

Abstract
LynF, an enzyme from the TruF family, O-prenylates tyrosines in proteins; subsequent Claisen rearrangements give C-prenylated tyrosine products. These reactions in tyrosines and model phenolic systems have been explored with DFT and SCS-MP2 calculations. Various ab initio benchmarks have been computed (CBS-QB3, MP2, SCS-MP2) to examine the accuracy of commonly used density functionals, such as B3LYP and M06-2X. Solvent effects from water were considered using implicit and explicit models. Studies of the ortho-C-prenylation and Claisen rearrangement of tyrosine, and the Claisen rearrangement of α,α-dimethylallyl (prenyl) coumaryl ether establish the energetics of these reactions in the gas phase and in aqueous solution.
Keywords: aromatic Claisen, prenylation, DFT, CPCM
INTRODUCTION
Recent reports establish the likelihood that C-prenylation of tyrosines involves an enzyme-catalyzed O-prenylation followed by a spontaneous Claisen rearrangement.[1] Because of our interest in Claisen rearrangements, we undertook a computational exploration of both the O-prenylation and C-prenylation processes of tyrosine with the biological alkylating agent 1,1-dimethylallyl (prenyl) pyrophosphate (DMAPP). Schmidt and coworkers showed by both NMR and kinetic measurements that O-prenylation of tyrosine was likely to be the first step in enzymatic C-prenylation.[1] We have explored possible alkylation mechanisms and the subsequent aromatic Claisen rearrangement of the O-prenylated intermediates. We have also explored the design and testing of an unnatural enzyme to catalyze a related aromatic Claisen rearrangement of prenyl coumaryl ether. We describe the computational study of these reactions and the influence of water on their rates. These studies include extensive benchmark of different computational methods.
BACKGROUND
The [3,3]-sigmatropic rearrangement of allyl vinyl or aryl ethers is important in synthetic organic chemistry,[2] and in biosynthesis; represented by the chorismate-prephenate rearrangement.[2d, 2f, 3] Simple examples are shown in Scheme 1.[4] Substituent effects and the stereochemical outcome of these reactions,[2c] solvent effects on the rates of reaction,[3a, b, 5] and the enzyme catalysis of a Claisen rearrangement by chorismate mutase,[3c, 6] and by catalytic antibodies[7] have attracted much attention.
Scheme 1.
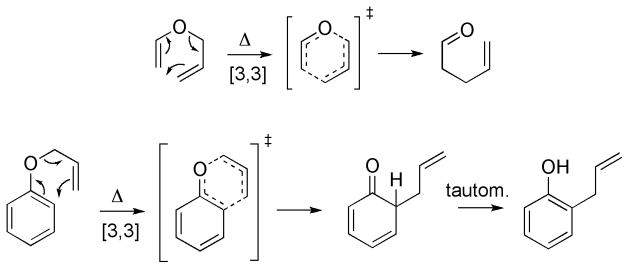
Claisen and aromatic Claisen rearrangements.
Solvent, particularly water, can lead to significant acceleration of the Claisen rearrangement.[2f, 3b, c, 5e, 8] After the first report by Carpenter in 1987,[3c] several theoretical calculations followed to identify the nature of the rate acceleration in water-based reactions.[3c, 5a, b, 5e, 8b, 9] Cramer and Truhlar proposed that aqueous acceleration is caused by electrostatic polarization and first-hydration-shell hydrophilic effects.[5a] Jorgensen investigated solvent effects on the chorismate to prephenate Claisen rearrangement: the reaction occurs 100-fold faster in water than methanol, which was attributed to greater stabilization of the transition state in water by specific interactions with first shell solvent molecules.[8b],[10] Severance and Jorgensen used quantum mechanical (QM) calculations to show that two water molecules hydrogen-bond with the ether oxygen atom of the transition state (TS) and accelerate the reaction.[5b] Density functional theory (DFT) calculations incorporating a polarizable continuum solvent model with two explicit solvent water molecules have been used to describe the decrease in the activation energy due to solvation.[11]
The aromatic Claisen rearrangement is typically performed in the temperature range 180–225°C.[4, 12] Sharpless observed that the rate of the aromatic Claisen rearrangement of an allyl naphthyl ether was accelerated in water relative to other organic solvents including toluene, dimethylformamide, acetonitrile and methanol.[13] Substantial rate accelerations were reported when insoluble reactants were stirred in aqueous suspensions, which has been denoted as “on water” conditions.[13] Zhang et al. investigated recently the reaction process and rate accelerations of the aromatic Claisen rearrangement on oil droplet/water interface.[14]
Many groups, especially Curran, Jacobsen and Kozlowski have studied hydrogen-bonding catalysis and have designed dual hydrogen-bonding organocatalysts such as ureas and thioureas,[15] positively charged catalysts based on guanidinium,[16] quinolinium thioamide,[17] and ammonium[2c, 5e] compounds. Jacobsen reported a diphenylguanidinium catalyst that promoted the Claisen rearrrangement of several substituted allyl vinyl ethers and β-ketoester derivatives.[16, 18] DFT calculations indicated that catalysis is achieved by the diphenylguanidinium catalyst through stabilization of the developed negative charge on the oxallyl fragment and to a secondary attractive interaction between the cationic allyl fragment and the π-system of the organocatalyst.[19] Kozlowski designed a bisamidinium catalyst salt for a Claisen rearrangement.[20]
The [3,3]-sigmatropic rearrangement has been observed in primary metabolism[2f] and may be catalyzed by enzymes.[21] Chorismate mutase is an excellent example, which accelerates the chorismate to prephenate rearrangement (see Scheme 2a) more than a millionfold.[6a, 6c, 9, 22] Both Cope and Claisen [3,3]-sigmatropic rearrangements in chorismate mutase have been computationally investigated.43, 59–65 Recently, the prenyltransferase LynF, from the TruF enzyme family, was characterized.[1] This enzyme is responsible for O-prenylation of tyrosine, serine and threonine in cyclic peptides. Schmidt et al. observed that O-prenylated tyrosine derivatives undergo facile Claisen rearrangement at physiological temperature (37°C) in aqueous buffers (see Scheme 2b).
Scheme 2.

Biological examples of Claisen rearrangements: a) reaction catalyzed by chorismate mutase and b) O-prenylation and subsequent aromatic Claisen rearrangement catalyzed by LynF.
Our group has been recently involved in the de novo computational design of a biocatalyst for the Claisen rearrangement of prenyl coumaryl ethers. We have used the same inside-out protocol as employed in the Kemp elimination,[23] retro-Aldol[24] and Diels-Alder cases.[25] We now report studies of the aromatic Claisen rearrangement in a series of reactions. First, a computational benchmark using different methodologies is reported for two prototypical Claisen and aromatic Claisen reactions for which the activation enthalpy is known. Second, the Claisen rearrangement of O-prenylated tyrosine, i.e. the mechanism by which TruF catalyzes the prenylation and the subsequent Claisen rearrangement, is studied in detail. Third, we studied the effect of implicit and explicit solvation in several aromatic Claisen rearrangements.
COMPUTATIONAL METHODOLOGY
All geometry optimizations were performed with Gaussian 09.[26] Optimizations of reactant, transition structure and product geometries were carried out with both B3LYP[27] and M06-2X39,40 with the 6-31G(d) basis set.[28] Frequency calculations were used to characterize the stationary points as minima or transition state structures. The transition states were also characterized by IRC calculations.[29] Single point energies with a variety of methods were calculated with B3LYP and PBE0[30] (also referred to as PBE1PBE) hybrid GGAs, the M06-2X hybrid-meta GGA and the B2PLYP[31] double hybrid functional (incorporating GGA exchange-correlation and second-order perturbative correlation) with the 6-311++G(d,p) basis set. Single point energy calculations with MP2[32] and the spin component scaled SCS-MP2[33] ab initio methods were also performed at the optimized B3LYP/6-31G(d) geometry, with correlation consistent cc-pVQZ[34] and cc-pVTZ[35] basis sets. Free energies were computed at 298K using unscaled zero point vibrational energies, unless otherwise specified. The effects of solvation on the reaction energetics were evaluated using a conductor-like polarizable continuum solvation model (CPCM).[36] The CBS-QB3 composite ab initio method, which is a five-step method starting with a B3LYP/6-311G(2d,d,p) geometry optimization and frequency calculation, followed by CCSD(T), MP4SDQ, and MP2 single-point calculations and a CBS extrapolation was used to benchmark calculations.[37] Of particular relevance to this study, the CBS-QB3 method has been found to give activation energies for a set of hydrocarbon pericyclic reactions with a mean absolute error of 2.3 kcal/mol. [38]
RESULTS AND DISCUSSION
1. Experimental and Computational Benchmarks
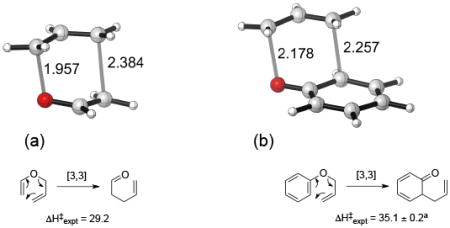
To gauge the performance of DFT calculations against more demanding ab initio computational methods, we first studied the prototypical Claisen and aromatic Claisen reactions of allyl vinyl ether and allyl phenyl ether, respectively. For these relatively small substrates we were able to compute the activation free energy barrier in vacuo using high-accuracy composite CBS-QB3 and SCS-MP2 calculations.
CBS-QB3 barrier heights yield sub kcal/mol accuracy with regard to the experimental (gas-phase) barriers (the barrier is underestimated by 0.8 and 1.0 kcal/mol in the Claisen and aromatic Claisen rearrangements, respectively, see Table 1). DFT functionals are well suited to study of the Claisen and aromatic Claisen rearrangement. B3LYP underestimates the activation barrier by 2 kcal/mol or less with 6-31G(d), whereas M06-2X overestimates ΔH‡ by ca. 3 kcal/mol. The hybrid GGA functional PBE0 gives the most accurate activation energies among DFT functionals. As is commonly observed, MP2 underestimates the barrier heights relative to all the other methods examined here (ca. −4 kcal/mol deviation respect to the experimental value), while the spin-component-scaled version of MP2 at the B3LYP optimized geometry gives values ca 1.5 kcal/mol above the experimental enthalpy. The SCS-MP2//M06-2X activation enthalpies are highly accurate, with a deviation respect to the experimental barrier of only ca. 0.3 kcal/mol. The B2PLYP functional also suffers from the same problems as MP2, underestimating the barrier height by ca. 3 kcal/mol. Taking CBS-QB3 and the experimental energy as a benchmark value, SCS-MP2 and PBE0 perform well in relation to the other methods tested. In a previous study, Grimme and coworkers performed a statistical assessment of quantum methods for describing the isomerization energies of a series of organic compounds.[40] The aromatic Claisen rearrangement was included in the set, and similarly to what is found here, SCS-MP2 was the most accurate method, followed by PBE0-D. Some of us have recently highlighted the importance of dispersion corrections in cycloaddition reactions.[41] We should mention here that in the case of the Claisen and aromatic Claisen rearrangements the introduction of dispersion corrections (D3)[42] yields larger errors on the activation enthalpies compared to the experimental barrier (see Supporting Information).
Table 1.
Enthalpy of activation at 298K for (a) prototypical Claisen and (b) aromatic Claisen rearrangements, showing optimized transition state geometries. Stationary points are optimized at the B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p) levels, and thermal and ZPE contributions to the free energy are computed at this level. For comparison, optimized B3LYP/6-31G(d) activation enthalpies are also included. Experimental (gas-phase values) for allyl vinyl ether and p-tolyl allyl ether used for comparison.[5c, 39]
|
|
||||
|---|---|---|---|---|---|
|
|
|
||||
| Error | Error | ||||
|
|
|
||||
| CBS-QB3a | 28.4 | −0.8 | CBS-QB3a | 34.1 | −1.0 |
| MP2/cc-pvQZb | 25.0 | −4.2 | MP2/cc-pvQZb | 32.1 | −3.0 |
| MP2/cc-pvQZc | 23.6 | −5.6 | MP2/cc-pvQZc | 29.7 | 0.5 |
| SCS-MP2/cc-pvQZb | 30.6 | 1.4 | SCS-MP2/cc-pvQZb | 36.7 | 1.6 |
| SCS-MP2/cc-pvQZc | 29.5 | 0.3 | SCS-MP2/cc-pvQZc | 34.9 | −0.2 |
| B3LYP/6-311++G(d,p) | 26.9 | −2.3 | B3LYP/6-311++G(d,p) | 33.5 | −1.6 |
| B3LYP/6-31G(d) | 27.2 | −2.0 | B3LYP/6-31G(d) | 34.8 | −0.3 |
| PBE0/6-311++G(d,p)b | 28.3 | −0.9 | PBE0/6-311++G(d,p)b | 34.9 | −0.2 |
| M06-2X/6-311++G(d,p)b | 31.8 | 2.6 | M06-2X/6-311++G(d,p)b | 38.7 | 3.6 |
| M06-2X/6-311++G(d,p) | 31.2 | 2.0 | M06-2X/6-311++G(d,p) | 37.4 | 2.3 |
| B2PLYP/6-311++G(d,p)b | 26.4 | −2.8 | B2PLYP/6-311++G(d,p)b | 32.0 | −3.1 |
|
|
|
||||
Experimental value for allyl p-tolyl ether[39]
Single point calculation at the B3LYP/6-311++G(d,p) optimized geometry, thermal corrections at B3LYP/6-311++G(d,p),
Single point calculation at the M06-2X/6-311++G(d,p) optimized geometry, thermal corrections at 6-311++G(d,p).
The aromatic Claisen reaction is computed to be slower than the alkylation reaction, as is found experimentally.[5c, 39] The larger barrier arises from a loss in aromaticity in the aromatic-Claisen TS. The difference in activation barrier between the two reactions is similar for all levels examined, with the aromatic Claisen rearrangement 5.6–7.1 kcal/mol higher in free energy, corresponding to a difference in rate constants between the two transformations of 4–5 orders of magnitude at 298K.
SCS-MP2 gives highly accurate activation barriers, especially at the M06-2X optimized geometries. In this study, reactants, transition states and products were optimized at B3LYP and M06-2X levels of theory and single point SCS-MP2 calculations at the optimized M06-2X geometry were performed.
2. Study of the Aromatic Claisen rearrangement in LynF
Prenyltransferases (PT) are enzymes responsible for catalyzing prenyl transfer to many systems. In general, PT catalyze electrophilic alkylation or electrophilic aromatic substitution in the case of the aliphatic and aromatic substrates. However, indirect mechanisms have also been proposed. C-prenylated phenols might also arise via O-prenylation followed by a Claisen rearrangement.[1, 43] NMR and kinetic studies showed that the sole enzymatic reaction catalyzed by LynF corresponds to O-prenylation on the tertiary terminus, followed by a spontaneous Claisen rearrangement to the C-prenylated phenol (pathway I in Scheme 3).[1] The rate constant for the Claisen rearrangement was estimated to be 0.23 h−1. The related O-allyl-Boc-L-tyrosine does not undergo the Claisen rearrangement in water at room temperature (see Scheme 4). This lack of reactivity is attributed to the gem-dimethyl substituent effect,[44] which accelerates the Claisen rearrangement. In addition, the stabilization of the partial positive charge on the allyl moiety in the transition state will be facilitated.
Scheme 3.
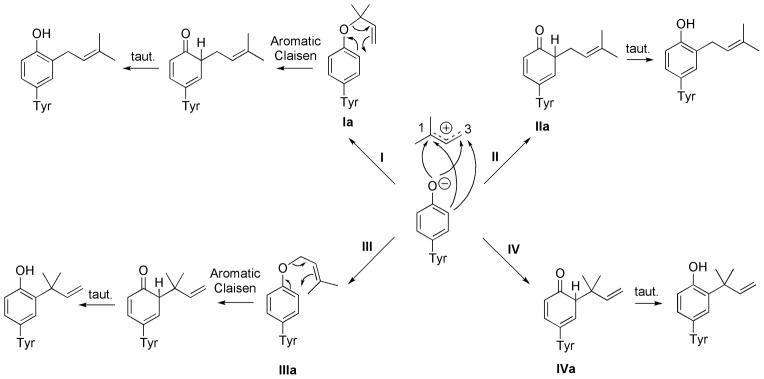
Four possible enzymatic mechanisms of tyrosine o-C-prenylation.
Scheme 4.
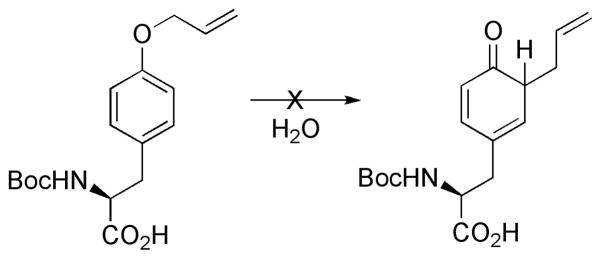
O-allyl-Boc-L-tyrosine does not undergo the Claisen rearrangement in water.
We explored the first step of the mechanism, where dimethylallyl pyrophosphate (DMAPP) acts as an alkylating agent reacting either at carbon-3 (pathways I and III) or carbon-1 (pathways II and IV). In some cases, an SN2 nucleophilic displacement is proposed with the diphosphate as a good leaving group, in others an SN1 process can operate where the DMAPP ionizes first giving the resonance-stabilized allylic carbocation.[45]
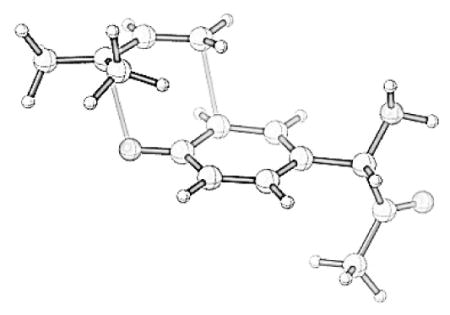
The enzymatic prenylation presumably involves deprotonation of the phenol of the tyrosine residue and strong pyrophosphate binding to facilitate ionization. These processes are not easy to model well in the absence of protein, and so we just explored the energies of each of the four possible prenylations in Scheme 3. Figure 1 shows the products of the reaction and the relative energies of the four reaction products. The two most stable products, Ia and IIIa correspond to the proposed products of O-prenylation. The Claisen rearrangement of IIIa is not expected to occur under the experimental conditions, since this is 6 kcal/mol lower in energy than Ia, and the rearrangement of IIIa would involve substantial steric hindrance. O-prenylation is thermodynamically favored because the aromaticity of the phenol is preserved. [44]
Figure 1.
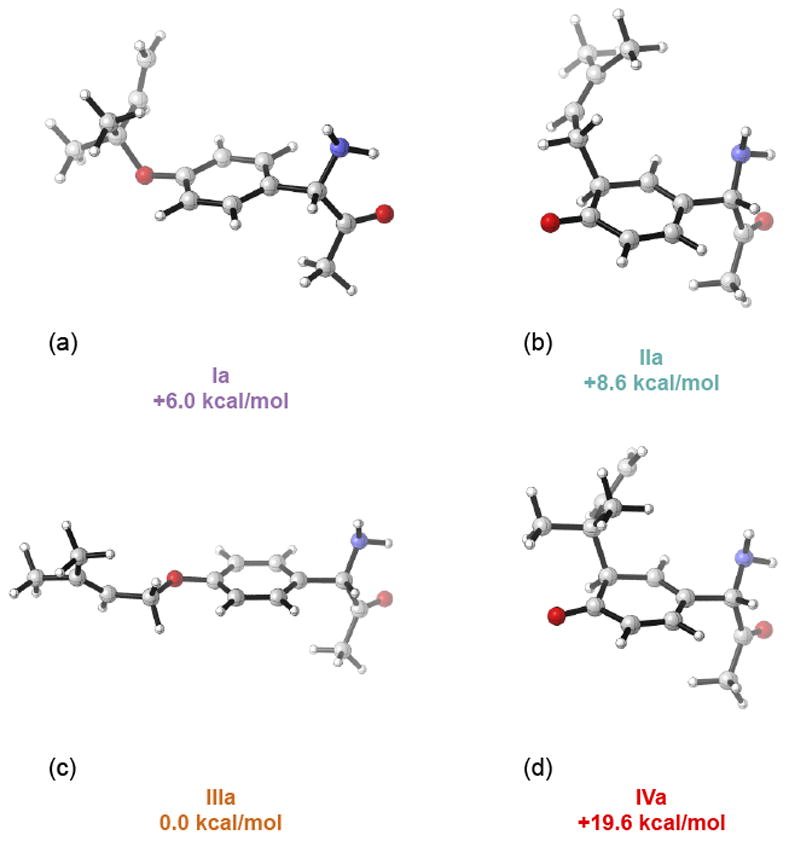
B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) optimized structures of Ia, IIa, IIIa and IVa.
After the formation of O-prenylated tyrosine at the more substituted terminus, the aromatic Claisen rearrangement is found experimentally to occur under physiological conditions. As can be seen in Table 2, the computed Gibbs free energies in gas phase are quite large. The SCS-MP2/cc-pVTZP//B3LYP/6-31G(d) calculations, which in the previous section yielded good estimates of the experimental barrier, predict a Gibbs free energy barrier for the aromatic Claisen of 32.9 kcal/mol. The activation barrier computed at B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) and M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) is 27.3 and 34.5 kcal/mol, respectively. Again, compared to the SCS-MP2 prediction, we observe an under or over estimation of the barrier with B3LYP and M06-2X, respectively. The activation barrier with the DFT functional that yielded the lowest error for the aromatic Claisen rearrangement (see Table 1) PBE0/6-311+G(d,p)//B3LYP/6-31G(d) is 29.4 kcal/mol. Using the corrections from Table 1, our best estimate of the barrier is 30 ± 1 kcal/mol. The corresponding rate constant is 1.6×10−6 s−1. This is not consistent with a facile Claisen rearrangement at physiological temperatures. However, as already pointed out in the introduction, water does significantly accelerate the aromatic Claisen rearrangement of prenyl aryl ethers.[13]
Table 2.
Computed free activation energies for the Claisen rearrangement of O-prenylated tyrosine at 310K including 0 and 2 explicit water molecules, and without or with CPCM corrections.
| number of explicit waters | |||
|---|---|---|---|
|
|
|||
| method/basis | 0 | 2 | |
| Gas Phase | B3LYP/6-311++G(d,p)a | 27.3 | 18.5 |
| B3LYP/6-31G(d) | 29.2 | 19.9 | |
| PBE0/6-311++G(d,p)a | 29.4 | 21.0 | |
| M06-2X/6-311++G(d,p)b | 34.5 | 26.5 | |
| SCS-MP2/cc-pVTZPb | 32.2 | 26.0 | |
| MP2/cc-pVTZPb | 27.4 | 21.4 | |
|
| |||
| CPCM | B3LYP/6-311++G(d,p)a | 24.1 | 17.5 |
| B3LYP/6-31G(d) | 26.8 | 19.5 | |
| PBE0/6-311++G(d,p)a | 26.7 | 20.4 | |
| M06-2X/6-311++G(d,p)b | 32.1 | 26.3 | |
| SCS-MP2/cc-pVTZPb | 30.9 | 26.4 | |
| MP2/cc-pVTZPb | 26.4 | 22.0 | |
Single point calculation at the B3LYP/6-31G(d) optimized geometry, thermal corrections at B3LYP/6-31G(d),
Single point calculation at the M06-2X/6-31G(d) optimized geometry, thermal corrections at M06-2X/6-31G(d).
Consequently, we have explored the influence of water on the rate of the reaction. We used a conductor-like polarizable continuum model (CPCM).82,83,100 The activation barriers predicted for water are lowered by approximately 2–3 kcal/mol in the case of B3LYP, M06-2X, PBE0, and SCS-MP2 (see Table 2). The activation barriers were also recomputed optimizing the system with two explicit waters. As mentioned in the introduction, this strategy of including two explicit waters to describe the decrease in the activation energy due to solvation has been used before.[11] The effect of including two explicit water molecules resulted in a barrier lowering of the the Claisen rearrangement of allyl vinyl ether of 6.3 kcal/mol. The experimental barrier lowering was found to be 3.5–4.7 kcal/mol, whereas PCM calculations gave a somewhat smaller barrier lowering of 0.6 kcal/mol. The new Gibbs free activation barriers including two water molecules in gas phase are approximately 8–9 kcal/mol lower in the case of B3LYP, PBE0, and M06-2X. The effect with SCS-MP2 is smaller with a reduction of the activation barrier of 6.2 kcal/mol. As found in Guest et al. study,[11] the activation barrier including two water molecules and also including CPCM bulk solvent effects are further reduced slightly. Taking into account systematic errors with different methods, we expect that the experimental value will be 21± 2 kcal/mol or 9 kcal/mol below the gas phase barrier. These calculations indicate that the use of explicit solvation is necessary to correctly account for the charge stabilization of the transition state of the reaction and demonstrated a very large effect of water on the rate of reaction. Even though the reported spontaneous Claisen rearrangement of O-prenylated peptides might seem surprising given the synthetic literature, these relatively low activation barriers computed in this study are indicating that the facile Claisen rearrangement is possible under the reported conditions (the calculated rate constant is 9.7×10−3 s−1 at 310 K).
In Figure 2, the B3LYP/6-31G(d) and M06-2X/6-31G(d) optimized transition state structures for the Claisen rearrangement of O-prenylated tyrosine are depicted. The B3LYP TSs are substantially more dissociative than the M06-2X TSs, especially for those cases where explicit solvation has been taken into account. For instance, CPCM calculations including two water molecules show that the C-C forming bond distances are 2.39 and 2.26 Å and the O-C breaking distances are 2.51 and 2.26 Å for B3LYP and M06-2X, respectively. The inclusion of two water molecules in the calculations leads to increases of both O-C breaking and C-C forming distances (ca. 0.15–0.20 Å and ca. 0.1 Å increase of the O-C and C-C distances, respectively). The hydrogen bond distances for the two explicit waters that stabilize the developed negative charge of the ether oxygen are ca. 1.79 Å and 1.81 Å in B3LYP and M06-2X calculations.
Figure 2.

B3LYP/6-31G(d) and M06-2X/6-31G(d) optimized structures in gas phase (a, c) and in CPCM water (b, d) including 0 and 2 explicit water molecules. Gibbs-free activation energies and distances are expressed in kcal/mol and Å, respectively. M06-2X values are represented in pink.
In summary, the O-prenylated tyrosine can undergo rapid rearrangement in water, or in a suitable environment involving hydrogen-bond donors situated to stabilize the partial negative charge on the ether oxygen in the transition state. In a nonpolar media, this rearrangement is predicted to be very slow at room temperature.
3. Study of the Aromatic Claisen rearrangement of prenyl coumaryl ether
Our group has been intensively involved in the de novo computational design of an aromatic Claisen biocatalyst. Using the so-called inside-out protocol,[23–25] we designed several enzymes to catalyze the aromatic Claisen rearrangement for the substrate shown in Scheme 5. However, during the course of the experiments, it became clear that the background reaction rate for the original substrate 1 was significantly faster than predicted by quantum mechanical calculations. In light of the results obtained for the Claisen rearrangement in O-prenylated tyrosine and the previous studies by Severance and Jorgensen, we explored the effect of implicit and explicit solvation in the coumarin 1.
Scheme 5.
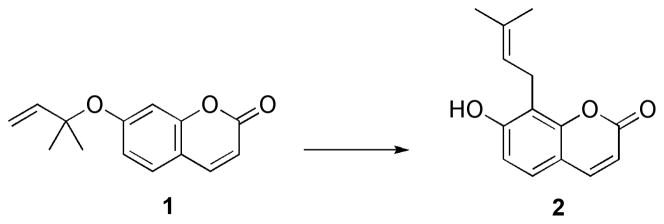
The aromatic Claisen rearrangement of prenyl coumaryl ether (1).
Table 3 lets the computational barrier heights calculated with different methods and numbers of explicit water molecules. We computed barriers with explicit water molecules in the gas phase, intended to mimic the effects of hydrogen bonding on TS stabilization, and with explicit water molecules surrounded by a continuum dielectric to mimic both hydrogen bonding and polarization/electrostatic interactions with bulk solvent. It can be seen that in the gas phase, the effect of adding an additional water molecule is to reduce the barrier as each additional hydrogen bond strengthens in the TS.[38a] However, it seems that for calculations modeling one explicit water and bulk solvation effects, the barrier height is substantially reduced by 3–5 kcal/mol (averaging 4 kcal/mol). The effect of adding a second water molecule is less profound with a decrease of 0–4 kcal/mol (averaging 2 kcal/mol). These results suggest that two explicit waters are necessary to capture the “missing” hydrogen bonding terms from a continuum dielectric model.
Table 3.
Computed activation free energies (in kcal/mol) including 0, 1 or 2 explicit water models for the prenyl coumaryl ether Claisen rearrangement. All energies are expressed in kcal/mol.
| number of explicit waters | ||||
|---|---|---|---|---|
|
|
||||
| method/basis | 0 | 1 | 2 | |
| Gas Phase | B3LYP/6-311++G(d,p)a | 25.1 | 19.7 | 15.9 |
| B3LYP/6-31G(d) | 27.1 | 21.3 | 17.5 | |
| PBE0/6-311++G(d,p)a | 27.4 | 22.0 | 18.9 | |
| M06-2X/6-311++G(d,p)b | 32.1 | 27.2 | 24.4 | |
| SCS-MP2/cc-pVTZPb | 30.8 | 26.9 | 25.0 | |
| MP2/cc-pVTZPb | 26.0 | 22.1 | 20.7 | |
|
| ||||
| CPCM | B3LYP/6-311++G(d,p)a | 21.7 | 17.0 | 14.2 |
| B3LYP/6-31G(d) | 24.6 | 19.5 | 16.4 | |
| PBE0/6-311++G(d,p)a | 24.6 | 20.0 | 17.4 | |
| M06-2X/6-311++G(d,p)b | 29.2 | 25.7 | 23.8 | |
| SCS-MP2/cc-pVTZPb | 29.2 | 26.3 | 25.0 | |
| MP2/cc-pVTZPb | 24.9 | 22.0 | 21.2 | |
Single point calculation at the B3LYP/6-31G(d) optimized geometry, thermal corrections at B3LYP/6-31G(d),
Single point calculation at the M06-2X/6-31G(d) optimized geometry, thermal corrections at M06-2X/6-31G(d)
Figure 3 shows B3LYP and M06-2X optimized geometries of the transition state of prenyl coumaryl ether (1) with none, one, and two explicit waters. Explicit solvent models were simulated at the quantum mechanical level by studying specific interactions in which one or two water molecules are hydrogen bonded to the ether oxygen atom. There is a progressive increase of the O-C breaking and C-C bonding distances when 1 and 2 water molecules are included in the calculations. The hydrogen bond distances when two water molecules are included in the calculations are ca. 1.80 and 1.83 Å.
Figure 3.

B3LYP/6-31G(d) and M06-2X/6-31G(d) optimized geometries of the transition state of prenyl coumaryl ether with (a) none, (b) one, (c) and two explicit waters optimized in gas phase (left column) and in CPCM water (right column). B3LYP distances are represented in angstroms and in black and M06-2X/6-31G(d) distances using pink color. B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p) activation free energies are represented in kcal/mol and using the same color scheme.
CONCLUSION
Models for the LynF, the enzyme that prenylates tyrosine residues in macrocyclic peptides, was explored computationally. The initial prenylation is found to lead to the most stable O-prenylated products, and subsequent Claisen rearrangement can occur in a hydrogen-bond or aqueous environment. Explicit solvation was crucial for activation barriers for the process in water. Significant contributions from H-bonding between protic solvents and the TS result in barrier lowering, which are not captured adequately by implicit solvation models. These effects have been observed in two biologically revelant aromatic Claisen rearrangements: the o-C-prenylation of tyrosine in LynF and the prenyl coumaryl ether used in our computationally designed aromatic Claisen biocatalyst.
Supplementary Material
Acknowledgments
We are grateful to the National Institute of General Medical Sciences, the National Institutes of Health Grant (GM-36700) and Defense Advanced Research Projects Agency for financial support of this research. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575. S.O. acknowledges the European Community for the postdoctoral fellowship PIOF-GA-2009-252856.
Footnotes
Supporting Information. Optimized Cartesian xyz coordinates (in Å) of all analyzed species.
References
- 1.McIntosh JA, Donia MS, Nair SK, Schmidt EW. J Am Chem Soc. 2011;133:13698–13705. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Lutz RP. Chem Rev. 1984;84:205–247. [Google Scholar]; b) Ziegler FE. Acc Chem Res. 1977;10:227–232. [Google Scholar]; c) Ziegler FE. Chem Rev. 1988;88:1423–1452. [Google Scholar]; d) Enders D, Knopp M, Schiffers R. Tetrahedron: Asymmetry. 1996;7:1847–1882. [Google Scholar]; e) Ito H, Taguchi T. Chem Soc Rev. 1999;28:43–50. [Google Scholar]; f) Majumdar KC, Alam S, Chattopadhyay B. Tetrahedron. 2008;64:597–643. [Google Scholar]
- 3.a) Coates RM, Rogers BD, Hobbs SJ, Curran DP, Peck DR. J Am Chem Soc. 1987;109:1160–1170. [Google Scholar]; b) Brandes E, Grieco PA, Gajewski JJ. J Org Chem. 1989;54:515–516. [Google Scholar]; c) Gajewski JJ, Jurayj J, Kimbrough DR, Gande ME, Ganem B, Carpenter BK. J Am Chem Soc. 1987;109:1170–1186. [Google Scholar]
- 4.Claisen L. Chem Ber. 1912;45:3157–3166. [Google Scholar]
- 5.a) Cramer CJ, Truhlar DG. J Am Chem Soc. 1992;114:8794–8799. [Google Scholar]; b) Severance DL, Jorgensen WL. J Am Chem Soc. 1992;114:10966–10968. [Google Scholar]; c) White WN, Wolfarth EF. J Org Chem. 1970;35:2196–2199. [Google Scholar]; d) Grieco PA, Brandes EB, McCann S, Clark JD. J Org Chem. 1989;54:5849–5851. [Google Scholar]; e) Gajewski JJ. Acc Chem Res. 1997;30:219–225. [Google Scholar]; f) Sehgal A, Shao L, Gao J. J Am Chem Soc. 1995;117:11337–11340. [Google Scholar]
- 6.a) Pawlak JL, Padykula RE, Kronis JD, Aleksejczyk RA, Berchtold GA. J Am Chem Soc. 1989;111:3374–3381. [Google Scholar]; b) Guilford WJ, Copley SD, Knowles JR. J Am Chem Soc. 1987;109:5013–5019. [Google Scholar]; c) Stewart J, Wilson DB, Ganem B. J Am Chem Soc. 1990;112:4582–4584. [Google Scholar]; d) Bartlett PA, Johnson CR. J Am Chem Soc. 1985;107:7792–7793. [Google Scholar]; e) Ganem B. Tetrahedron. 1978;34:3353–3383. [Google Scholar]
- 7.a) Hilvert D, Nared KD. J Am Chem Soc. 1988;110:5593–5594. [Google Scholar]; b) Jackson DY, Jacobs JW, Sugasawara R, Reich SH, Bartlett PA, Schultz PG. J Am Chem Soc. 1988;110:4841–4842. [Google Scholar]; c) Hilvert D, Carpenter SH, Nared KD, Auditor MT. Proc Natl Acad Sci USA. 1988;14:4953–4955. doi: 10.1073/pnas.85.14.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Acevedo O, Armacost K. J Am Chem Soc. 2010;132:1966–1975. doi: 10.1021/ja908680c. [DOI] [PubMed] [Google Scholar]; b) Repasky MP, Guimaraes CRW, Chandrasekhar J, Tirado-Rives J, Jorgensen WL. J Am Chem Soc. 2003;125:6663–6672. doi: 10.1021/ja021423z. [DOI] [PubMed] [Google Scholar]; c) Kincaid JF, Tarbell DS. J Am Chem Soc. 1939;61:3085–3089. [Google Scholar]; d) Chanda A, Fokin VV. Chem Rev. 2009;109:725–748. doi: 10.1021/cr800448q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganem B. Angew Chem Int Ed. 1996;35:936–945. [Google Scholar]
- 10.Copley SD, Knowles JR. J Am Chem Soc. 1987;109:5008–5013. [Google Scholar]
- 11.Guest JM, Craw JS, Vincent MA, Hillier IH. J Chem Soc, Perkin Trans 2. 1997;2:71–74. [Google Scholar]
- 12.Claisen L, Eisleb O. Justus Liebigs Ann Chem. 1913;401:21–119. [Google Scholar]
- 13.Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB. Angew Chem Int Ed. 2005;44:3275–3279. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]
- 14.Zheng Y, Zhang J. Journal of Physical Chemistry A. 2010;114:4325–4333. doi: 10.1021/jp908018u. [DOI] [PubMed] [Google Scholar]
- 15.Curran DP, Lung HK. Tetrahedron Lett. 1995;36:6647–6650. [Google Scholar]
- 16.Uyeda C, Jacobsen EN. J Am Chem Soc. 2008;130:9228–9229. doi: 10.1021/ja803370x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesh M, Seidel D. J Am Chem Soc. 2008;130:16464–16465. doi: 10.1021/ja8063292. [DOI] [PubMed] [Google Scholar]
- 18.Uyeda C, Rötheli AR, Jacobsen EN. Angew Chem Int Ed. 2010;49:9753–9756. doi: 10.1002/anie.201005183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Knowles RR, Jacobsen EN. Proc Natl Acad Sci USA. 2010;107:20678–20685. doi: 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Uyeda C, Jacobsen EN. J Am Chem Soc. 2011;133:5062–5075. doi: 10.1021/ja110842s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Annamalai VR, Linton EC, Kozlowski MC. Org Lett. 2008;11:621–624. doi: 10.1021/ol8026945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Claeyssens F, Ranaghan KE, Lawan N, Macrae SJ, Manby FR, Harvey JN, Mulholland AJ. Org Biomol Chem. 2011 doi: 10.1039/c0ob00691b. [DOI] [PubMed] [Google Scholar]; b) Lyne PD, Mulholland AJ, Richards WG. J Am Chem Soc. 1995;117:11345–11350. [Google Scholar]; c) Martí S, Andrés J, Moliner V, Silla E, Tuñón I, Bertrán J. J Phys Chem B. 2000;104:11308–11315. [Google Scholar]; d) Szefczyk B, Mulholland AJ, Ranaghan KE, Sokalski WA. J Am Chem Soc. 2004;126:16148–16159. doi: 10.1021/ja049376t. [DOI] [PubMed] [Google Scholar]; e) Štrajbl M, Shurki A, Kato M, Warshel A. J Am Chem Soc. 2003;125:10228–10237. doi: 10.1021/ja0356481. [DOI] [PubMed] [Google Scholar]; f) Guimarães CRW, Repasky MP, Chandrasekhar J, Tirado-Rives J, Jorgensen WL. J Am Chem Soc. 2003;125:6892–6899. doi: 10.1021/ja021424r. [DOI] [PubMed] [Google Scholar]; g) Hur S, Bruice TC. J Am Chem Soc. 2003;125:5964–5972. doi: 10.1021/ja0210648. [DOI] [PubMed] [Google Scholar]; h) Hur S, Bruice TC. J Am Chem Soc. 2003;125:1472–1473. doi: 10.1021/ja0293047. [DOI] [PubMed] [Google Scholar]; i) Hur S, Bruice TC. J Am Chem Soc. 2003;125:10540–10542. doi: 10.1021/ja0357846. [DOI] [PubMed] [Google Scholar]; j) Ishida T. J Am Chem Soc. 2010;132:7104–7118. doi: 10.1021/ja100744h. [DOI] [PubMed] [Google Scholar]
- 22.a) Andrews PR, Smith GD, Young IG. Biochemistry. 1973;12:3492–3498. doi: 10.1021/bi00742a022. [DOI] [PubMed] [Google Scholar]; b) Aemissegger A, Jaun B, Hilvert D. J Org Chem. 2002;67:6725–6730. doi: 10.1021/jo026096s. [DOI] [PubMed] [Google Scholar]; c) Lee A, Stewart JD, Clardy J, Ganem B. Chem Biol. 1995;2:195–203. doi: 10.1016/1074-5521(95)90269-4. [DOI] [PubMed] [Google Scholar]; d) Lee AY, Karplus PA, Ganem B, Clardy J. J Am Chem Soc. 1995;117:3627–3628. [Google Scholar]; e) Lee AY, Stewart JD, Clardy J, Ganem B. Chem Biol. 1995;2:195–203. doi: 10.1016/1074-5521(95)90269-4. [DOI] [PubMed] [Google Scholar]; f) Sogo SG, Widlanski TS, Hoare JH, Grimshaw CE, Berchtold GA, Knowles JR. J Am Chem Soc. 1984;106:2701–2703. [Google Scholar]
- 23.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 24.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, III, Hilvert D, Houk KN, Stoddard BL, Baker D. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, StClair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Vol. Gaussian, Inc; Pittsburgh, PA: 2009. [Google Scholar]
- 27.a) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; c) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 28.a) Hariharan PC, Pople JA. Theor Chim Acta. 1973;28:213–222. [Google Scholar]; b) Hehre WJ, Ditchfie R, Pople JA. J Chem Phys. 1972;56:2257–2261. [Google Scholar]
- 29.a) Gonzalez C, Schlegel HB. J Chem Phys. 1989;90:2154–2161. [Google Scholar]; b) Gonzalez C, Schlegel HB. J Phys Chem. 1990;94:5523–5527. [Google Scholar]
- 30.Adamo C, Barone V. J Chem Phys. 1999;110:6158–6169. [Google Scholar]
- 31.Grimme S. J Chem Phys. 2006;124:034108. doi: 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- 32.Moller C, Plesset MS. Phys Rev. 1934;46:618–622. [Google Scholar]
- 33.Grimme S. J Chem Phys. 2003;118:9095. [Google Scholar]
- 34.Woon DE, Dunning TH. J Chem Phys. 1993;98:1358–1371. [Google Scholar]
- 35.Kendall RA, Dunning TH, Harrison RJ. J Chem Phys. 1992;96:6796–6806. [Google Scholar]
- 36.a) Barone V, Cossi M. J Phys Chem A. 1998;102:1995–2001. [Google Scholar]; b) Cossi M, Rega N, Scalmani G, Barone V. J Comp Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 37.Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA. J Chem Phys. 2000;112:6532–6542. [Google Scholar]
- 38.a) Paton RS, Mackey JL, Kim WH, Lee JH, Danishefsky SJ, Houk KN. J Am Chem Soc. 2010;132:9335–9340. doi: 10.1021/ja1009162. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Paton RS, Steinhardt SE, Vanderwal CD, Houk KN. J Am Chem Soc. 2011;133:3895–3905. doi: 10.1021/ja107988b. [DOI] [PubMed] [Google Scholar]; c) Ess DH, Houk KN. J Phys Chem A. 2005;109:9542–9553. doi: 10.1021/jp052504v. [DOI] [PubMed] [Google Scholar]; d) Schwabe T, Grimme S. Phys Chem Chem Phys. 2007;9:3397–3406. doi: 10.1039/b704725h. [DOI] [PubMed] [Google Scholar]; e) Schwabe T, Grimme S. Acc Chem Res. 2008;41:569–579. doi: 10.1021/ar700208h. [DOI] [PubMed] [Google Scholar]; f) Zhao Y, Truhlar DG. J Chem Theory Comput. 2006;3:289–300. doi: 10.1021/ct6002719. [DOI] [PubMed] [Google Scholar]; g) Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 39.Schuler FW, Murphy GW. J Am Chem Soc. 1950;72:3155–3159. [Google Scholar]
- 40.Huenerbein R, Schirmer B, Moellmann J, Grimme S. Phys Chem Chem Phys. 2010;12:6940–6948. doi: 10.1039/c003951a. [DOI] [PubMed] [Google Scholar]
- 41.Osuna Sl, Swart M, Solà M. J Phys Chem A. 2011;115:3491–3496. doi: 10.1021/jp1091575. [DOI] [PubMed] [Google Scholar]
- 42.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 43.a) Quillinan AJ, Scheinmann F. J Chem Soc D. 1971:966–967. [Google Scholar]; b) Nicolaou KC, Li J. Angew Chem Int Ed. 2001;40:4264–4268. doi: 10.1002/1521-3773(20011119)40:22<4264::AID-ANIE4264>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]; c) Tisdale EJ, Slobodov I, Theodorakis EA. Org Biomol Chem. 2003;1:4418–4422. doi: 10.1039/b311833a. [DOI] [PubMed] [Google Scholar]
- 44.a) Goering HL, Jacobson RR. J Am Chem Soc. 1958;80:3277–3285. [Google Scholar]; b) Harfenist M, Thom E. J Org Chem. 1972;37:841–848. [Google Scholar]; c) Jung ME, Piizzi G. Chem Rev. 2005;105:1735–1766. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- 45.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. John Wiley & Sons, Ltd; Baffins Lane, West Sussex, England: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.