with specific environmental exposures is very important for better understanding of disease etiology and subsequently for disease treatment and prevention. The investigation of gene–environment interplay can also unravel the pathways involved in obesity and be beneficial for drug development and therapy. To date, studies of GEIs have been facing challenges and, thus, are limited compared with those examining only main genetic or environmental effects. Furthermore, the contribution of the epigenome to the establishment of obesity is largely unknown. Further GEI studies that are carefully designed can extend the list of genetic loci that exert effects in the presence of specific environmental exposures. The next generation of studies incorporating genetic–environment–epigenome information and utilizing new analytical approaches and environmental measurement technologies can improve understanding of the complex causes of obesity.
Acknowledgments
Dr. van Vliet-Ostaptchouk is supported by a Rubicon grant from the Netherlands Organization for Scientific Research (NWO file no. 825.10.035) and the Netherlands Consortium for Healthy Ageing (NCHA) (NCHA NGI grant no. 050-060-810).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser. 2000;894:i–xii, 1–253. [PubMed]
- 2.Kelly T, Yang W, Chen CS, et al. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 2008;32:1431–1437. doi: 10.1038/ijo.2008.102. [DOI] [PubMed] [Google Scholar]
- 3.Erlichman J, Kerbey AL, James WP. Physical activity and its impact on health outcomes. Paper 2: prevention of unhealthy weight gain and obesity by physical activity: an analysis of the evidence. Obes Rev. 2002;3:273–287. doi: 10.1046/j.1467-789X.2002.00078.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Beydoun MA. The obesity epidemic in the United States–gender, age, socioeconomic, racial/ethnic, and geographic characteristics: a systematic review and meta-regression analysis. Epidemiol Rev. 2007;29:6–28. doi: 10.1093/epirev/mxm007. [DOI] [PubMed] [Google Scholar]
- 5.Ogden CL, Carroll MD, Curtin LR, et al. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 6.Hill JO. Understanding and addressing the epidemic of obesity: an energy balance perspective. Endocr Rev. 2006;27:750–761. doi: 10.1210/er.2006-0032. [DOI] [PubMed] [Google Scholar]
- 7.Prentice AM. Early influences on human energy regulation: thrifty genotypes and thrifty phenotypes. Physiol Behav. 2005;86:640–645. doi: 10.1016/j.physbeh.2005.08.055. [DOI] [PubMed] [Google Scholar]
- 8.Speakman JR. Obesity: the integrated roles of environment and genetics. J Nutr. 2004;134:2090S–2105S. doi: 10.1093/jn/134.8.2090S. [DOI] [PubMed] [Google Scholar]
- 9.Ravussin E, Bouchard C. Human genomics and obesity: finding appropriate drug targets. Eur J Pharmacol. 2000;410:131–145. doi: 10.1016/S0014-2999(00)00811-6. [DOI] [PubMed] [Google Scholar]
- 10.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scuteri A, Sanna S, Chen WM, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Vliet-Ostaptchouk JV, Hofker MH, van der Schouw YT, et al. Genetic variation in the hypothalamic pathways and its role on obesity. Obes Rev. 2009;10:593–609. doi: 10.1111/j.1467-789X.2009.00597.x. [DOI] [PubMed] [Google Scholar]
- 14.Rankinen T, Zuberi A, Chagnon YC, et al. The human obesity gene map: the 2005 update. Obesity (Silver Spring) 2006;14:529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- 15.Day FR, Loos RJ. Developments in obesity genetics in the era of genome-wide association studies. J Nutrigenet Nutrigenomics. 2011;4:222–238. doi: 10.1159/000332158. [DOI] [PubMed] [Google Scholar]
- 16.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heid IM, Jackson AU, Randall JC, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilpelainen TO, Zillikens MC, Stancakova A, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–760. doi: 10.1038/ng.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas D. Gene--environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroder H, Fito M, Covas MI, et al. Association of fast food consumption with energy intake, diet quality, body mass index and the risk of obesity in a representative Mediterranean population. Br J Nutr. 2007;98:1274–1280. doi: 10.1017/S0007114507781436. [DOI] [PubMed] [Google Scholar]
- 21.Romaguera D, Norat T, Mouw T, et al. Adherence to the Mediterranean diet is associated with lower abdominal adiposity in European men and women. J Nutr. 2009;139:1728–1737. doi: 10.3945/jn.109.108902. [DOI] [PubMed] [Google Scholar]
- 22.Leite ML, Nicolosi A. Lifestyle correlates of anthropometric estimates of body adiposity in an Italian middle-aged and elderly population: a covariance analysis. Int J Obes (Lond) 2006;30:926–934. doi: 10.1038/sj.ijo.0803239. [DOI] [PubMed] [Google Scholar]
- 23.Knoops KT, de Groot LC, Kromhout D, et al. Mediterranean diet, lifestyle factors, and 10-year mortality in elderly European men and women: the HALE project. JAMA. 2004;292:1433–1439. doi: 10.1001/jama.292.12.1433. [DOI] [PubMed] [Google Scholar]
- 24.Koh-Banerjee P, Chu NF, Spiegelman D, et al. Prospective study of the association of changes in dietary intake, physical activity, alcohol consumption, and smoking with 9-y gain in waist circumference among 16 587 US men. Am J Clin Nutr. 2003;78:719–727. doi: 10.1093/ajcn/78.4.719. [DOI] [PubMed] [Google Scholar]
- 25.Lahti-Koski M, Pietinen P, Heliovaara M, et al. Associations of body mass index and obesity with physical activity, food choices, alcohol intake, and smoking in the 1982–1997 FINRISK Studies. Am J Clin Nutr. 2002;75:809–817. doi: 10.1093/ajcn/75.5.809. [DOI] [PubMed] [Google Scholar]
- 26.Bullo M, Garcia-Aloy M, Martinez-Gonzalez MA, et al. Association between a healthy lifestyle and general obesity and abdominal obesity in an elderly population at high cardiovascular risk. Prev Med. 2011;53:155–161. doi: 10.1016/j.ypmed.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 27.Speakman JR, Levitsky DA, Allison DB, et al. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech. 2011;4:733–745. doi: 10.1242/dmm.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bouchard C. Gene-environment interactions in the etiology of obesity: defining the fundamentals. Obesity (Silver Spring) 2008;16(Suppl 3):S5–S10. doi: 10.1038/oby.2008.528. [DOI] [PubMed] [Google Scholar]
- 30.Ordovas JM, Shen J. Gene-environment interactions and susceptibility to metabolic syndrome and other chronic diseases. J Periodontol. 2008;79:1508–1513. doi: 10.1902/jop.2008.080232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreasen CH, Andersen G. Gene-environment interactions and obesity–further aspects of genomewide association studies. Nutrition. 2009;25:998–1003. doi: 10.1016/j.nut.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Qi L, Cho YA. Gene-environment interaction and obesity. Nutr Rev. 2008;66:684–694. doi: 10.1111/j.1753-4887.2008.00128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franks PW. Gene x environment interactions in type 2 diabetes. Curr Diab Rep. 2011;11:552–561. doi: 10.1007/s11892-011-0224-9. [DOI] [PubMed] [Google Scholar]
- 34.Wareham NJ, Young EH, Loos RJ. Epidemiological study designs to investigate gene-behavior interactions in the context of human obesity. Obesity (Silver Spring) 2008;16(Suppl 3):S66–S71. doi: 10.1038/oby.2008.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Kim CM, Kim HJ, et al. Interactive effects of main genotype, caloric intakes, and smoking status on risk of obesity. Asia Pac J Clin Nutr. 2011;20:563–571. [PubMed] [Google Scholar]
- 36.Lagou V, Liu G, Zhu H, et al. Lifestyle and socioeconomic-status modify the effects of ADRB2 and NOS3 on adiposity in European-American and African-American adolescents. Obesity (Silver Spring) 2011;19:595–603. doi: 10.1038/oby.2010.224. [DOI] [PubMed] [Google Scholar]
- 37.Ruiz JR, Larrarte E, Margareto J, et al. Role of beta-adrenergic receptor polymorphisms on body weight and body composition response to energy restriction in obese women: preliminary results. Obesity (Silver Spring) 2011;19:212–215. doi: 10.1038/oby.2010.130. [DOI] [PubMed] [Google Scholar]
- 38.Phillips CM, Goumidi L, Bertrais S, et al. Gene-nutrient interactions and gender may modulate the association between ApoA1 and ApoB gene polymorphisms and metabolic syndrome risk. Atherosclerosis. 2011;214:408–414. doi: 10.1016/j.atherosclerosis.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Mattei J, Demissie S, Tucker KL, et al. The APOA1/C3/A4/A5 cluster and markers of allostatic load in the Boston Puerto Rican Health Study. Nutr Metab Cardiovasc Dis. 2011;21:862–870. doi: 10.1016/j.numecd.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Moreno C, Ordovas JM, Smith CE, et al. APOA5 gene variation interacts with dietary fat intake to modulate obesity and circulating triglycerides in a Mediterranean population. J Nutr. 2011;141:380–385. doi: 10.3945/jn.110.130344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corella D, Tai ES, Sorli JV, et al. Association between the APOA2 promoter polymorphism and body weight in Mediterranean and Asian populations: replication of a gene-saturated fat interaction. Int J Obes (Lond) 2011;35:666–675. doi: 10.1038/ijo.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iqbal Kring SI, Barefoot J, Brummett BH, et al. Associations between APOE variants and metabolic traits and the impact of psychological stress. PLoS One. 2011;6:e15745. doi: 10.1371/journal.pone.0015745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palla L, Higgins JP, Wareham NJ, et al. Challenges in the use of literature-based meta-analysis to examine gene-environment interactions. Am J Epidemiol. 2010;171:1225–1232. doi: 10.1093/aje/kwq051. [DOI] [PubMed] [Google Scholar]
- 44.Dedoussis GV, Manios Y, Kourlaba G, et al. An age-dependent diet-modified effect of the PPARgamma Pro12Ala polymorphism in children. Metabolism. 2011;60:467–473. doi: 10.1016/j.metabol.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 45.Corella D, Arregui M, Coltell O, et al. Association of the LCT-13910 C > T polymorphism with obesity and its modulation by dairy products in a Mediterranean population. Obesity (Silver Spring) 2011;19:1707–1714. doi: 10.1038/oby.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards TL, Velez Edwards DR, Villegas R, et al. HTR1B, ADIPOR1, PPARGC1A, and CYP19A1 and obesity in a cohort of Caucasians and African Americans: an evaluation of gene-environment interactions and candidate genes. Am J Epidemiol. 2012;175:11–21. doi: 10.1093/aje/kwr272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jourdan C, Kloiber S, Nieters A, et al. Gene-PUFA interactions and obesity risk. Br J Nutr. 2011;106:1263–1272. doi: 10.1017/S0007114511001541. [DOI] [PubMed] [Google Scholar]
- 48.Du H, Vimaleswaran KS, Angquist L, et al. Genetic polymorphisms in the hypothalamic pathway in relation to subsequent weight change–the DiOGenes study. PLoS One. 2011;6:e17436. doi: 10.1371/journal.pone.0017436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erez G, Tirosh A, Rudich A, et al. Phenotypic and genetic variation in leptin as determinants of weight regain. Int J Obes (Lond) 2011;35:785–792. doi: 10.1038/ijo.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruiz JR, Larrarte E, Margareto J, et al. Preliminary findings on the role of PLIN1 polymorphisms on body composition and energy metabolism response to energy restriction in obese women. Br J Nutr. 2011;106:486–490. doi: 10.1017/S0007114511000432. [DOI] [PubMed] [Google Scholar]
- 51.Santos JL, De la Cruz R, Holst C, et al. Allelic variants of melanocortin 3 receptor gene (MC3R) and weight loss in obesity: a randomised trial of hypo-energetic high- versus low-fat diets. PLoS One. 2011;6:e19934. doi: 10.1371/journal.pone.0019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmad T, Lee IM, Pare G, et al. Lifestyle interaction with fat mass and obesity-associated (FTO) genotype and risk of obesity in apparently healthy U.S. women. Diabetes Care. 2011;34:675–680. doi: 10.2337/dc10-0948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corella D, Arnett DK, Tucker KL, et al. A high intake of saturated fatty acids strengthens the association between the fat mass and obesity-associated gene and BMI. J Nutr. 2011;141:2219–2225. doi: 10.3945/jn.111.143826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demerath EW, Lutsey PL, Monda KL, et al. Interaction of FTO and physical activity level on adiposity in African-American and European-American adults: the ARIC Study. Obesity (Silver Spring) 2011;19:1866–1872. doi: 10.1038/oby.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hubacek JA, Pikhart H, Peasey A, et al. FTO variant, energy intake, physical activity and basal metabolic rate in Caucasians. The HAPIEE study. Physiol Res. 2011;60:175–183. doi: 10.33549/physiolres.932066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moleres A, Ochoa MC, Rendo-Urteaga T, et al. Dietary fatty acid distribution modifies obesity risk linked to the rs9939609 polymorphism of the fat mass and obesity-associated gene in a Spanish case–control study of children. Br J Nutr. 2012;107:533–538. doi: 10.1017/S0007114511003424. [DOI] [PubMed] [Google Scholar]
- 57.Kilpelainen TO, Qi L, Brage S, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med. 2011;8:e1001116. doi: 10.1371/journal.pmed.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dedoussis GV, Yannakoulia M, Timpson NJ, et al. Does a short breastfeeding period protect from FTO-induced adiposity in children? Int J Pediatr Obes. 2011;6:e326–e335. doi: 10.3109/17477166.2010.490269. [DOI] [PubMed] [Google Scholar]
- 59.Xi B, Wang C, Wu L, et al. Influence of physical inactivity on associations between single nucleotide polymorphisms and genetic predisposition to childhood obesity. Am J Epidemiol. 2011;173:1256–1262. doi: 10.1093/aje/kwr008. [DOI] [PubMed] [Google Scholar]
- 60.Scherag A, Kleber M, Boes T, et al. SDCCAG8 obesity alleles and reduced weight loss after a lifestyle intervention in overweight children and adolescents. Obesity (Silver Spring) 2012;20:466–470. doi: 10.1038/oby.2011.339. [DOI] [PubMed] [Google Scholar]
- 61.Orkunoglu-Suer FE, Harmon BT, Gordish-Dressman H, et al. MC4R variant is associated with BMI but not response to resistance training in young females. Obesity (Silver Spring) 2011;19:662–666. doi: 10.1038/oby.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qi Q, Bray GA, Smith SR, et al. Insulin receptor substrate 1 gene variation modifies insulin resistance response to weight-loss diets in a 2-year randomized trial: the Preventing Overweight Using Novel Dietary Strategies (POUNDS LOST) trial. Circulation. 2011;124:563–571. doi: 10.1161/CIRCULATIONAHA.111.025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Delahanty LM, Pan Q, Jablonski KA, et al. Genetic predictors of weight loss and weight regain after intensive lifestyle modification, metformin treatment, or standard care in the diabetes prevention program. Diabetes Care. 2012;35:363–366. doi: 10.2337/dc11-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.•• Li S, Zhao JH, Luan J, et al. Physical activity attenuates the genetic predisposition to obesity in 20,000 men and women from EPIC-Norfolk prospective population study. PLoS Med. 2010;7. This is the first study to examine whether genetic predisposition to obesity risk assessed by a GRS is modified by lifestyle. The results of the analysis suggest that individuals who are genetically predisposed to obesity would benefit more from elevated physical activity levels. [DOI] [PMC free article] [PubMed]
- 65.Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–68. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 66.Campion J, Milagro FI, Martinez JA. Individuality and epigenetics in obesity. Obes Rev. 2009;10:383–392. doi: 10.1111/j.1467-789X.2009.00595.x. [DOI] [PubMed] [Google Scholar]
- 67.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 68.Dolinoy DC, Jirtle RL. Environmental epigenomics in human health and disease. Environ Mol Mutagen. 2008;49:4–8. doi: 10.1002/em.20366. [DOI] [PubMed] [Google Scholar]
- 69.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 70.Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–1350. doi: 10.1001/jama.299.11.1345. [DOI] [PubMed] [Google Scholar]
- 71.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 72.Van den Veyver IB. Genetic effects of methylation diets. Annu Rev Nutr. 2002;22:255–282. doi: 10.1146/annurev.nutr.22.010402.102932. [DOI] [PubMed] [Google Scholar]
- 73.Barres R, Zierath JR. DNA methylation in metabolic disorders. Am J Clin Nutr. 2011;93:897S–900S. doi: 10.3945/ajcn.110.001933. [DOI] [PubMed] [Google Scholar]
- 74.Rubin BS, Murray MK, Damassa DA, et al. Perinatal exposure to low doses of bisphenol a affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:675–680. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Janesick A, Blumberg B. Obesogens, stem cells and the developmental programming of obesity. Int J Androl. 2012. [DOI] [PMC free article] [PubMed]
- 76.Herrera BM, Keildson S, Lindgren CM. Genetics and epigenetics of obesity. Maturitas. 2011;69:41–49. doi: 10.1016/j.maturitas.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gerken T, Girard CA, Tung YC, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Widiker S, Karst S, Wagener A, et al. High-fat diet leads to a decreased methylation of the Mc4r gene in the obese BFMI and the lean B6 mouse lines. J Appl Genet. 2010;51:193–197. doi: 10.1007/BF03195727. [DOI] [PubMed] [Google Scholar]
- 79.Milagro FI, Campion J, Garcia-Diaz DF, et al. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem. 2009;65:1–9. doi: 10.1007/BF03165964. [DOI] [PubMed] [Google Scholar]
- 80.Fujiki K, Kano F, Shiota K, et al. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009;7:38. doi: 10.1186/1741-7007-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, Zhu H, Snieder H, et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010;8:87. doi: 10.1186/1741-7015-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Franks PW, Ling C. Epigenetics and obesity: the devil is in the details. BMC Med. 2010;8:88. doi: 10.1186/1741-7015-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hellman A, Chess A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin. 2010;3:11. doi: 10.1186/1756-8935-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet. 2012;13:153–162. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- 85.Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 2011;152:2228–2236. doi: 10.1210/en.2010-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bell JT, Saffery R. The value of twins in epigenetic epidemiology. Int J Epidemiol. 2012. [DOI] [PubMed]
- 87.•• Bookman EB, McAllister K, Gillanders E, et al. Gene-environment interplay in common complex diseases: forging an integrative model-recommendations from an NIH workshop. Genet Epidemiol. 2011. This is an important paper that discusses approaches for identifying genetic and/or environmental risk factors for complex disease, and formulates requirements and study design for the investigation of GEIs. [DOI] [PMC free article] [PubMed]
- 88.Eckhardt F, Beck S, Gut IG, et al. Future potential of the human epigenome project. Expert Rev Mol Diagn. 2004;4:609–618. doi: 10.1586/14737159.4.5.609. [DOI] [PubMed] [Google Scholar]