

Infectious and parasitic disease is estimated to be the second leading cause of death worldwide and is becoming increasingly problematic due to the steady rise in drug resistant pathogens.[1] The increase in resistance has also coincided with decreasing numbers of antibiotics brought to the market for the past few decades.[2] Naturally, it is paramount to global human health that new antibiotics are developed, particularly those with novel modes of action and/or unique chemical structures. Herein we present the discovery of sphaerimicin A, a sulfated hybrid polyketide-nucleoside that can be considered to fit both of these descriptors.
Peptidoglycan cell wall plays an essential role in the viability of bacteria, and as a result the inhibition of its biosynthesis has proven to be revolutionary for treating bacterial infections.[3] Nearly all bacteria rely minimally on twelve conserved enzymes to install the cell wall, and intriguingly, the majority of these enzymes have yet to be successfully targeted by commercial antibiotics (Figure S1). One of this majority is bacterial phospho-N-acetylmuramyl-pentapeptide translocase (translocase I, annotated as MraY), which initiates the lipid cycle of peptidoglycan biosynthesis by catalyzing the transfer of phospho-N-acetylmuramic acid-pentapeptide from UDP-N-acetylmuramic acid-pentapeptide to undecaprenyl phosphate, releasing UMP to generate undecaprenyl-disphospho-N-acetylmuramic acid-pentapeptide, or Lipid I. Within the past decade and shortly after connecting the mraY gene product with the translocase activity,[4] several potent, natural product inhibitors have been discovered using activity-based screens.[5] Most of the inhibitors are structurally categorized as uridine-based nucleosides wherein the canonical ribofuranose is modified at the C-5′ position via a C-C bond to generate a so-called high-carbon sugar nucleoside.[5] These high-carbon sugar nucleosides are further divided into subgroups based on the core scaffold, which includes those containing the nonproteinogenic amino acid 5′-C-glycyluridine (GlyU) exemplified by 1–5 or those with a uridine-5′-carboxamide (CarU) core exemplified by 6–8 (Figure 1).[6]
Figure 1.

Representative congeners for two structural groups of bacterial translocase I inhibitors that have been isolated from various actinomycetes. Nucleoside antibiotics that contain A) a 5′-C-glycyluridine (GlyU) core or B) a uridine-5′-carboxamide (CarU) core.
The biosynthetic gene clusters for 1–5 have all been identified.[7] Bioinformatic analysis uncovered a shared open reading frame (orf) encoding a gene product with closest sequence similarity to serine hydroxymethyltransferase (SHMT, EC 2.1.2.1) (Table S1), and the respective orf was shown to be essential for the biosynthesis of 1 and 5 upon gene inactivation.[7a,e] We have functionally assigned this SHMT-like enzyme involved in the biosynthesis of 1 as a pyridoxal-5-phosphate-dependent L-Thr:uridine-5′-aldehyde (9) transaldolase that generates acetaldehyde and (5′S,6′S)-GlyU (10) (Figure 2A).[8] Rather unexpectedly, a homologous orf encoding the transaldolase was also uncovered within the biosynthetic gene cluster of 6 and 7 (Table S1).[9] Although not biochemically confirmed, this realization suggested that CarU biosynthesis also proceeds via 9 and 10 as intermediates.
Figure 2.

Characterization of the new family of L-Thr:9 transaldolases. A) Reaction catalyzed by the SHMT-like transaldolase enzymes generating a shared intermediate (5′S,6′S)-10. B) HPLC confirmation of the reaction catalyzed by SphJ including a) control reaction without L-Thr at 4 h using IMAC-purified SphJ expressed from the codon optimized gene; b) 4 hr reaction; c) 12 h reaction; d) reaction at 12 h using IMAC-purified SphJ expressed from the native gene; and e) reaction at 12 h using SphJ(K248A). C) Stereochemical assignment of the SphJ product by phosgene modification including f) control sample analyzed in (a); g) reaction components analyzed in (c); h) reaction using LipK; and i) synthetic (5′S,6′S)-10. Indicated are retention times for products of phosgene modification of synthetic 10 diastereomers. A260: absorbance at 260 nm.
Based on the integral role of the SHMT-like transaldolase in the biosynthesis of 10-containing inhibitors of translocase I and the likely requirement in the biosynthesis of CarU-containing inhibitors, we set out to identify similar transaldolases and hence potential novel nucleoside antibiotics from our strain collection. Sequence alignment of the transaldolases revealed blocks of conserved amino acids (Figure S2) not observed in bona fide SHMTs (Figure S3), which prompted us to develop a PCR-based strategy for screening. In contrast to mining by whole genome sequencing or activity-based screens by using a variety of fermentation conditions, we considered the PCR methodology the preferred approach due to the vast amount of strains that were targeted. Moreover, we focused on a rarely-explored actinomycetes, none of which have been subjected to whole genome sequencing, since it was thought that such strains would increase the possibility of discovering compounds having novel chemical structures. Thus, degenerate primers were designed (Table S2), and in contrast to results using well-characterized actinomycetes whose genomes have been sequenced and that do not produce high-carbon nucleosides, the designed primer pairs successfully and specifically amplified DNA fragments of the expected size and sequence from the genomic DNA of all the producing strains for both 10- and CarU-containing nucleoside antibiotics (Figure S4).
With the primers in hand, PCR screening was performed with a library of ~2,500 strains, and DNA products of the expected size were obtained from a single strain, Sphaerisporangium sp. SANK 60911. Sequencing identified the amplified DNA fragment as the desired transaldolase gene (sphJ) having 51% amino acid sequence identity with LipK, the characterized transaldolase involved in the biosynthesis of 1 (Table S3).[8] The sphJ gene was expressed in E. coli to confirm the expected transaldolase activity, and in contrast to the expression construct containing the native gene sequence, a protein of the expected size was apparent following IMAC when using a construct incorporating the sphJ gene that was optimized for expression in E. coli (Figure S5). Using our previously developed HPLC and UV/Vis spectroscopic assays, IMAC-purified SphJ converted 9 to 10 with a specific activity of 2.1 × 10−2 μmol•min−1•mg−1, ~ 7-fold lower than LipK under identical conditions (Figure 2B).[8] Continued purification of SphJ by anion exchange yielded a protein of increased specific activity (4.9 × 10−2 μmol•min−1•mg−1) (Figure S5), yet several attempts to obtain a homogenous sample by additional purification steps were unsuccessful. Therefore, a mutant protein SphJ(K248A) was prepared since it was previously established that the corresponding Lys in LipK is essential for activity;[8] as expected, SphJ(K248A) partially purified by IMAC was unable to generate 10 (Figures 2B and S5). Finally, HPLC analysis following phosgene modification of crude reaction mixtures in comparison to the LipK-catalyzed reaction and synthetic diastereomers of 10 revealed (5′S,6′S)-10 as the product of SphJ (Figure 2C).[8,10]
After assigning SphJ as an L-Thr:9 transaldolase, we used sphJ as a probe to clone the entire genetic locus. Sequencing of four overlapping cosmids yielded 57-kb contigious DNA consisting of 34 putative orfs including sphJ (Figure S6 and Table S3). Eight orfs (sphE-L) were identified whose gene products have 32–51% sequence identity to those encoded within the biosynthetic gene cluster of 1, and one orf (sphT) whose gene product has 48% sequence identity to Mur29, a protein of unknown function involved in 5 biosynthesis (Table S3). In addition, two orfs (sphU and sphV) encoding putative type I modular polyketide synthases and another orf (sphW) encoding a free-standing condensation domain found in nonribosomal peptide synthetases were uncovered. We chose to monitor the expression of three potentially key biosynthetic genes: sphJ; sphV; and sphE, a lipL homolog whose gene product has been characterized as an α-ketoglutarate:UMP dioxygenase in 1 biosynthesis,[11] to identify conditions that promote production of the corresponding metabolite. A growth condition using solid media (YMA) was found to have acceptable levels of expression of all three orfs (Figure 3A), and in contrast to cultures that lacked the desired expression, acetone extracts of cultures grown on YMA were shown to inhibit the activity of recombinant bacterial translocase I in vitro. HPLC analysis of these same extracts revealed four UV-active products with absorption near 260 nm that we termed sphaerimicin A–D (11–14) (Figure 3B). The physicochemical properties of 11–14 are summarized in Table S4.
Figure 3.

Discovery of the sphaerimicins. A) Expression analysis of the putative sphaerimicin biosynthetic genes under different cultivation conditions including a) YMA solid media compared to b) CNZ4, c) AP-1, and d) 172F liquid media. B) HPLC analysis of the extracts obtained from the indicated culture broths. A260: absorbance at 260 nm. C) Structure of 11 and hydrolysis products (15 and 16).
Due to production limitations, only 11 was purified on a large-scale for complete structural elucidation (Figure 3C). The UV absorption spectrum of 11 revealed maxima at 237 and 261 nm, characteristic of dienes and nucleosides, respectively. HR-ESI-MS yielded an [M-H]− ion at m/z 973.4316 (Figure S7), consistent with the molecular formula of C44H70N4O18S (calcd. 973.4333); MS/MS gave product ions for decarboxylated-11 (m/z 929), sulfate (HOSO3−: m/z 97) and uracil (C4H3N2O2−: m/z 111) (Figure S8), suggesting that 11 is an O-sulfated uracil-based nucleoside antibiotic with a carboxylate functionality. Similar MS/MS fragmentation ions were observed with 12–14 (Figure S7). As detailed in Table S5, the 1H, 13C NMR and heteronuclear 2D spectra of 11 in DMSO-d6 (Figures S9–S13) revealed CH3 x 7, CH2 x 8, CH x 24, C=O x 4 and >C= x 1 signals including five olefinic protons, two anomeric carbons, several O- and aliphatic methines, overlapping aliphatic methylenes and six methyl groups. This suggested that the modified nucleoside possesses a branched aliphatic side chain. Mild alkaline hydrolysis of 11 using 1N NaOH indeed gave two products—a nucleoside core (15) and acyl side chain (16)—consistent with a side chain linked via a standard ester bond (Figure 3C).
To simplify the structural elucidation of 11, a thorough spectroscopic analysis was performed with 15 and 16, and partial stereochemical assignments were facilitated in part by comparison to NMR assignments for 1–5 and various simplified, synthetic derivatives.[12] HMBC and HSQC-TOCSY analysis of 15 (Figures S14–16) revealed key 1H-13C and 1H-15N long-range correlations (Figure 4A) that were consistent with a ribosylated 10 as found in 1–5 and the fusion of the C-5″ amine to a dihydroxylated piperidine to give an unusual 14-membered macro-heterocycle containing two bridges. The 13C chemical shifts of C-5″, 2‴, 3‴ and 6‴ were also consistent with adjacency to a nitrogen and hence the structure shown for 15 (Figures 3C and 4A). The ESI-MS spectrum of 16 yielded an [M-H]−ion at m/z 365 (C23H41O3−)that, upon MS/MS analysis, generated a product ion at m/z 321 indicating 16 contains a carboxylate (Figure S8). Through analysis of the 1H-13C HMBC and HSQC-TOCSY spectra (Figures S17 and S18), the chemical structure of 16 was subsequently deduced as a branched and highly reduced, linear polyketide.
Figure 4.
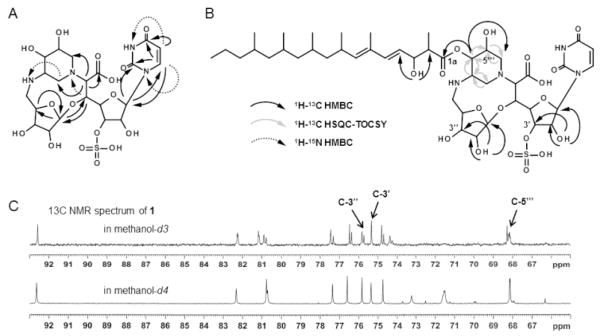
NMR analysis of sphaerimicin A. Key 1H-13C and 1H-15N long range correlations of A) 15 and B) 11. C) Deuterium shifts analysis of 11.
The final structural assignments were concluded based on interpretation of the NMR spectra for 11. The connectivity of 15 and 16 was established by 1H-13C long-range coupling from H-4‴ to C-1a (Figure 4B). The configuration of Δ4a was determined to be E based on the 1H-1H coupling constant between H-4a and 5a (15.5 Hz) followed by the clear NOE and ROE correlations between H-4a and 19a (Figures S19 and S20). Likewise, NOE and ROE correlations between H-5a and 7a as well as H-8a and 19a clearly revealed that the stereochemistry of Δ6a was also E. Finally, three exchangeable protons were observed upon obtaining the 1H NMR spectrum of 11 in DMSO-d6. They were assigned to 2′, 2″ and 3a-hydoxyl residues by the observed 1H-13C long-range couplings from OH-2′ to C-1′, 2′ and 3′, OH-2″ to C-1″, 2″ and 3″, and OH-3a to C-2a and 4a (Figure 4B). Consequently, candidate positions for O-sulfation were narrowed to C-3′, 3″ or 5‴, and hence deuterium shifts of the 13C signals were investigated. Among the three, only the signal assigned to C-3′ (δ 75.4) remained unchanged (Figure 4C). MS/MS analyses of the m/z 607 and 625 product ions of 11 also supported sulfation at the hydroxyl of C-3′ (Figure S21). In total the final chemical structure of 11 was deduced as shown in Figure 3C.
As expected, 11–14 were all potent inhibitors of bacterial translocase I with IC50 values between 12–65 ng/mL (Figure 5), which is comparable to reported values obtained with other inhibitors. Although ineffective against Gram-negative bacteria, which is potentially a result of TolC-mediated efflux as observed with other nucleoside inhibitors of bacterial translocase I,[5] compound 11 displayed promising antibacterial activity against Gram-positive bacteria of utmost medical importance (Table 1). Moving forward, it will be of interest to investigate the role of the several unique structural features of 11 in structure-activity relationship (SAR) studies. For instance, sulfation appears to be common for the 10-containing nucleoside antibiotics, yet the regiochemistry is different in 11 compared to 1 and 3 that contain a 2′ or 2″ sulfate group, respectively. In the latter examples this sulfation event has a pronounced negative effect on the antimicrobial activity suggesting that desulfo-11 may have an even better antimicrobial profile than 11. An additional unique feature of 11 is the fused ribose and piperidine that leads to a secondary amine at C-5″ of the ribosyl moiety. Prior SAR studies with simplified 2 derivatives suggested the C-5″ primary amine can be replaced by certain secondary amines without a significant impact on in vitro inhibitory activity,[13] and the discovery of 11 indicates the same is likely true for the overall antibacterial activity for 10-containing nucleoside antibiotics, which was not previously tested.
Figure 5.
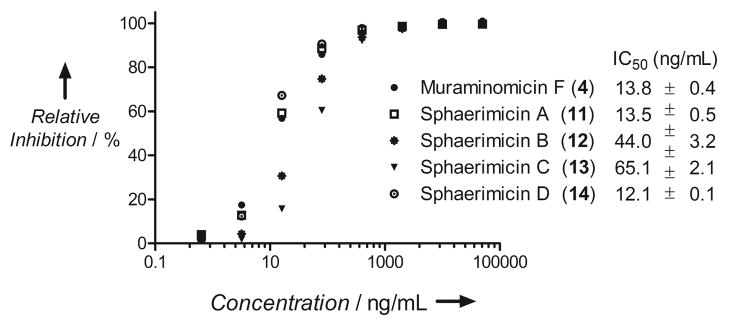
Inhibition of bacterial translocase I. For comparison 4 was used.
Table 1.
Antibacterial spectrum of 11
| Test Strain | Minimum inhibitory concentration (μg/ml) | ||
|---|---|---|---|
| Sphaerimicin A | Chloramphenicol* | Linezolid* | |
| Streptococcus pneumoniae ATCC 496193THB3 | 1 | 2 | 1 |
| Streptococcus pneumoniae ATCC 49619 | 16 | 2 | 1 |
| Streptococcus pyogenes ATCC 12344 | 16 | 2 | 1 |
| Staphylococcus aureus ATCC 6538P | 4 | 8 | 2 |
| Staphylococcus aureus 10925 | 8 | 16 | 2 |
| Staphylococcus epidermidis ATCC 14990 | 8 | 4 | 1 |
| Enterococcus faecalis ATCC 29212 | 2 | 8 | 2 |
| Enterococcus faecium ATCC 19434 | 2 | 8 | 4 |
| Moraxella catarrhalis ATCC 25238 | 4 | 1 | 4 |
| Haemophilus influenzae ATCC 49247 | 8 | 1 | 8 |
| Haemophilus influenzae Rd | 32 | 1 | 8 |
| Escherichia coli ATCC 47076 | >128 | 8 | >128 |
| Escherichia coli ATCC 25922 | >128 | 4 | >128 |
| Klebsiella pneumoniae ATCC 13883 | >128 | 4 | >128 |
| Enterobacter cloacae ATCC 13047 | >128 | 8 | >128 |
| Serratia marcescens ATCC 13880 | >128 | 8 | 128 |
| Proteus vulgaris ATCC 13315 | >128 | 4 | 8 |
| Pseudomonas aeruginosa ATCC 15692 | >128 | 64 | >128 |
| Stenotrophomonas maltophilia ATCC 13637 | >128 | 4 | >128 |
| Acinetobacter baumannii ATCC 19606 | >128 | 128 | 128 |
Chloramphenicol and linezolid were used as a standard in this assay.
In conclusion, we have discovered a novel 10-containing nucleoside antibiotic by using a gene-guided approach, a strategy we envision can be easily applied to other strain collections or metagenomic libraries. This strategy yielded a bacterial translocase I inhibitor 11 with several unusual structural features including a unique piperidine ring system fused to an aminoribose, a 3′-sulfate group, and a branched, highly reduced polyketide side chain. The results now pave the way to explore in greater detail how these structural features are installed and, more importantly, the application of 11 and the congeners 12–14 as new antibiotics to counteract the ever-increasing limitations of today’s antibiotic arsenal.
Supplementary Material
Footnotes
This work was supported by NIH grant AI087849 to S.V.L.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Dr. Masanori Funabashi, Email: funabashi.masanori.ea@rdn.daiichisankyo.co.jp, Natural Product Research Group, Discovery Science and Technology Department, Drug Discovery and Biomedical Technology Unit, Daiichi Sankyo RD Novare Co., Ltd., Tokyo 134-8630 (Japan)
Dr. Satoshi Baba, New Modality Research Laboratories, R&D Division, Daiichi Sankyo Co., Ltd., Tokyo 140-8710 (Japan)
Dr. Toshio Takatsu, Analytical Chemistry Research Group, Center for Pharmaceutical and Biomedical Analysis, Daiichi Sankyo RD Novare Co., Ltd., Tokyo 134-8630 (Japan)
Masaaki Kizuka, Natural Product Research Group, Discovery Science and Technology Department, Drug Discovery and Biomedical Technology Unit, Daiichi Sankyo RD Novare Co., Ltd., Tokyo 134-8630 (Japan).
Yasuo Ohata, Analytical Chemistry Research Group, Center for Pharmaceutical and Biomedical Analysis, Daiichi Sankyo RD Novare Co., Ltd., Tokyo 134-8630 (Japan).
Dr. Masahiro Tanaka, Natural Product Research Group, Discovery Science and Technology Department, Drug Discovery and Biomedical Technology Unit, Daiichi Sankyo RD Novare Co., Ltd., Tokyo 134-8630 (Japan)
Dr. Koichi Nonaka, Biologics Technology Research Laboratories, R&D Division, Daiichi Sankyo Co., Ltd., Gunma 370-0503 (Japan)
Anatol P Spork, Department of Chemistry, University of Paderborn, Paderborn 33098 (Germany).
Prof. Dr. Christian Ducho, Department of Chemistry, University of Paderborn, Paderborn 33098 (Germany)
Wei-Chen Leyla Chen, Department of Pharmaceutical Sciences College of Pharmacy, University of Kentucky 789 S. Limestone Street, Lexington, KY 40536 (USA).
Prof. Dr. Steven G Van Lanen, Email: svanlanen@uky.edu, Department of Pharmaceutical Sciences College of Pharmacy, University of Kentucky 789 S. Limestone Street, Lexington, KY 40536 (USA)
References
- 1.World Health Organization. The Global Burden of Disease: 2004 Update. 2004. p. 146. [Google Scholar]
- 2.a) Wright GD. Chem Biol. 2012;19:3. doi: 10.1016/j.chembiol.2011.10.019. [DOI] [PubMed] [Google Scholar]; b) Fischbach MA, Walsh CT. Science. 2009;325:1089. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kohanski MA, Dwyer DJ, Collins JJ. Nat Rev Microbiol. 2010;8:423. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Bugg TDH, Walsh CT. Nat Prod Rep. 1992;9:199. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]; b) Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. FEMS Microbiol Rev. 2008;32:208. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]; c) Bugg TD, Braddick D, Dowson CG, Roper DI. Trends in Biotechnol. 2011;29:163. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Iketa M, Wachi M, Jung HK, Ishnino F, Matsuhashi M. J Bacteriol. 1991;173:1021. doi: 10.1128/jb.173.3.1021-1026.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winn M, Goss RJ, Kimura K, Bugg TD. Nat Prod Rep. 2010;27:279. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 6.a) Fujita Y, Kizuka M, Funabashi M, Ogawa Y, Ishikawa T, Nonaka K, Takatsu T. J Antibiot. 2011;64:495. doi: 10.1038/ja.2011.38. [DOI] [PubMed] [Google Scholar]; b) Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T, Akamatsu Y. J Antibiot. 2005;58:327. doi: 10.1038/ja.2005.41. [DOI] [PubMed] [Google Scholar]; c) Kimura K, Ikeda Y, Kagami S, Yoshihama M, Ubukata M, Esumi Y, Osada H, Isono K. J Antibiot. 1998;51:647. doi: 10.7164/antibiotics.51.647. [DOI] [PubMed] [Google Scholar]; d) Muramatsu Y, Fujita Y, Aoyagi A, Kizuka M, Takatsu T, Miyakoshi S. Patent WO 2004046368. Sankyo Co., Ltd; 2004; e) McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. J Am Chem Soc. 2002;124:10260. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]; f) Muramatsu Y, et al. J Antibiot. 2003;56:243. doi: 10.7164/antibiotics.56.243. [DOI] [PubMed] [Google Scholar]; g) Muramatsu Y, Ohnuki T, Ishii MM, Kizuka M, Enokita R, Shunichi M, Takatsu T, Inukai M. J Antibiot. 2004;57:639. doi: 10.7164/antibiotics.57.639. [DOI] [PubMed] [Google Scholar]; h) Murakami R, Fujita Y, Kizuka M, Kagawa T, Muramatsu Y, Miyakoshi S, Takatsu T, Inukai M. J Antibiot. 2007;60:690. doi: 10.1038/ja.2007.88. [DOI] [PubMed] [Google Scholar]
- 7.a) Funabashi M, Baba S, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Van Lanen SG. Chem Bio Chem. 2010;11:184. doi: 10.1002/cbic.200900665. [DOI] [PubMed] [Google Scholar]; b) Kaysser L, Siebenberg S, Kammerer B, Gust B. Chem Bio Chem. 2010;11:191. doi: 10.1002/cbic.200900637. [DOI] [PubMed] [Google Scholar]; c) Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B, Gust B. J Biol Chem. 2009;284:14987. doi: 10.1074/jbc.M901258200. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chi X, Baba S, Tibrewal N, Funabashi M, Nonaka K, Van Lanen SG. Med Chem Comm. 2013;4:239. doi: 10.1039/C2MD20245J. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cheng L, Chen W, Zhai L, Xu D, Huang T, Zhou X, Deng Z. Mol Biosyst. 2011;7:920. doi: 10.1039/c0mb00237b. [DOI] [PubMed] [Google Scholar]
- 8.Barnard-Britson S, Chi X, Nonaka K, Spork AP, Tibrewal N, Goswami A, Pahari P, Ducho C, Rohr J, VanLanen SG. J Am Chem Soc. 2012;134:18514. doi: 10.1021/ja308185q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Funabashi M, Nonaka K, Yada C, Hosobuchi M, Masuda N, Shibata T, Van Lanen SG. J Antibiot. 2009;62:325. doi: 10.1038/ja.2009.38. [DOI] [PubMed] [Google Scholar]; b) Funabashi M, Yang Z, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Chi X, Van Lanen SG. Nat Chem Biol. 2010;6:581. doi: 10.1038/nchembio.393. [DOI] [PubMed] [Google Scholar]
- 10.Spork AP, Ducho C. Synlett. 2013;24:343. [Google Scholar]
- 11.Yang Z, et al. J Biol Chem. 2011;286:7885. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Spada MR, Ubukata M, Isono K. Heterocycles. 1992;34:1147. [Google Scholar]; b) Dini C, Collette P, Drochon No, Guillot JC, Lemoine G, Mauvais P, Aszodi J. Bioorg Med Chem Lett. 2000;10:1839. doi: 10.1016/s0960-894x(00)00349-8. [DOI] [PubMed] [Google Scholar]; c) Hirano S, Ichikawa S, Matsuda A. Angew Chem Int Ed. 2005;44:1854. doi: 10.1002/anie.200462439. [DOI] [PubMed] [Google Scholar]; d) Hirano S, Ichikawa S, Matsuda A. Tetrahedron. 2007;63:2798. [Google Scholar]; e) Hirano S, Ichikawa S, Matsuda A. J Org Chem. 2008;73:569. doi: 10.1021/jo702264e. [DOI] [PubMed] [Google Scholar]
- 13.Dini C, Drochon N, Feteanu S, Guillot JC, Peixoto C, Aszodi J. Bioorg Med Chem Lett. 2001;11:529. doi: 10.1016/s0960-894x(00)00715-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.