

Abstract
Activation of the kappa opioid receptor (KOR) system mediates negative emotional states and considerable evidence suggests that KOR and their natural ligand, dynorphin, are involved in ethanol dependence and reward. The central amygdala (CeA) plays a major role in alcohol dependence and reinforcement. Dynorphin peptide and gene expression are activated in the amygdala during acute and chronic administration of alcohol, but the effects of activation or blockade of KOR on inhibitory transmission and ethanol effects have not been studied. We used the slice preparation to investigate the physiological role of KOR and interaction with ethanol on GABAA receptor-mediated synaptic transmission. Superfusion of dynorphin or U69593 onto CeA neurons decreased evoked inhibitory postsynaptic potentials (IPSPs) in a concentration-dependent manner, an effect prevented by the KOR antagonist norbinaltorphimine (norBNI). Applied alone, norBNI increased GABAergic transmission, revealing a tonic endogenous activity at KOR. Paired-pulse analysis suggested a presynaptic KOR mechanism. Superfusion of ethanol increased IPSPs and pretreatment with KOR agonists diminished the ethanol effect. Surprisingly, the ethanol-induced augmentation of IPSPs was completely obliterated by KOR blockade. Our results reveal an important role of the dynorphin/KOR system in the regulation of inhibitory transmission and mediation of ethanol effects in the CeA.
Keywords: opioid, kappa, alcohol, synaptic, GABA, amygdala
1. Introduction
Alcohol impacts many aspects of neuronal communication, including synaptic transmission, ion channels and intracellular signaling cascades. Synaptic transmission is a sensitive substrate for ethanol actions (Siggins et al., 2005). GABAergic transmission is affected by alcohol in several brain structures and contributes to the behavioral effects of alcohol as well as the development of alcohol dependence (Koob, 2004; Siggins et al., 2005; Weiner and Valenzuela, 2006). The central amygdala (CeA) plays a critical role in alcohol dependence (Davis et al., 1994; Koob, 2008) and behavioral studies have implicated CeA GABAergic transmission in the regulation of alcohol intake (Hyytia and Koob, 1995; Koob, 2003). In ex vivo recordings from CeA slices, ethanol increases GABAergic transmission by acting principally at a presynaptic site to augment GABA release (Nie et al., 2004; Roberto et al., 2003).
The endogenous peptide dynorphin activates kappa opioid receptors (KOR) in the brain (Chavkin et al., 1982), and the dynorphin/KOR system modulates affective-like states (Knoll and Carlezon, Jr., 2010). Several studies suggest that multiple physiological and behavioral effects of ethanol involve KOR (Dar, 1998; Matsuzawa et al., 1999; Pohorecky et al., 1989), and mice lacking KOR exhibit decreased alcohol consumption (Kovacs et al., 2005). Ethanol treatment upregulates dynorphin and its precursor prodynorphin as well as KOR mRNA in the CeA (D’Addario et al., 2011; Lam et al., 2008), and ethanol also increases prodynorphin levels in the nucleus accumbens and prefrontal cortex (D’Addario et al., 2011; Marinelli et al., 2006). Dynorphin peptide and gene expression are activated in the amygdala during acute and chronic administration of alcohol and the KOR antagonist nor-binaltorphimine (norBNI) reduces ethanol self-administration in alcohol-dependent animals (Walker and Koob, 2008). However, little is known about the role of the dynorphin/KOR system in regulating the cellular effects of ethanol.
Prodynorphin neurons are abundant in the CeA (Marchant et al., 2007), and KOR immunoreactivity is localized in the CeA and medial amygdala, with no specific staining in the lateral, basolateral and cortical amygdaloid nuclei (Mansour et al., 1996). Within the CeA, KOR-like immunoreactive fibers are localized predominantly in the medial division, identifying this area as a prime target for electrophysiological recordings. The synaptic network in CeA is mostly GABAergic and projection neurons also are GABAergic (Sah et al., 2003), thus we focused on inhibitory transmission in the medial division of the CeA in this study.
We report here that KOR activation reduced GABAergic synaptic responses whereas KOR blockade increased it, revealing a tonic endogenous KOR activity that suppresses inhibition in the CeA. Dynorphin diminished but did not prevent the ethanol-elicited increase of GABAergic transmission, whereas KOR antagonism blocked the effect of ethanol. We conclude that dynorphin reduces inhibitory transmission and KOR activation tonically controls neuronal activity in the CeA. Dynorphin also antagonizes the effect of ethanol in the CeA of the alcohol-naïve rat brain, suggesting an important role of the dynorphin/KOR system to regulate CeA tone during challenge with ethanol.
2. Materials and Methods
2.1 Slice preparation
All experimental protocols were consistent with guidelines issued by the National Institutes of Health and were approved by The Scripps Research Institute’s Institutional Animal Care and Use Committee. We prepared CeA slices as previously described (Roberto et al., 2003) from male Sprague-Dawley rats (120–200 g) that were anesthetized with halothane (3%) and decapitated. In brief, the brains were rapidly removed and placed into ice-cold artificial cerebrospinal fluid (ACSF) gassed with 95% O2 and 5% CO2. Transverse slices 400 μm thick were cut on a Vibratome Series 3000 (Technical Products International, St. Louis, MO), incubated in an interface configuration for about 30 min, and then completely submerged and superfused at a constant flow rate of 2–4 ml/min with warm (31° C), gassed ACSF of the following composition in mM: NaCl, 130; KCl, 3.5; NaH2PO4, 1.25; MgSO4·7H2O, 1.5; CaCl2, 2.0; NaHCO3, 24; glucose, 10. We added drugs to the ACSF from stock solutions to obtain known concentrations in the superfusate. The inner recording chamber had a total volume of 0.8 ml, so drug concentrations reach 90% of the nominal concentration within 2 min.
2.2 Recordings
We recorded neurons in the medial subdivision of the CeA with sharp micropipettes filled with 3M KCl (impedance range of 60–90 MΩ) using current-clamp mode. We held most neurons near their resting membrane potential (RMP), and the RMP observed in control conditions was maintained throughout the experiment. Data were acquired with an Axoclamp-2 preamplifier (Axon Instruments, Foster City, CA) and stored for later analysis using pClamp software (Axon Instruments). Pharmacologically-isolated GABA receptor-mediated inhibitory postsynaptic currents (IPSPs) were evoked by stimulating locally within the CeA through a bipolar stimulating electrode while superfusing the slices with the glutamate receptor blockers 6-cyano-7-nitroquinoxaline-2,3-dione (DNQX, 20 μM) and DL-2-amino-5-phosphonovalerate (APV, 30 μM). We also applied the GABAB receptor antagonist CGP 55845 (1 μM) to isolate the GABAA receptor-mediated component of the IPSP.
2.3 Electrophysiological protocols
To determine the experimental response parameters for each cell, we performed an input-output protocol consisting of a range of current stimulations (typically between 50 and 250 mA; 0.125 Hz), starting at the threshold current required to elicit an IPSP up to the strength required to elicit the maximum amplitude. The stimulus strength eliciting 50% of the maximum response was chosen to conduct experiments and maintained throughout the entire duration of the experiment. Stability of IPSPs was established by stimulating for at least 15 min prior to beginning experiments. The synaptic responses were quantified by averaging two consecutive responses (30 sec apart, i.e. 1 data point/min) and calculating the IPSP amplitude with Clampfit software (Axon Instruments). We examined paired-pulse facilitation (PPF), a phenomenon whereby a secondary evoked synaptic response is increased by a preceding primary stimulation of equal intensity (Siggins et al., 2005; Thomson, 2000), in each neuron using 100 msec inter-stimulus intervals; we calculated the paired-pulse ratio as the second IPSP amplitude over that of the first IPSP.
2.4 Drugs
Drugs were added to the superfusate in known concentrations. U69593 and nor-binaltorphimine (norBNI) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and dissolved in dimethylsulfoxide. Dimethylsulfoxide final concentration was 0.05–0.1%, which did not affect the studied synaptic responses in control experiments. Dynorphin A [1–17], CTAP and bicuculline were purchased from Tocris (Ellisville, MO), and ethanol from Remet (La Mirada, CA). We purchased all other chemicals from Sigma-Aldrich (St Louis, MO).
2.5 Statistics
We took measures every minute before drug superfusion (control) and during drug superfusion. All values are expressed as mean ± s.e.m. We performed statistical analysis using GraphPad Prism version 5.00 (GraphPad Software, San Diego California USA). In the case of a single drug application, we used Student’s paired t-test. One-way repeated measures ANOVA with the Tukey post hoc test were performed in a multiple drug application. Statistical significance was set at p < 0.05.
3. Results
We recorded intracellularly from medial CeA neurons using sharp micropipettes and we evoked pharmacologically isolated GABAA–mediated synaptic responses via local stimulations through an electrode placed near the recording site (see Roberto et al., 2010). The average resting membrane potential (RMP) of our neuronal sample was −77.0 ± 0.6 mV (n = 60).
3.1 Kappa opioid receptor ligands decrease inhibitory transmission at a presynaptic site
We first assessed the effect of dynorphin (dynorphin A [1–17]), an endogenous kappa receptor agonist, on GABAergic transmission. Superfusion of 1 μM dynorphin consistently decreased Inhibitory PostSynaptic Potentials (IPSPs) in 80% of the neurons exposed to the peptide (12 of 15.) The depressant effect of dynorphin developed immediately after the start of application and reached a maximum effect after 9 minutes of superfusion (Fig. 1A,B). Upon washout of the peptide, IPSP amplitude recovered to control level within 20 minutes (4 of 4 experiments). On average (mean of 4 points after steady level reached and just before change of condition), IPSP amplitudes were significantly decreased to 79 ± 4% of control (n = 12, t = 6.568). The dynorphin decrease of IPSP amplitude persisted upon extended application of the peptide for 25 minutes (n = 2 at 1 μM; n = 2 at 2 μM), demonstrating a lack of short term desensitization of KOR in our preparation. Another neuron did not respond and another 2 showed a slight IPSP increase (10–15 %) upon exposure to dynorphin. We used the selective KOR antagonist nor-binaltorphimine (norBNI) (Portoghese et al., 1987) to confirm that dynorphin decreased IPSPs by activating KOR. In the presence of 0.2 μM norBNI, dynorphin did not affect IPSPs which remained at 98 ± 4% of pre-dynorphin level (n = 4; Fig 1C). The dynorphin-elicited decrease of inhibitory transmission was concentration-dependent. We observed no effect of 0.1 μM dynorphin (101 ± 4% of control, n = 3), whereas IPSPs were decreased to 96 ± 5% of control with 0.2 μM dynorphin (n = 3), to 91 ± 5% with 0.5 μM (n = 4) and 77 ± 5% with 2 μM (n = 6). We used a sigmoidal (logistic) fit to analyze the concentration-response relationship (Fig. 2A). The apparent EC50 for dynorphin to decrease IPSPs was 0.5 μM. We conclude that the endogenous peptide dynorphin concentration-dependently decreases inhibitory transmission in CeA by activating KOR.
Figure 1. Kappa receptor ligands decrease GABAA-mediated synaptic transmission.

A. Representative recordings showing IPSPs elicited before (Control) and during superfusion of 1 μM dynorphin (Dyn) in a CeA neuron. Dynorphin diminished the evoked IPSP with recovery to control level after washout. Recordings were performed at RMP, −82 mV for this neuron. Stimulus artifact indicated by arrows; traces identified with numbers are magnified and superimposed on the right for comparison. B. Graph average of dynorphin effect on IPSP amplitudes over time. Dynorphin (1 μM; application indicated with bar) was added at t = 0 and the maximum effect was obtained after 9 min of superfusion. C. In the presence of the KOR antagonist norBNI (0.2 μM), dynorphin did not affect IPSP amplitude. RMP was −79 mV. D. Superfusion of U69 at concentrations of 0.5 μM (open circles) or 1 μM (closed squares) decreased IPSPs to the same extent as dynorphin and with a similar time course.
Figure 2. Dynorphin activates kappa receptors to decrease IPSPs at a presynaptic site.

A. Concentration-response curve (logistic fit) of the dynorphin effect. Dynorphin decreased IPSPs by a maximum of 23%, with an apparent EC50 of 0.50 μM (dashed line) and a near maximum effect at 1 μM. U69 also depicted (open squares) for comparison: near-maximum effect was obtained at 0.5 μM. B. Upper panel: IPSP recordings obtained with the PPF paradigm. Dynorphin (1 μM) diminished P1 with little effect on P2, increasing the paired-pulse ratio (P2/P1). RMP was −76. Arrows indicate stimulation artifact. Lower left: recordings from upper panel superimposed and normalized to P1 to magnify the increase of PPF. For equivalent P1 amplitudes, P2 (identified with numbers) was larger in the presence of dynorphin. Lower right: bar graph average depicting the PPF change relative to control condition: dynorphin (1 μM) and U69 (1 μM) increased PPF by 28% and 26% respectively.
We also determined the effect of the highly selective synthetic KOR agonist U69593 (U69) (Raynor et al., 1994) on GABAA–mediated transmission. These experiments were designed to confirm KORs as the locus of action for dynorphin effects and as such, we utilized only two concentrations typical of recently published electrophysiological studies in brain slices (Lemos et al., 2012; Li et al., 2012). Upon superfusion of 0.5 μM U69, IPSP amplitude decreased to 78 ± 6% of control (n = 4; Fig. 1D). A concentration of 1 μM U69 did not elicit a larger effect, decreasing IPSPs to 76 ± 6% of control (n = 4; Fig. 1D, 2A). Combining those two concentrations, U69 significantly decreased IPSP amplitude to 77 ± 5% of control (n = 8, t = 4.478). Another neuron did not respond to U69. Thus, the synthetic (U69) and endogenous (dynorphin) KOR agonists reduced inhibitory transmission by 20–25% in CeA. Prolonged application of 1 μM U69 (25 minutes; n = 2) did not elicit a desensitization of the response.
To determine if dynorphin decreased inhibitory transmission by acting at a presynaptic site in CeA, we used a paired-pulse facilitation (PPF) protocol of IPSPs evoked by delivering two stimuli of the same intensity separated by 100 ms. At this interval, the second IPSP (P2) is larger than the first IPSP (P1), and an increase of the paired-pulse ratio (P2/P1) reflects a decrease of transmitter release (see Siggins et al., 2005). Analysis of PPF revealed that dynorphin (1 μM) significantly increased the paired-pulse ratio to 128 ± 6% of control (n = 11, t = 4.372; Fig. 2B), from 1.11 ± 0.06 before dynorphin to 1.42 ± 0.08 in the presence of the peptide. In 4 of 4 experiments where washout of the peptide was performed, PPF values returned to control level (not shown). We also conducted a PPF protocol during superfusion of U69. U69 (0.5–1 μM) significantly increased the paired-pulse ratio of IPSPs to 126 ± 8% of control (n = 8, t = 3.409; Fig. 2B). These data indicate that KOR agonists decreased GABAA receptor-mediated inhibitory transmission by decreasing the release of GABA at a presynaptic site.
3.2 The kappa receptor ligands diminish the effect of ethanol on GABAergic transmission
Ethanol increases GABAergic transmission in CeA with a maximally effective concentration of 44 mM (Roberto et al., 2003). We first confirmed these results and found that superfusion of 44 mM ethanol significantly increased IPSP amplitude to 148 ± 5% of control (n = 8, t = 8.159; Fig. 3A). PPF analysis indicated that ethanol decreased the paired-pulse ratio to 66 ± 6% of control (n = 8; not shown), confirming a presynaptic site of action for ethanol-induced increase in GABAergic transmission.
Figure 3. Kappa receptor ligands diminish the action of ethanol.
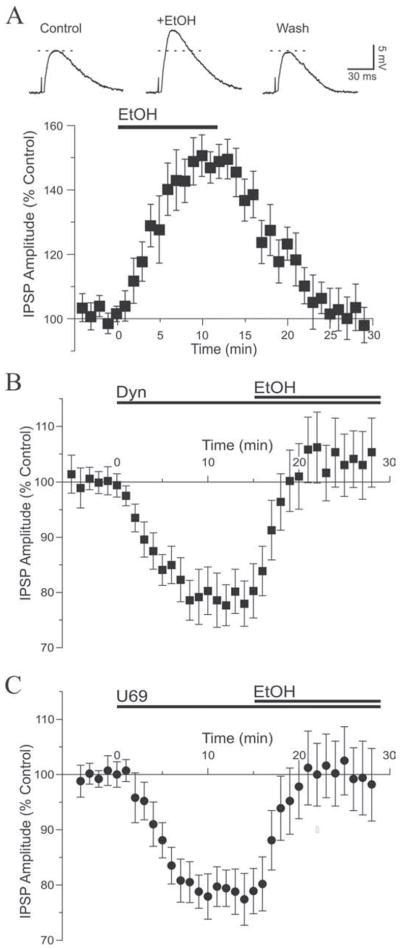
A. Top: superfusion of ethanol (EtOH) onto this neuron increased the amplitude of the evoked IPSP with recovery to control level upon washout. RMP was −80 mV. Bottom: pooled data showing the time course of the effect of ethanol, which on average increased inhibitory transmission by 48%. B. Dynorphin (1–2 μM; t = 0) decreased IPSPs to 79% of control. Addition of 44 mM ethanol (t = 15) reversed the dynorphin effect, increasing IPSPs by a net 32%. C. We observed a similar pattern with U69: U69 alone (0.5–1 μM) decreased IPSPs to 81% of control and addition of ethanol reversed the U69 effect, increasing IPSPs by a net 29%.
We next assessed the interaction of ethanol with KOR agonists. In this set of experiments we activated KOR with dynorphin or U69 and subsequently applied ethanol in the continued presence of the ligands. Two concentrations of dynorphin (1 and 2 μM) and U69 (0.5 and 1 μM) were used to ensure that maximum effect was obtained. Superfusion of 1 μM dynorphin decreased IPSPs to 80 ± 6% of control and subsequent addition of 44 mM ethanol reversed the dynorphin effect and increased IPSP amplitude to 106 ± 7% of pre-dynorphin level (n = 5). Superfusion of 2 μM dynorphin decreased IPSPs to 78 ± 7% of control, and subsequent addition of 44 mM ethanol reversed the dynorphin effect and increased IPSP amplitude to 101 ± 10% of pre-dynorphin level (n = 5). Because results obtained with both concentrations were similar, the data from both sets of neurons were combined and showed that 1–2 μM dynorphin significantly decreased IPSPs to 79 ± 4% of control (n = 10; F (2,9) = 14.43). Addition of ethanol significantly increased IPSPs to 104 ± 6% of pre-dynorphin level (n = 10; Fig. 3B), reversing the dynorphin effect. The net ethanol effect relative to dynorphin levels (dynorphin values being control values) was a 32 ± 6% increase of IPSP amplitude, and the magnitude of this increase was significantly dampened relative to ethanol application in the absence of dynorphin (unpaired t-test: t = 2.972, df = 15). In addition, the paired-pulse ratio was increased by 1–2 μM dynorphin to 125 ± 8% of control and significantly reversed to 96 ± 10% of control by subsequent addition of ethanol (n = 9; F(2,8) = 4.37; not shown), a net 23 ± 7% decrease elicited by ethanol relative to dynorphin values. These data confirm that dynorphin and ethanol interact at a presynaptic site to differentially affect GABA release and modulate inhibitory transmission.
Similar results were obtained with application of 0.5–1 μM U69. Superfusion of 0.5 μM U69 decreased IPSPs to 81 ± 6% of control and subsequent addition of 44 mM ethanol reversed the effect of U69, increasing IPSPs to 105 ± 7% of pre-U69 level (n = 4). Superfusion of 1 μM U69 decreased IPSPs to 77 ± 6% of control. Subsequent addition of 44 mM ethanol reversed the U69 effect and increased IPSP amplitude to 99 ± 9% of pre-U69 level (n = 4). Combining the data, U69 (0.5–1 μM) significantly decreased IPSPs to 78 ± 5% of control (n = 8; F(2,7) = 10.31). Subsequent addition of ethanol significantly increased IPSPs to 100 ± 7% of pre-U69 level (n = 8; Fig. 3C), reversing the U69 effect. The net ethanol effect relative to U69 levels was a 29 ± 7% increase of IPSP amplitude, an effect significantly dampened from the effect of ethanol alone (unpaired t-test: t = 2.186, df = 14). Also, the paired-pulse ratio was increased to 125 ± 9% of control value by U69 then reversed to 104 ± 10% of control by application of ethanol (n = 7; U69 versus ethanol did not reach significance), a net 17 ± 8% decrease elicited by ethanol.
3.3 A tonic kappa opioid activity in CeA revealed by blockade of the receptor
We then investigated a possible role for endogenously formed KOR ligands to regulate basal synaptic transmission in CeA by applying norBNI alone onto slices. Superfusion of 0.2 μM norBNI markedly augmented IPSPs in 75% of the neurons (12 of 16; Fig. 4A). On average, norBNI significantly increased inhibitory transmission to 136 ± 4% of pre-drug value after 10 min of application (n = 12, t = 6.438; Fig. 4B). The KOR antagonist concomitantly decreased the paired-pulse ratio in a significant manner to 77 ± 8% of control value (n = 12, t = 3.215; Fig. 4C), from 1.16 ± 0.07 before norBNI to 0.89 ± 0.09 in the presence of the antagonist, indicating increased GABA release upon blockade of KOR. The augmentation of IPSPs upon blockade of KOR by norBNI is likely due to the prevention of basal activity exerted by endogenously formed dynorphin. These data suggest that endogenously formed KOR ligands tonically regulate inhibitory transmission in CeA by decreasing the release of GABA.
Figure 4. Kappa receptor blockade reveals a tonic endogenous opioidergic activity.
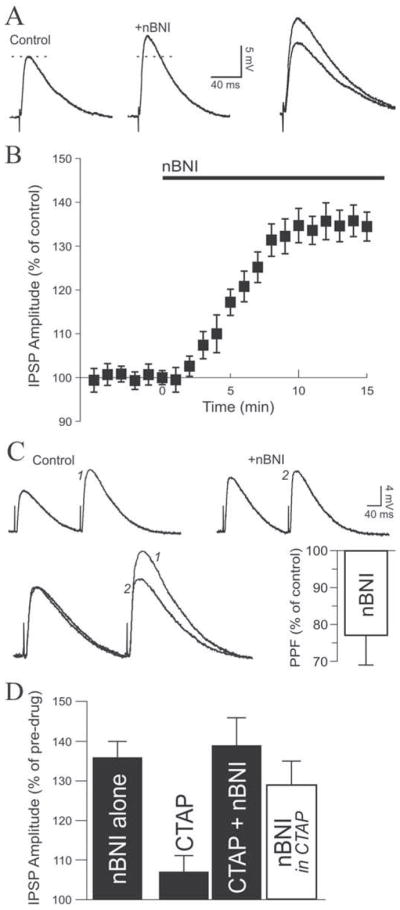
A. In this representative neuron, norBNI (0.2 μM) applied alone markedly increased IPSP amplitude by 34%. RMP was −77 mV. B. Average time course of the effect of 0.2 μM norBNI. C. Top: in this CeA neuron, a PPF paradigm revealed that norBNI increased the first IPSP but had little effect on the second IPSP. Bottom left: traces superimposed and scaled to the first IPSP, magnifying the PPF decrease elicited by norBNI. Bottom right: on average, norBNI decreased PPF to 77% of control, suggesting a tonic presynaptic effect of an endogenous KOR ligand to decrease GABA release. RMP was −78 mV. D. Applied alone, norBNI increase IPSPs by 36%. In the presence of the mu opioid receptor antagonist CTAP, norBNI still increased inhibitory transmission by a net 29%.
Because norBNI may also act as an antagonist at mu opioid receptors (MOR) (Munro et al., 2012) and the endogenous dynorphin could bind to MOR (Raynor et al., 1994), we performed additional experiments by combining the application of norBNI with the selective MOR antagonist CTAP to further implicate the endogenous dynorphin/KOR system. Bath application of 1 μM CTAP increased IPSPs to 107 ± 5% of control, and further addition of 0.2 μM norBNI further increased IPSP amplitude to 139 ± 7% of pre-CTAP values (n = 5, Fig. 4D). Thus, norBNI was able to increase IPSPs by an additional 29 ± 6% relative to CTAP values (calculated with CTAP values being control values), suggesting that KOR mediate dynorphin effects on inhibitory transmission in CeA.
3.4 Kappa receptor antagonism prevents the effect of ethanol on GABAergic transmission
Because norBNI and ethanol displayed comparable effects, we sought to determine the influence of KOR blockade on the ethanol-induced augmentation of IPSPs by first applying norBNI and subsequently adding ethanol in the continued presence of norBNI. Superfusion of 0.2 μM norBNI significantly increased IPSP amplitudes to 134 ± 6% of control (n = 7; F (2,6) = 16.26), and subsequent addition of 44 mM ethanol together with norBNI did not significantly affect IPSP amplitudes which remained at 131 ± 8% of control level (n = 7; Fig. 5A). Thus, norBNI completely blocked the action of ethanol. Consistent with a lack of effect of ethanol in the presence of norBNI, the norBNI-elicited decrease of the paired-pulse ratio remained unchanged from 75 ± 10% (norBNI) to 79 ± 12% of control upon application of ethanol (n = 7; F (2,6) = 4.678; not shown). Another 2 neurons that showed no response to norBNI were exposed to ethanol. Ethanol largely increased IPSPs in both neurons (157% and 165% of control; not shown).
Figure 5. Ethanol and norBNI reciprocally block each others’ effect on GABA transmission.

A. Top: in this neuron, superfusion of norBNI increased the IPSP but subsequent addition of ethanol in the continued presence of norBNI had no further effect. RMP was −77 mV. Bottom: graph depicting the complete lack of ethanol effect with KORs blocked. B. Top: reciprocally, ethanol applied first increased the IPSP amplitude but further addition of norBNI was without effect. Bottom: graph average showing the average effect of the sequence ethanol then norBNI. C. Bar graph summarizing the influence of the dynorphin/KOR system on the effect of ethanol in CeA neurons. The effect of ethanol was significantly diminished by activation of KORs but completely obliterated by KOR blockade. D. The ethanol-induced decrease of the paired-pulse ratio was also affected by the KOR ligands and obliterated by the KOR antagonist, consistent with a presynaptic of interaction.
Reversing the order of drug application yielded similar results. Superfusion of 44 mM ethanol significantly increased IPSP amplitude to 145 ± 7% of control (n = 5; F (2,4) = 69.14), and subsequent addition of 0.2 μM norBNI together with ethanol did not significantly affect IPSP amplitudes which remained at 148 ± 8% of control level (n = 5; Fig. 5B). The ethanol-elicited decrease of the paired-pulse ratio remained unchanged upon application of norBNI (63 ± 9% to 59 ± 12%, n = 5; not shown). Thus, norBNI prevented but did not reverse the action of ethanol on inhibitory transmission and there was no additive effect between ethanol and norBNI to increase GABA transmission, regardless of the order of drug application.
The effect of the KOR system in modulating ethanol effects on GABA transmission in CeA is summarized on Figure 5C,D. In control condition, ethanol alone increased IPSPs by 48%. In the presence of dynorphin or U69 the action of ethanol was diminished, increasing GABAergic transmission by 32% and 29% respectively. However, the ethanol effect was obliterated when neurons were pretreated with norBNI. The use of paired-pulse paradigms suggest that these effects took place at a presynaptic level. The paired pulse ratio was decreased by 34% with ethanol alone, 23% when ethanol was applied in the presence of dynorphin and 17% in the presence of U69, a range consistent with drug effects observed on IPSP amplitude.
4. Discussion
Our results reveal an important role for the dynorphin/KOR system to regulate synaptic transmission and alcohol effects in the CeA, a brain nucleus implicated in the etiology of drug and alcohol dependence. Activation of KOR with its natural ligand dynorphin or with the synthetic selective agonist U69 decreased inhibitory transmission and diminished the effect of ethanol by acting on GABA release. Blockade of KOR unmasked a tonic activity at KOR by dynorphin and also prevented the ethanol effect at a presynaptic site. Because dynorphin may affect other opioid receptors such as MOR and the synthetic agonist U69 is highly selective for KORs (Raynor et al., 1994), we used the selective U69 to verify the involvement of KOR and eliminate the participation of endogenous degradation mechanisms. Conversely, we used dynorphin throughout this study to ensure that the endogenous form of the KOR ligand reproduced effects obtained with the synthetic ligand.
4.1 KOR agonists decrease inhibitory transmission and diminish the effect of ethanol
The dynorphin/U69-induced decrease of GABAergic synaptic transmission we describe compares well with studies performed in other brain regions. Facilitating KOR signaling also decreases GABAergic responses by 15–50% in globus pallidus (Ogura and Kita, 2000), nucleus accumbens (Hjelmstad and Fields, 2003), ventral tegmental area (Ford et al., 2007), bed nucleus stria terminalis (Li et al., 2012) and hypothalamus (Pennock and Hentges, 2011), as well as in the CeA (Kang-Park et al., 2013). Interestingly, activation of presynaptic KOR in the bed nucleus stria terminalis decrease GABA released from projecting CeA neurons (Li et al., 2012). In the CeA, data from knockout mice have identified a role for delta opioid receptors to also modulate ethanol effects (Kang-Park et al., 2007), further implicating the opioid system in ethanol actions in CeA.
Prodynorphin and KOR immunoreactivity are abundant in the CeA (Mansour et al., 1996; Marchant et al., 2007). Activation of postsynaptic KOR in the CeA activates potassium currents in 8 to 17% of the recorded neurons (Chieng et al., 2006; Zhu and Pan, 2004), suggesting that only 1 out of 6 to 12 neurons express KOR. In our study, 80% of the neurons responded to dynorphin and 89% responded to U69 by a decrease of inhibitory transmission. Because the dynorphin effect occurred presynaptically, the presence of KOR on the recorded neuron is not necessary, whereas neurons displaying postsynaptic effects must express the receptor. Such KOR feature has also been reported in the globus pallidus where 25% of the recorded neurons showed a KOR-induced postsynaptic effect whereas 100% showed a KOR-induced decrease of GABAergic synaptic responses (Ogura and Kita, 2000). Thus CeA neurons possessing KOR influence the inhibitory synaptic network, and a large majority of neurons recorded in the medial division of the CeA were modulated by the dynorphin/KOR system, presumably on presynaptic terminals.
We showed that dynorphin decreased GABA release and opposed ethanol augmentation of GABA release from CeA neurons. It is important to note that endogenous levels of dynorphin are likely lower than exogenously applied concentrations, and endogenous dynorphin effects are likely smaller than those described in this study. Although one might anticipate desensitization of KOR upon superfusion of large concentrations of exogenous dynorphin (Liu-Chen, 2004), the effect of dynorphin did not weaken after up to 25 minutes of application. We conclude that under normal physiological conditions, ethanol effects on GABAergic transmission in CeA are modulated by endogenous dynorphin levels and actions at KOR, certainly in the ex vivo brain slice and likely also in the CeA of the alcohol-drinking organism.
4.2 A kappa opioid tone actively regulates inhibitory transmission in CeA
We observed that blockade of KOR augmented inhibitory responses in CeA, suggesting active production of an endogenous ligand acting at KOR. Because dynorphin and norBNI also act at MOR (Munro et al., 2012; Raynor et al., 1994), we verified that the tonic activity we report was specific to KOR activation by co-applying the MOR antagonist CTAP together with norBNI. Application of norBNI alone does not affect GABAergic activity in the bed nucleus of the stria terminalis, globus pallidus or ventral tegmental area, (Ford et al., 2007; Li et al., 2012; Ogura and Kita, 2000) but serotonin release is modulated by tonic KOR activity in septal neurons (Rutz et al., 2007) and increased neuronal firing in vivo has been reported in supraoptic neurons upon administration of norBNI (Brown et al., 1998), indicative of tonic dynorphin/KOR activity regulating neuronal activity in other brain regions.
We hypothesize that basal KOR activity in CeA is generated by an endogenous dynorphin tone that diminishes local GABA release and dampens inhibition, perhaps increasing activity of CeA output neurons. The CeA is the principal amygdaloid output nucleus that has a major influence on the activity of downstream effector regions responsible for producing physiological and behavioral responses to environmental stimuli (Cassell et al., 1999; Sah et al., 2003). Because CeA projections are mostly GABAergic, tonic KOR depression of inhibitory transmission in CeA likely results in inhibition of downstream targets. Reciprocally, KOR antagonist-induced increase of inhibitory transmission in CeA likely disinhibits downstream target structures.
4.3 The effect of ethanol is occluded by a kappa receptor antagonist
We report that blockade of KOR elicits effects similar to ethanol on GABAergic transmission in CeA. Interestingly, norBNI occluded or prevented the effect of ethanol. This finding differs from recent results reported from CeA slice recordings in mice that showed increased facilitation of GABAergic transmission by ethanol in slices from KOR knockout mice, and in slices from wild-type mice pre-treated with norBNI (Kang-Park et al., 2013). In that study, the facilitatory effect of ethanol alone on GABAergic transmission was significantly lower (14%) than values we have reported in rats (40–50% in this study as well as Roberto et al. 2003 and 2010), suggesting a differential ethanol sensitivity and/or mechanism of action in CeA of mice and rats. In addition, KOR distribution and KOR modulation of GABAergic transmission in CeA may also differ between mice and rats, since the Kang-Park et al. (2013) study reported that approximately 50% of recorded CeA neurons did not respond to KOR antagonism (25% in our study) and 40% did not respond to KOR agonism (15% in our study).
The blockade of the effect of ethanol by norBNI observed in our study could be due to a ceiling effect implicating a common pool of GABA or a common site of action on the GABA release machinery. This possibility is consistent with the results we obtained by reversing the order of application. Indeed ethanol prevented the effect of norBNI, indicating a reciprocal blockade by the two drugs and pointing to a common pool of transmitter targeted by norBNI and ethanol. On the other hand, ethanol produced larger increases in GABAergic transmission than norBNI, but the norBNI effect was not augmented by subsequent application of ethanol, contrary to what one would expect if both drugs are targeting a common pool of GABA. Furthermore, a prior study from our lab showed that CB1 receptor antagonism increased inhibitory transmission to a level similar to that observed with norBNI in the present study (Roberto et al., 2010), an effect that was augmented by subsequent application of ethanol. This suggests there may be a fundamental difference in the way that KOR and other receptors (e.g., CB1) interact with ethanol-activated cascades at the presynaptic GABA terminal in CeA.
Another possibility is the requirement of active or functioning KOR in order to permit ethanol actions on GABAergic transmission. The dynorphin/KOR system could be part of the cascade of events leading to increased GABA release by ethanol, for example KOR-induced activation of kinase cascades (Bruchas and Chavkin, 2010) or the unlocking of a particular pool of GABA. Intracellular signaling cascades downstream of KOR activation may interact with cascades activated by receptors for Corticotropin-Releasing Factor, a peptide that participates in ethanol actions in CeA, and that is intimately tied to dynorphin/KOR system activity (Bruchas et al., 2010; Heilig and Koob, 2007; Nie et al., 2004). However, ethanol effects on GABAergic transmission were not diminished by subsequent KOR blockade with norBNI, weakening the argument that functional KORs are required for ethanol effects. Further experiments are necessary to elucidate the exact mechanism of this KOR antagonist blockade or prevention of ethanol effects.
4.4 Physiological relevance
The CeA is part of a conceptual macrostructure called the extended amygdala that is profoundly implicated in alcohol and drug reinforcement and dependence (Koob 2003, 2008). The dynorphin/KOR system plays a key role in stress and addiction processes (Bruchas et al., 2010; Wee and Koob, 2010), and antagonism of KOR selectively reduces alcohol self-administration in alcohol-dependent rats (Walker et al., 2012). We showed here that the dynorphin/KOR system tonically influences CeA activity. An active dynorphin/KOR tone has not been observed in other limbic structures such as the bed nucleus of the stria terminalis, basolateral amygdala or nucleus accumbens (Huge et al., 2009; Mu et al., 2011; Li et al., 2012). It remains to be seen whether chronic alcohol produces neuroadaptations in this KOR tone in CeA during the transition to dependence.
Because acute ethanol increases dynorphin release in CeA (Lam et al., 2008), we hypothesize that ethanol consumption by non-dependent animals stimulates dynorphin release that functions as a brake to limit ethanol-induced enhancement of GABAergic transmission in CeA, thereby preventing disinhibition of downstream targets of CeA. Indeed, dynorphin activation suppresses drug reward and drug self-administration in non-dependent animals (Shippenberg et al., 2007) including alcohol (Nestby et al., 1999), suggesting that its physiological role is to limit excessive drug seeking (Shippenberg et al., 2007). It is reasonable to hypothesize that KOR effects on GABAergic transmission would be substantially altered in the CeA of an alcohol-dependent rat. The transition to alcohol and drug dependence produces hyperactivation of brain KOR systems that promote negative affect and subsequent seeking of alcohol and drugs for their negative reinforcing effects (Koob, 2008; Wee and Koob, 2010). Indeed, recent data from our group shows that pharmacological manipulations of KORs produces opposite effects on GABAergic transmission in CeA of rats with in vivo history of long-access versus short-access to cocaine (Kallupi et al., 2013), suggesting the same may be true for other drugs of abuse, including alcohol.
4.5 Conclusion
The dynorphin/KOR system has emerged as a potential target for the treatment of alcohol and drug dependence. We showed here that KOR ligands decrease inhibitory transmission in CeA neurons and interfere with the physiological effects of ethanol. We also uncovered a dynorphin/KOR tone that influences neuronal activity in the CeA, a brain structure that plays a major role in alcohol dependence and reinforcement. Further studies will determine the influence of chronic alcohol treatment on the cellular effects elicited by the dynorphin/KOR system and its interactions with other neuromodulators (e.g., Corticotropin-Releasing Factor) implicated in CeA signaling in the alcohol-dependent organism.
Highlights.
We studied dynorphin/ethanol interactions in slices of the rat central amygdala
Kappa opioid receptor agonists decreased GABA transmission
Endogenous Kappa agonists tonically inhibit the central amygdala network
Dynorphin decreased the augmenting effect of ethanol on GABA transmission
Kappa receptor antagonism blocked the effect of ethanol on GABA transmission
Acknowledgments
This work was supported by National Institutes of Health funding from NIAAA (AA018400, AA013517, AA06420 and AA016985). This is publication number 24011 from The Scripps Research Institute.
Footnotes
Disclosure/Conflicts of interest
The authors have no financial interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Brown CH, Ludwig M, Leng G. kappa-opioid regulation of neuronal activity in the rat supraoptic nucleus in vivo. J Neurosci. 1998;18:9480–9488. doi: 10.1523/JNEUROSCI.18-22-09480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassell MD, Freedman LJ, Shi C. The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci. 1999;877:217–241. doi: 10.1111/j.1749-6632.1999.tb09270.x. [DOI] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Chieng BC, Christie MJ, Osborne PB. Characterization of neurons in the rat central nucleus of the amygdala: cellular physiology, morphology, and opioid sensitivity. J Comp Neurol. 2006;497:910–927. doi: 10.1002/cne.21025. [DOI] [PubMed] [Google Scholar]
- D’Addario C, Caputi FF, Rimondini R, Gandolfi O, Del BE, Candeletti S, Romualdi P. Different alcohol exposures induce selective alterations on the expression of dynorphin and nociceptin systems related genes in rat brain. Addict Biol. 2011;18:425–433. doi: 10.1111/j.1369-1600.2011.00326.x. [DOI] [PubMed] [Google Scholar]
- Dar MS. Involvement of kappa-opioids in the mouse cerebellar adenosinergic modulation of ethanol-induced motor incoordination. Alcohol Clin Exp Res. 1998;22:444–454. [PubMed] [Google Scholar]
- Davis M, Rainnie DG, Cassell MD. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Huge V, Rammes G, Beyer A, Zieglgansberger W, Azad SC. Activation of kappa opioid receptors decreases synaptic transmission and inhibits long-term potentiation in the basolateral amygdala of the mouse. Eur J Pain. 2009;13:124–129. doi: 10.1016/j.ejpain.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Hyytia P, Koob GF. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol. 1995;283:151–159. doi: 10.1016/0014-2999(95)00314-b. [DOI] [PubMed] [Google Scholar]
- Kallupi M, Wee S, Edwards S, Whitfield TW, Jr, Oleata CS, Luu G, Schmeichel BE, Koob GF, Roberto M. Kappa opioid receptor-mediated dysregulation of gamma-aminobutyric acidergic transmission in the central amygdala in cocaine addiction. Biol Psychiatry. 2013;74:520–528. doi: 10.1016/j.biopsych.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang-Park MH, Kieffer BL, Roberts AJ, Siggins GR, Moore SD. Presynaptic delta opioid receptors regulate ethanol actions in central amygdala. J Pharmacol Exp Ther. 2007;320:917–925. doi: 10.1124/jpet.106.112722. [DOI] [PubMed] [Google Scholar]
- Kang-Park MH, Kieffer BL, Roberts AJ, Siggins GR, Moore SD. Kappa opioid receptors in the central amygdala regulate ethanol actions at presynaptic GABAergic sites. J Pharmacol Exp Ther. 2013;346:130–137. doi: 10.1124/jpet.112.202903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Eur Neuropsychopharmacol. 2003;13:442–452. doi: 10.1016/j.euroneuro.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role for GABA mechanisms in the motivational effects of alcohol. Biochem Pharmacol. 2004;68:1515–1525. doi: 10.1016/j.bcp.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs KM, Szakall I, O’Brien D, Wang R, Vinod KY, Saito M, Simonin F, Kieffer BL, Vadasz C. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol Clin Exp Res. 2005;29:730–738. doi: 10.1097/01.alc.0000164361.62346.d6. [DOI] [PubMed] [Google Scholar]
- Lam MP, Marinelli PW, Bai L, Gianoulakis C. Effects of acute ethanol on opioid peptide release in the central amygdala: an in vivo microdialysis study. Psychopharmacology (Berl) 2008;201:261–271. doi: 10.1007/s00213-008-1267-8. [DOI] [PubMed] [Google Scholar]
- Lemos JC, Roth CA, Messinger DI, Gill HK, Phillips PE, Chavkin C. Repeated stress dysregulates kappa-opioid receptor signaling in the dorsal raphe through a p38alpha MAPK-dependent mechanism. J Neurosci. 2012;32:12325–12336. doi: 10.1523/JNEUROSCI.2053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB, Stuber GD, Kash TL. Presynaptic Inhibition of Gamma-Aminobutyric Acid Release in the Bed Nucleus of the Stria Terminalis by Kappa Opioid Receptor Signaling. Biol Psychiatry. 2012;71:725–732. doi: 10.1016/j.biopsych.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chen LY. Agonist-induced regulation and trafficking of kappa opioid receptors. Life Sci. 2004;75:511–536. doi: 10.1016/j.lfs.2003.10.041. [DOI] [PubMed] [Google Scholar]
- Mansour A, Burke S, Pavlic RJ, Akil H, Watson SJ. Immunohistochemical localization of the cloned kappa 1 receptor in the rat CNS and pituitary. Neuroscience. 1996;71:671–690. doi: 10.1016/0306-4522(95)00464-5. [DOI] [PubMed] [Google Scholar]
- Marchant NJ, Densmore VS, Osborne PB. Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: evidence of two distinct endogenous opioid systems in the lateral division. J Comp Neurol. 2007;504:702–715. doi: 10.1002/cne.21464. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Lam M, Bai L, Quirion R, Gianoulakis C. A microdialysis profile of dynorphin A(1–8) release in the rat nucleus accumbens following alcohol administration. Alcohol Clin Exp Res. 2006;30:982–990. doi: 10.1111/j.1530-0277.2006.00112.x. [DOI] [PubMed] [Google Scholar]
- Matsuzawa S, Suzuki T, Misawa M, Nagase H. Different roles of mu-, delta- and kappa-opioid receptors in ethanol-associated place preference in rats exposed to conditioned fear stress. Eur J Pharmacol. 1999;368:9–16. doi: 10.1016/s0014-2999(99)00008-4. [DOI] [PubMed] [Google Scholar]
- Mu P, Neumann PA, Panksepp J, Schluter OM, Dong Y. Exposure to cocaine alters dynorphin-mediated regulation of excitatory synaptic transmission in nucleus accumbens neurons. Biol Psychiatry. 2011;69:228–235. doi: 10.1016/j.biopsych.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro TA, Berry LM, Van’t Veer A, Beguin C, Carroll FI, Zhao Z, Carlezon WA, Jr, Cohen BM. Long-acting kappa opioid antagonists nor-BNI, GNTI and JDTic: pharmacokinetics in mice and lipophilicity. BMC Pharmacol. 2012;12:5. doi: 10.1186/1471-2210-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestby P, Schoffelmeer AN, Homberg JR, Wardeh G, De Vries TJ, Mulder AH, Vanderschuren LJ. Bremazocine reduces unrestricted free-choice ethanol self-administration in rats without affecting sucrose preference. Psychopharmacology (Berl) 1999;142:309–317. doi: 10.1007/s002130050894. [DOI] [PubMed] [Google Scholar]
- Nie ZG, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science. 2004;303:1512–1514. doi: 10.1126/science.1092550. [DOI] [PubMed] [Google Scholar]
- Ogura M, Kita H. Dynorphin exerts both postsynaptic and presynaptic effects in the globus pallidus of the rat. J Neurophysiol. 2000;83:3366–3376. doi: 10.1152/jn.2000.83.6.3366. [DOI] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci. 2011;31:281–288. doi: 10.1523/JNEUROSCI.4654-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohorecky LA, Patel V, Roberts P. Effects of ethanol in an open field apparatus: modification by U50488H and WIN 44441-3. Physiol Behav. 1989;45:273–287. doi: 10.1016/0031-9384(89)90129-7. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor-binaltorphimine, potent and selective kappa-opioid receptor antagonists. Life Sci. 1987;40:1287–1292. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Bajo M, Siggins GR, Parsons LH, Schweitzer P. The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology. 2010;35:1962–1972. doi: 10.1038/npp.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA. 2003;100:2053–2058. doi: 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz S, Riegert C, Rothmaier AK, Jackisch R. Presynaptic modulation of 5-HT release in the rat septal region. Neuroscience. 2007;146:643–658. doi: 10.1016/j.neuroscience.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez de AM, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie ZG. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Walker BM, Koob GF. Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BM, Valdez GR, McLaughlin JP, Bakalkin G. Targeting dynorphin/kappa opioid receptor systems to treat alcohol abuse and dependence. Alcohol. 2012;46:359–370. doi: 10.1016/j.alcohol.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther. 2006;111:533–554. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Zhu W, Pan ZZ. Synaptic properties and postsynaptic opioid effects in rat central amygdala neurons. Neuroscience. 2004;127:871–879. doi: 10.1016/j.neuroscience.2004.05.043. [DOI] [PubMed] [Google Scholar]