

Abstract
Cancer cell cytotoxicity-guided fractionation of the acetone extracts of two cultured marine-derived Streptomyces strains belonging to the MAR4 group yielded six new napyradiomycins, compounds A–F (1–6), together with three known compounds, napyradiomycins B2–B4 (7–9). Napyradiomycins 1–4 are new members of the napyradiomycin “C type“ meroterpenoids that possess a linear monoterpene moiety bridging between C-7 and C-10a. Compound 4 has an additional tetrahydropyran ring fused to the phenol moiety. Compounds 5–9 are related to the napyradiomycin “B type” meroterpenoids. The structures of all new compounds were assigned by interpretation of 1D and 2D NMR, MS and other spectroscopic data. The relative configurations were assigned based upon interpretation of ROESY 2D NMR experiments. The cytotoxicity of 1–9 against the human colon carcinoma cell line HCT-116, and their antibacterial activities against Methicillin-resistant Staphylococcus aureus (MRSA) are presented.
Keywords: napyradiomycins, cancer cell cytotoxicity, antibiotics, mixed biosynthesis, meroterpenoids, halogenated terpenoids, dihydronaphthoquinones
Introduction
Actinomycete bacteria are commonly found in the soil and are characterised by complex genomes possessing numerous secondary metabolite biosynthetic clusters. Among the actinomycetes, those of the genus Streptomyces produce over two-thirds of the clinically useful antibiotics of natural origin. By the 1970s, the discovery of new antibiotics from Streptomyces strains was in substantial decline with fewer new molecules being reported.1 By the 1990s, the overall enthusiasm for microbial antibiotic discovery had begun to decline. Because drug resistance is a consequence of antibiotics misuse and pathogen evolution, the discovery of new antibiotics is, more than ever, viewed as an essential activity. Since the existence of true marine actinomycetes was revealed in 1984, unique bioactive natural products such as marinopyrroles,2 heronapyrroles,3 ansalactam,4 ammosamides5 and salinosporamide A6 have been reported from marine strains indicating a new direction for microorganism-based drug discovery.
One group of antibacterial agents, the napyradiomycins, was first isolated from the soil-derived bacterium Chainia rubra G802-AF1 in 1986.6–8 As time passed, a series of congeners were isolated from different actinomycetes, such as Streptomyces antimycoticus,9 S. aculeolatus,10 and other Streptomyces strains,11,12 and also from a marine-derived strain.13
The napyradiomycins are meroterpenoids composed of polyketide and terpenoid components, and they are formed by a process shown to involve halogen-induced cyclization.14 This class of metabolites is well-known to possess significant antibacterial activities.6–11 For example, in a recent pharmacological study,15 napyradiomycin derivatives demonstrated potent bactericidal activity against contemporary and clinical relevant methicillinresistant Staphylococcus aureus (MRSA) strains, indicating they might be of some utility. In this paper, we report the isolation of six new napyradiomycin analogs, compounds 1–6, and three known derivatives, napyradiomycins B2–B4 (7–9)7,8,10, from the culture extracts of two marine-derived Streptomyces strains. These strains share >99% 16S rRNA gene sequence identity and belong to the MAR4 group of Streptomyces, which are best known for the production of meroterpenoid and other hybrid isoprenoid secondary metabolites.15 We also report their evaluation against Methicillin-resistant Staphylococcus aureus (MRSA) and their inhibitory effects against the human colon adenocarcinoma cell line (HCT-116).
Results and Discussion
Streptomyces strains CNQ-329 and CNH-070 were isolated from marine sediments collected in California. These strains were cultured in a saltwater-based medium in 10 and 5 L scale, respectively. After 7 days of fermentation, cultures were resin extracted (20g/L, XAD-7), the resin was collected by filtration through cheesecloth and then extracted with acetone. The acetone was removed from the extract under vacuum, and the residue was partitioned between H2O and EtOAc to yield separate organic extracts for each strain. Repeated flash and HPLC chromatographic purification of these extracts afforded six new napyradiomycin derivatives (1–6), together with three known compounds, napyradiomycins B2–B4 (7–9, Figure 1).7,10 The structures of all compounds were established by interpretation of spectral data, especially 2D NMR. The relative configurations of the new compounds 1–6 were assigned by interpretation of ROESY NMR data.
Figure 1.

Structures of the napyradiomycins 1–9 isolated and identified.
Napyradiomycin 1 was obtained as a white powder and was assigned the molecular formula, C25H27ClO6 (12 degrees of unsaturation), by evaluation of HRESIMS (m/z 459.1596 [M+H]+) and NMR data. The IR spectrum of 1 showed the presence of hydroxyl (3422 cm−1), conjugated carbonyl (1705 and 1649 cm−1) and ether (1056 cm−1) functional groups. These data and the UV spectrum of 1 suggested that this compound posessed the classic dihydronaphthoquinone moiety as typically observed in the napyradiomycin class metabolites. The 1H NMR spectrum of 1 (Table 1) in DMSO-d6 exhibited three methyl singlets (δ 1.02, 1.14, and 1.42), two aromatic methine singlets (δ 6.81 and 6.88), two olefinic methine signals (δ 4.26 and 5.27), one pairs of oxygenbearing methylene signals (δ 3.91 and 3.94), and a characteristic peri-proton singlet (δ 12.69) derived from a hydroxyl group. 13C NMR (Table 2) and HSQC NMR spectral data for 1 showed 25 carbon signals, consisting of two carbonyls (δ 187.7 and 194.8), four olefinic methines (δ 106.4, 117.5, 123.9 and 136.0), eight olefinic quaternary carbons (δ 112.9, 120.4, 134.7, 135.9, 136.4, 138.9, 161.3, and 163.2), one oxymethine carbon (δ 67.6), two oxygenated quaternary carbons (δ 76.2 and 83.4), one aliphatic methine (δ 59.4), four aliphatic methylenes (δ 22.9, 24.0, 38.8, and 42.2), and three methyls (δ 14.2, 20.3, and 27.0). These assignments accounted for eight of the twelve degrees of unsaturation, indicating that 1 was composed of a tetracyclic ring system.
Table 1.
1H NMR Spectroscopic Data (500 MHz) for 1–3a
| C# | 1 | 2 | 3 |
|---|---|---|---|
| 2-Me | 1.02 s | 0.95 s | 1.02 s |
| 2-Me | 1.42 s | 1.29 s | 1.44 s |
| 3 | 4.95 s | 3.74 t (6.5) | 4.99 d (1.5) |
| 4 | 6.81 s | 5.16 d (6.8) | 6.83 d (1.5) |
| 6-OH | 12.69 s | 13.00 s | 12.88 s |
| 9 | 6.88 s | 6.72 s | 6.85 s |
| 11a | 2.35 dd (8.4, 13.1) | 2.38 dd (9.3, 13.3) | 2.31 dd (6.4, 13.3) |
| 11b | 2.43 dd (9.8, 13.1) | 2.57 dd (8.7, 13.3) | 2.55 dd (11.1, 13.3) |
| 12 | 4.26 t (8.4) | 4.33 t (8.5) | 4.38 dd (6.4, 11.1) |
| 14 | 1.47 m | 1.54 m | 1.35 m |
| 1.58 m | 1.54 m | 1.89 m | |
| 15 | 1.37 m | 1.44 m | 0.02 m |
| 1.59 m | 1.71 m | 1.10 m | |
| 16 | 5.27 t (7.5) | 5.26 t (8.4) | 3.74 dd (2.2, 8.3) |
| 18 | 3.24 m | 3.24 m | 3.49 d (13.6) |
| 3.33 m | 3.24 m | 3.57 d (13.6) | |
| 19 | 3.91 d (13.8) | 3.89 (12.7) | 5.01 s |
| 3.94 d (13.8) | 3.96 d (12.7) | 5.09 s | |
| 20 | 1.14 s | 1.17 s | 1.15 s |
Chemical shifts are in ppm; J values in Hz are in parentheses; Measured in DMSO-d6.
Table 2.
13C NMR Spectroscopic Data (75 MHz) for 1–3a
| C# | 1 | 2 | 3 |
|---|---|---|---|
| 2 | 76.2 C | 75.1 C | 76.1 C |
| 2a-Me | 20.3 CH3 | 24.8 CH3 | 20.2 CH3 |
| 2b-Me | 27.0 CH3 | 25.5 CH3 | 27.0 CH3 |
| 3 | 59.4 CH | 65.8 CH | 59.4 CH |
| 4 | 136.0 CH | 135.0 CH | 136.1 CH |
| 4a | 136.4 C | 137.4 C | 136.5 C |
| 5 | 187.7 C | 188.5 C | 187.8 C |
| 5a | 112.9 C | 111.7 C | 112.2 C |
| 6 | 161.3 C | 161.1 C | 161.8 C |
| 7 | 120.4 C | 119.8 C | 118.9 C |
| 8 | 163.2 C | 161.1 C | 164.4 C |
| 9 | 106.4 CH | 107.3 CH | 106.5 CH |
| 9a | 134.7 C | 137.1 C | 134.4 C |
| 10 | 194.8 C | 196.5 C | 194.8 C |
| 10a | 83.4 C | 83.0 C | 82.8 C |
| 11 | 42.2 CH2 | 42.4 CH2 | 42.4 CH2 |
| 12 | 117.5 CH | 118.2 CH | 116.3 CH |
| 13 | 138.9 C | 137.8 C | 141.0 C |
| 14 | 38.8 CH2 | 38.9 CH2 | 37.0 CH2 |
| 15 | 24.0 CH2 | 24.2 CH2 | 36.1 CH2 |
| 16 | 123.9 CH | 124.3 CH | 73.8 CH |
| 17 | 135.9 C | 136.4 C | 149.9 C |
| 18 | 22.9 CH2 | 22.7 CH2 | 28.3 CH2 |
| 19 | 67.6 CH2 | 67.9 CH2 | 111.5 CH2 |
| 20 | 14.2 CH3 | 14.1 CH3 | 15.0 CH3 |
Assignments were made on the basis of HMQC and HMBC data; Measured in DMSO-d6.
The overall 1H and 13C NMR data for 1 confirmed that the compound posessed a dihydronaphthoquinone moiety with two terpenoid substituents. The dihydronaphthoquinone ring was constructed on the basis of HMBC correlations from H-9 (δ 6.88) to C-5a (δ 112.9), C-6 (δ 161.3), C-8 (δ 163.2), C-9a (δ 134.7), and C-10 (δ 194.8), and from peri-proton (OH at C-6; δ 12.69) to C-5a, C-6, and C-7 (δ 120.4), as well as from H-4 to C-4a (δ 136.4), C-5 (δ 187.7) and C-10a (δ 83.4) (Figure 2). In addition, HMBC correlations from the two geminal methyl groups (δ 1.02, and 1.42) to C-2 (δ 76.2) and C-3 (δ 59.4), and from H-4 (δ 6.81) to C-2, and C-3 indicated the presence of a dehydropyran ring with a chlorine substituent at C-3. These NMR features, and HMBC correlations of H-4 with C-4a, C-5, and C-10a, revealed this partial structure was positioned at C-4a of the dihydronaphthoquinone ring, similar to the structure of napyradiomycin B2.7 COSY NMR data (Figure 2) derived from 1 exhibited two proton spin systems of H2-11 (δ 2.35 and 2.43)/H-12 (δ 4.26) and H2-14 (δ 1.47 and 1.58)/H2-15 (δ 1.37 and 1.59)/H-16 (δ 5.27). These two proton sequences could be connected by interpretation of HMBC correlations from the methyl group protons (C-20, δ 1.14) with C-12 (δ 117.5), C-13 (δ 138.9), and C-14 (δ 38.8). Moreover, HMBC correlations of H-19 (δ 3.91 and 3.94) with C-16 (δ 123.9), C-17 (δ 135.9), and C-18 (δ 22.9) were used to establish a bridging monoterpenoid link from C-11 to C-19. Further data illustrating this link between C-10a and C-7 was defined by HMBC correlations from H-11 to C-4a, C-10, and C-10a and from H-18 to C-6, C-7, and C-8. On the basis of these definitive NMR features, the planar structure of 1 was established.
Figure 2.
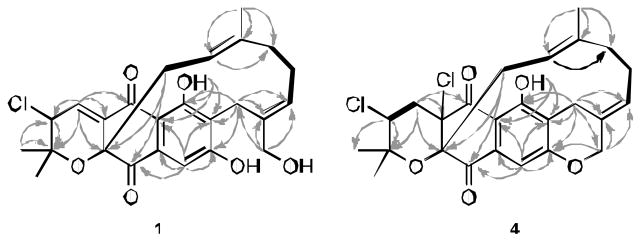
HMBC (arrows) and COSY NMR (bold bonds) correlations observed for 1 and 4.
The relative configuration of 1 was determined on the basis of ROESY correlations as illustrated in Figure 3. Considering the results from biosynthetic studies of napyradiomycin derivatives,13 H2-11 is often β-oriented. The ROESY NMR spectrum of 1 showed correlations between H-11 and H-3 indicating that they are on the same face of the molecule. Moreover, the presence of ROESY cross-peaks between H-11/H-12, H-11/C-13-Me, and the absence of H-12/C-13-Me allowed the E-geometry of the double bond at C-12 to be assigned. Clear ROESY correlations between H2-19 and H-16 allowed the geometry of the double bond at C-16 to also be assigned as E. On the basis of the data described above, the relative configuration of 1 was established as shown.
Figure 3.
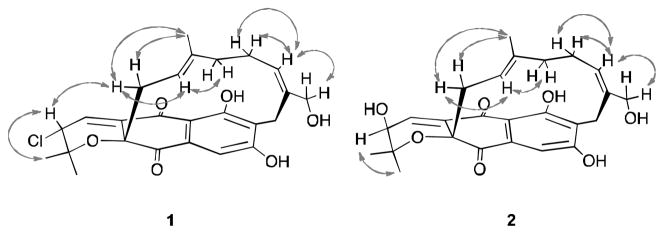
Selected NMR ROESY proton interactions observed for 1 and 2.
Napyradiomycin 2 was assigned the molecular formula C25H28O7, indicating twelve degrees of unsaturation, on the basis of HRESIMS (m/z 463.1723 [M+Na]+) and 13C NMR spectral data. IR absorption bands revealed the presence of hydroxyl (3407 cm−1), conjugated carbonyl (1700, 1649 cm−1), and ether (1056 cm−1) functionalities in 2. The 1H, 13C NMR and UV data for this compound were analogous to those from 1, suggesting it was closely related. On comparing the 13C NMR spectra of these two compounds, it was noted that the C-3 chemical shifts of 2 and 1 were δ 65.8 and 59.4, respectively. This suggested that a chlorine substituent at C-3 was replaced by a hydroxyl group. The molecular formula was also consistent with this conclusion.
The hydroxyl replacement was confirmed by the observation of HMBC correlations from both H-2-Me (δ 0.95 and 1.29) and H-4 (δ 6.97) to C-3, and COSY correlations between H-3 (δ 3.74) and H-4. The relative configuration and geometry of 2 was determined on the basis of ROESY experiments (Figure 3). The presence of ROESY correlations between H-3 and both C-2-Mes and the absence of any ROESY correlation between H-3 and H-11 indicated that the hydroxyl group attached at C-3 was on the β-face of the molecule. The E-geometries of two double bonds at C-12 and C-16 were assigned on the same ROESY NMR evidence as in 1. On the basis of this reasoning, structure 2 was assigned.
Napyradiomycin 3 analyzed for C25H27ClO6, a molecular formula identical to 1, on the basis of HRESIMS (m/z 481.1386 [M+Na]+) data. The UV, IR, and NMR spectral data of 3 were highly analogous to those of 1. Comparing the 1H and 13C NMR spectra of 3 and 1 illustrated the presence of an exocyclic methylene (δH 5.01 and 5.09; δC 111.5) and an oxymethine (δH 3.74; δC 73.8) carbon in 3 instead of olefinic methine (δH 5.27; δC 123.9) and oxygen-bearing methylene (δH 3.91 and 3.94; δC 64.6) carbons in 1. These two carbons were assigned as C-16 and C-19 on the basis of HMBC correlations from H2-19 to C-16, C-17 (δ 149.9), and C-18 (δ 28.3), and from H-16 to C-17 and C-19. The NOESY spectrum of 3 (Figure 4) exhibited correlations between H-11/H-3, and H-11/H-12/H-14, indicating the β-orientation of H-3 and E-geometry of the double bond at C-12, the same as 1. It was noted that H2-15 (δ 0.02 and 1.10) were quite shielded as a result of the proximate aromatic ring. This finding suggested that the C-15 protons are α-oriented. The ROESY correlations of H-16 (δ 3.74) with H2-15 revealed that the hydroxyl group attached at C-16 was β-oriented. Similar shielding does not appear to occur in napyradiomycins 1 and 2, presumable because the olefinic bond nearby in within the ring modifying the conformation of the bridging terpenoid chain. From the data obtained the napyradiomycin 3 was confidently assigned.
Figure 4.
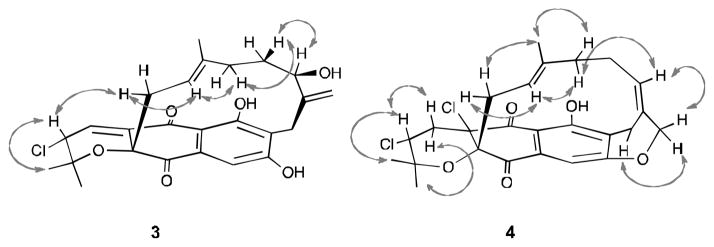
Selected NMR ROESY proton interactions observed for 3 and 4.
Napyradiomycin 4 was obtained as a yellow amorphous powder that analyzed for C25H26Cl2O5 (12 unsaturations), a molecular formula infered from HRESIMS (m/z 499.1105 [M+Na]+) data. The similarity of the UV and IR spectral data of 4 and 1 suggested these compounds to be analogs. HMBC NMR correlations from H-3 (δ 4.37) to C-2 (δ 78.3), C-2-Me (δ 21.9 and 29.5), and C-4a (δ 75.2) and from H2-4 (δ 2.33 and 2.70) to C-2, C-3 (δ 60.0), C-4a, and C-5 (δ 185.2) indicated the presence of the familiar pyran ring and two chlorine atoms substituted at C-3 and C-4a.6 The 13C NMR spectrum of 4 illustrated twelve sp2 carbons and lacked the common C-4/C-4a double bond in comparison to 1. However, based upon an additional degree of unsaturation napyradiomycin 4 was concluded to possess an additional ring. HMBC NMR correlations from H2-19 (δ 4.53 and 4.83) to C-8 (δ 156.8), and from H2-18 (δ 2.99 and 3.32) to C-6 (δ 159.6), C-7 (δ 123.9), C-8, C-17 (δ 132.6) and C-19 (δ 75.4), revealed the presence of a second tetrahydropyran ring fused to the phenolic ring (Figure 2). The relative configuration of 4 was determined by interpretation of ROESY NMR data (Figure 4). The results of the ROESY NMR experiment were identical to that from 1, suggesting the same configuration at C-3 and the same geometry of the double bond at C-12. The double bond at C-16 was assigned with E-geometry as a result of the observation of ROESY NMR correlations between H-15/H-16/H-19 and the absence of ROESY correlation between H-16/H2-18. It’s noteworthy that 4 shares a similar planar structure to Napyradiomycin SR,9 except that the geometry of the double bond at C-16 is reversed.
Napyradiomycin 5 analyzed for the molecular formula C25H28BrClO5 by HRESIMS analysis. The UV, IR, 1H and 13C NMR data were quite similar to those of Napyradiomycin B2 (7)10, suggesting that 5 is a close analog of 7. The major difference between them is that a chlorine atom is replaced by a bromine atom in 5. The halogenated methine (δH 4.38; δC 70.6) in 1 shifted to (δH 4.65; δC 67.6) in 5. This methine was assigned at C-16 on the basis of HMBC NMR correlations from H-16 to C-15 (δ 35.9) and C-17-Me (δ 15.9 and 27.6). The HRESIMS of 6 revealed a pseudomolecular ion peak at m/z 495.1566 [M+Na]+ corresponding to the molecular formula C25H32Cl2O6. This compound had similar spectral features to Napyradiomycin B4 (8), except it lacked the chlorine substituent.8 The COSY NMR spectrum showed correlations between H-3 (δ 3.93)/H2-4 (δ 2.49 and 2.11)/H-4a (δ 3.15. This strongly suggested that the chlorine substitution was replaced by hydrogen. This was confirmed by interpretation of HMBC correlations from H-4a to C-5 (δ 199.1), C-5a (δ 109.3), C-10 (δ 193.9), and C-10a (δ 81.2). On the basis of the above deductions, the structures of napyradiomycins 5 and 6 were confidently assigned.
Conclusions
Napyradiomycins 1–9 were tested for their in vitro inhibitory activity against Methicillin-resistant Staphylococcus aureus (MRSA) and their cytotoxicities against the human colon denocarcinoma cell line (HCT-116); the results are shown in Table 5. Compounds 1 and 8 are the most active analogs against MRSA (16 and 2 mg/mL), respectively. In the cytotoxicity test, napyradiomycins 1 and 9 showed the strongest activity (IC50 = 4.19 and 1.41 μg/mL), while compounds 2 and 6 were less active. From these data, it would appear that replacing a chloring atom with a hydroxyl group at C-3, or with a hydrogen at position C-4a, results in diminished activity.
Table 5.
Cytotoxicities and Antibacterial Activities of 1–9
| compound | HCT-116 | MRSA inhibition |
|---|---|---|
|
| ||
| IC50 (μg/mL) | MIC (μg/mL) | |
| Napyradiomycin 1 | 4.19 | 16 |
| Napyradiomycin 2 | >20 | 64 |
| Napyradiomycin 3 | >20 | >64 |
| Napyradiomycin 4 | 16.1 | >64 |
| Napyradiomycin 5 | 4.81 | >64 |
| Napyradiomycin 6 | 9.42 | >64 |
| Napyradiomycin B2 (7) | 3.18 | 32–64 |
| Napyradiomycin B3 (8) | 0.19 | 2 |
| Napyradiomycin B4 (9) | 1.41 | 32 |
Experimental Section
General Methods
Optical rotations were measured using a Rudolph Research Autopol III polarimeter with a 1 cm cell. UV spectra were recorded in a Varian Cary UV-visible spectrophotometer with a 1 cm path length cell, and IR spectra were obtained with a Thermo Nicolet IR100 FTIR spectrophotometer. 1H, 13C, and 2D NMR spectral data were obtained in CDCl3 or DMSO-d6 on Varian Unity 300 and 500 MHz NMR spectrometers. High-resolution ESI-TOF mass spectra are provided by the mass spectrometry facility on the Department of Chemistry and Biochemistry at the University of California, San Diego, La Jolla, CA. Low-resolution LC/MS data were measured using a Hewlett-Packard series 1100 LC/MS system with a reversed-phase C18 column (Phenomenex Luna, 4.6 mm ×100 mm, 5 μm) at a flow rate of 0.7 mL/min. A Betasil Phenyl column (5 μm, 250-10, Keystone Scientific Inc.) was also used for RP- HPLC (Shimadzu, SCL-10A; flow rate 3.5 mL/min, SPD-M10A DAD detection).
Collection, Identification, Cultivation and Extraction of Strain CNQ- 329
Strain CNQ-329 was isolated on A1 agar medium (10 g starch, 4 g yeast extract, 2 g peptone, and 18 g agar in 750 ml of seawater and 250 ml distilled water) from a marine sediment sample collected in San Diego, California. The 16S rRNA gene sequence for this strain has been deposited with GenBank (accession number KC261610). It shares 99.0% sequence identity with the type strain for Streptomyces aculeolatus (accession number NR_041166). Strain CNQ-329 was cultured in ten replicate 2.5 L Ultra Yield flasks (Thomson Scientific) each containing 1 L of fermentation medium A1 without agar for 7 days at 27 °C while shaking at 200 rpm. At the end of the fermentation period, 20 g/L Amberlite XAD-7 adsorbent resin was added into each flask, and shaking was continued for two additional hours at 100 rpm. The resin was then collected by filtration through cheesecloth, washed with deionized water, and eluted twice with acetone. The acetone solution was concentrated to afford an organic extract.
Collection, Identification, Cultivation and Extraction of Strain CNH- 070
Strain CNH-070 was isolated on A1 agar medium (10 g starch, 4 g yeast extract, 2 g peptone, and 18 g agar in 750 ml seawater and 250 ml distilled water) from a marine sediment sample collected at the mouth of San Elijo Lagoon in Encinitas, California at a depth of 1 meter in 1993. The 16S rRNA gene sequence for this strain has been deposited with GenBank (accession number KC261625). The strain shares 99.7% sequence identity with the S. aculeolatus type strain. Strain CNH-070 was cultured in five replicate 2.5 L Ultra Yield flasks (Thomson Scientific Co.) each containing 1 L of fermentation medium A1 without agar for 7 days at 27 °C while shaking at 200 rpm. At the end of the fermentation period, 20 g/L Amberlite XAD-7 adsorbent resin was added to each flask, and shaking was continued for two additional hours at 100 rpm. The resin was then collected by filtration through cheesecloth, washed with deionized water, and eluted twice with acetone. The acetone solution was concentrated to afford an organic extract.
Isolation of Compounds 1–9
The acetone extract of strain CNQ-329 was partitioned between EtOAc and H2O (1:1) to obtain an EtOAc-soluble layer. After evaporating the organic solvent to afford a EtOAc extract (830.0 mg). This extract was subjected to fractionation using a Si gel flash column (nhexane–EtOAc–MeOH, gradient from 10:1:0 to 0:0:1) to furnish fractions A-I. Fraction C (75.1 mg) was separated by preparative TLC (iso-octane–CH2Cl2–MeOH, 10:10:1) to furnish subfractions C1–C4. Subfraction C1 was subjected to RP-HPLC (Phenomenex C-18, 10 mm ×250 mm, flow = 2.0 mL/min, MeCN–H2O, gradient from 85:15 to 95:5) to give 7 (3.0 mg) and 5 (1.9 mg). Fraction D (87.1 mg) was subjected to RP-HPLC (Betasil Phenyl, 10 mm ×250 mm, flow = 3.5 mL/min, MeCN–H2O, 70:30 to 90:10) to afford 1 (12.0 mg) and 4 (26.0 mg). Fraction E (107.4 mg) was separated by RP-HPLC (Betasil Phenyl, 10 mm ×250 mm, flow = 3.5 mL/min, MeCN–H2O, gradient from 65:35 to 90:10) to give a subfraction E-10 (11.1 mg). Subfraction E-10 was further purified by RP-HPLC (Betasil Phenyl, 10 mm ×250 mm, flow = 3.5 mL/min, MeCN–H2O, gradient from 55:45 to 80:20) to yield 3 (3.8 mg). Separation of fraction F (62.0 mg) by RP-HPLC (Betasil Phenyl, 10 mm ×250 mm, flow = 3.5 mL/min, MeCN–H2O, gradient from 55:45 to 80:20) yielded subfraction F- 1 (18.6 mg). Consequently, subfraction F1 was purified by RP-HPLC (Betasil Phenyl, 10 mm ×250 mm, flow = 3.5 mL/min, MeCN–H2O, 40:60) to obtain 2 (2.6 mg).
The acetone extract of strain CNH-070 was partitioned between EtOAc and H2O (1:1) to obtain an EtOAc-soluble layer, which after solvent removal gave an EtOAc extract (1096.2 mg). This extract was subjected to fractionation using a Si gel open column (n-hexane–EtOAc–MeOH, gradient from 10:1:0 to 0:0:1) to furnish fractions A–I. ). Fraction C (247.7 mg) was subjected to RP-HPLC (Betasil Phenyl, 10 mm ×250 mm, flow = 4.0 mL/min, MeCN–H2O, 75:25 to 90:10) to afford 6 (2.8 mg), 7 (19.7 mg), 8 (18.5 mg) and 9 (2.0 mg).
Napyradiomycin 1 (12.0 mg): yellow oil; [α]D 30 (c 10, CH2Cl2); IR (film) vmax 3422, 2929, 1705, 1649, 1608, 1572, 1442, 1371, 1269, 1212, 1149, 1056 cm−1; UV λmax (log ε) 282 (3.26), 340 (2.97), 372 (2.94); 1H NMR (DMSO-d6) and 13C NMR (DMSO-d6) spectroscopic data, see Tables 1 and 2, respectively; HRESIMS m/z 459.1596 ([M+H]+), calcd for C25H28ClO6, 459.1574).
Napyradiomycin 2 (2.6 mg): yellow oil; [α]D −151 (c 1.0, MeOH); IR (film) 3394, 2928, 1705, 1647, 1608, 1440, 1370, 1298, 1217, 1152, 1056 cm−1; UV λ max (log ε) 282 (3.10), 338 (2.80), 372 (2.75); 1H NMR (DMSOd6) and 13C NMR (DMSO-d6) spectroscopic data, see Tables 1 and 2, respectively; HRESIMS m/z 463.1723 ([M+Na]+, calcd for C25H28O7Na, 463.1733).
Napyradiomycin 3 (3.8 mg): yellow oil; [α]D −181 (c 1.0, MeOH); IR (film) vmax 3407, 2921, 1700, 1649, 1605, 1446, 1375, 1293 cm−1; UV λ max (log ε) 282 (3.17), 340 (2.82), 372 (2.79); 1H NMR (DMSO-d 6) and 13C NMR (DMSO-d6) spectroscopic data, see Tables 1 and 2, respectively; HRESIMS m/z 481.1386 ([M+Na]+, calcd for C25H27ClO6Na, 481.1394).
Napyradiomycin 4 (2.6 mg): yellow amorphous powder; [α]D −70 (c 10, CH2Cl2); IR (film) vmax 3419, 2927, 1680, 1630, 1569, 1431, 1373, 1304, 1201, 1135 cm−1; UV λ max (log ε) 266 (3.16), 315 (2.75), 366 (2.44); 1H NMR (DMSO-d6) and 13C NMR (DMSO-d6) spectroscopic data, see Tables 3 and 4, respectively; HRESIMS m/z 499.1105 ([M+Na]+, calcd for C25H26Cl2O5Na, 499.1055).
Table 3.
1H NMR Spectroscopic Data (500 MHz) for 4–6a
| C# | 4 | 5 | 6b |
|---|---|---|---|
| 2-Me | 1.29 s | 0.98 s | 1.33 s |
| 2-Me | 1.36 s | 1.46 s | 1.51 s |
| 3 | 4.37 dd (3.3, | 5.27 s | 3.93 dd |
| 4a | 2.33 dd (3.3, | 6.73 s | 2.11 m |
| 2.70 dd (12.1, | 2.49 m | ||
| 4b | 3.15 dd | ||
| 6-OH | 11.16 s | 12.55 s | 12.20 s |
| 7 | 6.63 s | 6.71 s | |
| 9 | 6.75 s | 6.94 s | 7.48 s |
| 11 | 2.22 m | 2.09 m | 1.93 m |
| 2.19 m | 2.03 m | ||
| 12 | 4.64 dd (6.1, | 2.01 m | 1.46 m |
| 14 | 1.64 m | 1.74 m | 1.90 m |
| 1.95 m | 2.01 m | 1.90 m | |
| 15 | 1.96 m | 1.83 m | 1.75 m |
| 1.96 m | 2.17 m | 1.92 m | |
| 16 | 5.47 d (10.4) | 4.65 dd (4.0, 12.0) | 3.53 dd |
| 17a-Me | 0.58 s | 0.43 s | |
| 17b-Me | 1.06 s | 0.72 s | |
| 18 | 2.99 d (15.3) 3.32 m b |
||
| 19 | 4.53 dd (3.5, 4.83 d (9.0) |
||
| 20 | 0.98 s | 4.18 s, 4.72s | 1.24 s |
Chemical shifts are in ppm; J values in Hz are in parentheses; Measured in DMSO-d6.
Measured in CDCl3.
Table 4.
13C NMR Spectroscopic Data (75 MHz) for 4–6a
| C# | 4 | 5 | 6b |
|---|---|---|---|
| 2 | 78.3 C | 75.5 C | 80.8 C |
| 2-Me | 21.9 CH3 | 20.3 CH3 | 21.2 CH3 |
| 29.5 CH3 | 26.5 CH3 | 28.9 CH3 | |
| 3 | 60.0 CH | 59.3 CH | 61.1 CH |
| 4 | 41.5 CH2 | 136.9 CH | 33.8 CH2 |
| 4a | 75.2 C | 135.6 C | 57.9 C |
| 5 | 185.2 C | 187.4 C | 199.1 C |
| 5a | 116.9 C | 110.0 C | 109.3 C |
| 6 | 159.6 C | 164.9 C | 165.3 C |
| 7 | 123.9 C | 107.8 CH | 109.6 CH |
| 8 | 156.8 C | 165.7 C | 165.4 C |
| 9 | 106.2 CH | 108.0 CH | 108.8 CH |
| 9a | 135.2 C | 135.9 C | 135.6 C |
| 10 | 195.5 C | 194.0 C | 193.9 C |
| 10a | 86.0 C | 82.1 C | 81.2 C |
| 11 | 43.7 CH2 | 36.8 CH2 | 40.6 CH2 |
| 12 | 118.3 CH | 46.5 CH | 51.4 CH |
| 13 | 137.9 C | 146.0 C | 71.4 C |
| 14 | 40.1 CH2 | 35.7 CH2 | 40.9 CH2 |
| 15 | 24.8 CH2 | 35.9 CH2 | 30.2 CH2 |
| 16 | 135.1 CH | 67.5 CH | 70.8 CH |
| 17 | 132.6 | C 41.8C | 40.5 C |
| 17-Me | 15.9 CH3 | 15.9 CH3 | |
| 27.6 CH3 | 28.4 CH3 | ||
| 18 | 26.7 CH2 | ||
| 19 | 75.4 CH2 | ||
| 20 | 15.4 CH3 | 108.4 CH2 | 24.2 CH3 |
Assignments were made using HMQC and HMBC data; Measured in DMSO-d6.
Napyradiomycin 5 (1.9 mg): yellow oil; [α]D −251 (c 1.0, CH2Cl2); IR (film) vmax 3421, 2925, 1695, 1486, 1451, 1380, 1285, 1213, 1124 cm−1; UV λ max (log ε) 255 (3.49), 310 (3.18), 360 (3.24); 1H NMR (DMSO-d6) and 13C NMR (DMSO-d6) spectroscopic data, see Tables 3 and 4, respectively; HRESIMS m/z 545.0725 ([M+Na]+, calcd for C25H28BrClO5Na, 545.0706).
Napyradiomycin 6 (2.8 mg): red, amorphous powder; [α]D −70 (c 10, CH2Cl2); IR (film) vmax 3404, 2928, 1704, 1625, 1462, 1378, 1271, 1175, 1121 cm−1; UV λ max (log ε) 247 (4.20), 266 (4.03), 301 (4.03), 356 (3.74); 1H NMR (CDCl3) and 13C NMR (CDCl3) spectroscopic data, see Tables 3 and 4, respectively; HRESIMS m/z 498.1566 ([M]+, calcd for C25H32Cl2O6, 498.1576).
Supplementary Material
Acknowledgments
This research is a result of financial support from the National Institutes of Health (USA), National Cancer Institute, under grant R37 CA44848 (to WF) and, in part, from the NOAA California Sea Grant College Program, Project R/NMP-100 (Grant NA100AR4170060 to P.R.J.) through NOAA’S National Sea Grant College Program, U.S. Dept. of Commerce. The statements, findings, conclusions and recommendations are those of the author(s) and do not necessarily reflect the views of California Sea Grant or the U.S. Dept. of Commerce.
Footnotes
Dedicated to the memory of our good friend and close colleague Ernesto Fattorusso
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.xxxxxxxxx.
Supporting Information (see footnote on the first page of this article): 1H and 13C spectra and tabulated 1H and 13C data for all new compounds.
References
- 1.Watve MG, Tickoo R, Jog MM, Bhole BD. Arch Microbiol. 2001;176:386–390. doi: 10.1007/s002030100345. [DOI] [PubMed] [Google Scholar]
- 2.Hughes CC, Prieto-Davo A, Jensen PR, Fenical W. Org Lett. 2008;10:629–631. doi: 10.1021/ol702952n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raju R, Piggott AM, Diaz LXB, Khalil Z, Capon RJ. Org Lett. 2010;12:5158–5161. doi: 10.1021/ol102162d. [DOI] [PubMed] [Google Scholar]
- 4.Wilson MC, Nam SJ, Gulder TAM, Kauffman CA, Jensen PR, Fenical W, Moore BS. J Am Chem Soc. 2011;133:1971– 1977. doi: 10.1021/ja109226s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes CC, MacMillan JB, Gaudêncio SP, Jensen PR, Fenical W. Angew Chem Int Ed Engl. 2009;48:728–732. doi: 10.1002/anie.200804107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shiomi K, Iinuma H, Hamada M, Naganawa H, Manabe M, Matsuki C, Takeuchi T, Umezawa H. J Antibiot. 1986;39:487–493. doi: 10.7164/antibiotics.39.487. [DOI] [PubMed] [Google Scholar]
- 7.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Isshiki K, Takeuchi T, Umezawa H, Iitaka Y. J Antibiot. 1986;39:494–501. doi: 10.7164/antibiotics.39.494. [DOI] [PubMed] [Google Scholar]
- 8.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Isshiki K, Takeuchi T, Umezawa H, Iitaka Y. J Antibiot. 1987;40:1213–1219. doi: 10.7164/antibiotics.40.1213. [DOI] [PubMed] [Google Scholar]
- 9.Motohashi K, Sue M, Furihata K, Ito S, Seto H. J Nat Prod. 2008;71:595–601. doi: 10.1021/np070575a. [DOI] [PubMed] [Google Scholar]
- 10.Gomi S, Ohuchi S, Sasaki T, Itoh J, Sezaki M. J Antibiot. 1987;40:740–749. doi: 10.7164/antibiotics.40.740. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda DS, Mynderse J, Baker P, Berry D, Boeck L, Yao RC, Mertz FP, Nakatsukasa WM, Mabe J, Ott J, Counter FT, Ensminger PW, Allen NE, Alborn WE, Hobbs JN. J Antibiot. 1990;43:623–633. doi: 10.7164/antibiotics.43.623. [DOI] [PubMed] [Google Scholar]
- 12.Soria-Mercado IE, Prieto-Davo A, Jensen PR, Fenical W. J Nat Prod. 2005;68:904–910. doi: 10.1021/np058011z. [DOI] [PubMed] [Google Scholar]
- 13.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC, Moore BS. J Biol Chem. 2007;282:16362–16368. doi: 10.1074/jbc.M611046200. [DOI] [PubMed] [Google Scholar]
- 14.Haste NM, Farnaes L, Perera VR, Fenical W, Nizet V, Hensler ME. Mar Drugs. 2011;9:680–689. doi: 10.3390/md9040680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallagher KA, Fenical W, Jensen PR. Curr Opin Biotechnol. 2010;21:794–800. doi: 10.1016/j.copbio.2010.09.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.