

Abstract
Background
Patients with diabesity have a significantly increased risk of developing cardiovascular disease. Therefore, therapy addressing the multiple metabolic abnormalities linked with diabesity and leading to further reduction of cardiovascular risk is highly desirable. Activation of the TGR5 receptor holds therapeutic potential for diabesity. In the present study, we evaluated the efficacy of TRC210258, a novel TGR5 agonist, in clinically relevant animal models of diabesity.
Methods
A novel small molecule, TRC210258 (N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo (1, 2-a) pyrimidine-3-carboxamide), was synthesized. The in vitro TGR5 receptor activation potential of TRC210258 was assessed by cyclic adenosine monophosphate (cAMP) assay and cAMP-responsive element reporter assay using cells overexpressing the human TGR5 receptor. The effect of TRC210258 on glucagon-like peptide-1 release was evaluated in vitro using a human enteroendocrine cell line. The effect of TRC210258 on energy expenditure and glycemic control was evaluated in high-fat diet-induced obese mice. Additionally, the effect of TRC210258 on dyslipidemic parameters was determined in high fat-fed hamsters.
Results
TRC210258 demonstrated potent TGR5 agonist activity, with enhanced glucagon-like peptide-1 release and energy expenditure. Treatment with TRC210258 resulted in better glycemic control and improved parameters of dyslipidemia such as plasma triglyceride, low-density lipoprotein cholesterol, and non-high-density lipoprotein cholesterol levels. Treatment with TRC210258 also improved emerging dyslipidemic cardiovascular risk parameters, including remnant cholesterol and triglyceride clearance.
Conclusion
This study highlights the potential of TRC210258, a novel TGR5 agonist, to improve dyslipidemic cardiovascular risk beyond glycemic control in patients with type 2 diabetes.
Keywords: TGR5, diabesity, atherogenic dyslipidemia, triglyceride clearance, remnant cholesterol, cardiovascular risks
Introduction
The common clinical association between type 2 diabetes mellitus and obesity is well captured in the term “diabesity”.1–4 Diabesity is the continuum of progressive abnormal biology, which ranges from mild insulin resistance to full-blown type 2 diabetes. It is noteworthy that patients with diabesity have a significantly high risk of developing cardiovascular disease.2,5 Diabesity and cardiovascular disease share many common risk factors, including central obesity, hyperinsulinemia, hyperglycemia, elevated blood pressure, and dyslipidemic features. The most common dyslipidemic features of diabesity include low levels of high-density lipoprotein (HDL) cholesterol, elevated triglycerides, and increased low-density lipoprotein (LDL) cholesterol. In view of this association, the ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial was designed to assess the impact of intensive therapy to control glycemia, blood pressure, and lipids on cardiovascular outcome. The results of this research suggest that, in spite of intensive interventions for glycemic and lipid control, a residual risk of cardiovascular events remains in patients with type 2 diabetes.6 These findings substantiate the possible role of concomitant pathologic features other than blood pressure, hyperglycemia, and LDL cholesterol in the residual cardiovascular risk found in these patients. Emerging evidence suggests that, in addition to LDL cholesterol, there are other atherogenic dyslipidemic parameters in patients with type 2 diabetes that contribute to this residual cardiovascular risk. These include low HDL cholesterol levels and elevated triglycerides, as well as non-HDL cholesterol.7,8 Recently, it has also been demonstrated that remnant cholesterol has a causal link with ischemic heart disease, and it has been suggested that remnant cholesterol be considered as a residual risk factor for cardiovascular disease.9 Therefore, there is a need to include these additional atherogenic dyslipidemic components as therapeutic targets beyond the current recommendations for controlling LDL cholesterol, blood pressure, and glycemia in such patients.10 In view of all these evidence, therapy addressing the multiple metabolic abnormalities linked to diabesity and leading to further reduction of cardiovascular risk is highly desirable.
It has been shown recently that activation of the TGR5 receptor signaling pathway counteracts the metabolic dysfunction associated with diabesity.11–15 TGR5 has been identified as a lithocholic acid-activated receptor.12,16 The natural bile acid ligands of the TGR5 receptor, ie, lithocholic acid and taurocholic acid, have been shown to affect energy homeostasis via TGR5-mediated effects on energy expenditure and glucose disposal.11 Experimental evidence has demonstrated the role of the TGR5 receptor in the release of glucagon-like peptide-1 (GLP1) and peptide YY from intestinal L-cells17,18 and deiodinase type II (D2) activation in skeletal muscle and brown adipose tissue.11 In Kupffer cells and monocytes, activation of TGR5 leads to a reduction in the release of cytokines such as interleukin-6 and tumor necrosis factor alpha.16,19 Based on these evidence, TGR5 is emerging as a potential therapeutic target for the management of cardiometabolic disorders including diabetes, obesity, dyslipidemia, and atherosclerosis.
In the present study, we investigated the efficacy of TRC210258, a novel TGR5 agonist, in clinically relevant animal models of diabesity. We conducted a series of in vivo experiments to assess the efficacy of TRC210258, as follows: (A) the effect of TRC210258 on energy expenditure in high-fat diet-induced obese (DIO) mice as an in vivo functional measure of TGR5 activation; (B) the effect of 4 weeks of treatment with TRC210258 on glycemic parameters in DIO mice; and (C) the effect of 5 weeks of treatment with TRC210258 on lipid parameters in DIO hamsters. The effect of TRC210258 on parameters of dyslipidemia was evaluated in a high-fat DIO hamster model, which has relevance to the human lipid metabolism and lipid profile.20,21
Materials and methods
Chemistry of TRC210258
Novel TGR5 agonists were designed and synthesized. Around 400 new chemical entities were synthesized and screened for TGR5 activation under in vitro conditions. TRC210258 was identified as a novel lead, with the chemical name of N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide (Figure 1). TRC210258 was synthesized using commercially available diethyl bro-momalonate as the starting material. This was condensed with 2-amino pyrimidine to obtain the ethyl-2-hydroxy imidazo [1, 2-a] pyrimidine-3-carboxylate derivative, which was then chlorinated with phosphorus chloride and treated with 4-fluorophenol to obtain the phenoxy derivative. Further alkaline hydrolysis gave the carboxylic acid derivative. This was further condensed with 4-chloroaniline using 1-(3-dimetylaminopropyl)-3-ethylcarbodiimide hydrochloride and 1-hydroxy benzotriazole to obtain the carboxamide derivative, which was treated with iodomethane to obtain TRC210258 with a high-performance liquid chromatography purity of more than 99%.
Figure 1.
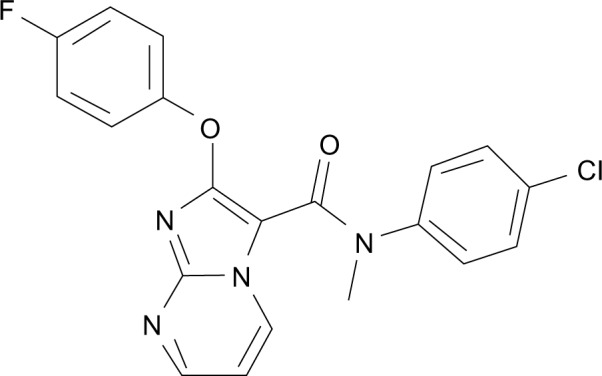
Chemical structure of TRC210258: N-(4-chlorophenyl)-2-(4-fluoro phenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide.
In vitro studies
TGR5-mediated formation of cyclic adenosine monophosphate
Chinese Overy Hamster-K1 (CHO-K1) cells (American Type Culture Collection, Manassas, VA, USA, catalog number CCL-61) were transiently transfected with human TGR5 expression vector (Origene, Rockville, MD, USA). After 24 hours of transfection, the cells were incubated with different concentrations of TRC210258. After 1 hour of incubation, cAMP levels were measured in cell lysates using a Time resolved fluorescence resonance energy transfer (TR-FRET)-based assay kit (Lance Assay; Invitrogen, Carlsbad, CA, USA) with EnVision® (Perkin Elmer, Waltham, MA, USA). The results are expressed as the EC50 value in nM.
TGR5-mediated cAMP-responsive element reporter activation
CHO-K1 cells were transiently transfected with human TGR5 receptor expression vector along with a cAMP-responsive element-driven luciferase reporter vector (CRE-Luc vector; Clontech, Mountain View, CA, USA). B-galactosidase was used as a normalization vector for transfection efficiency. After 24 hours of transfection, the cells were treated with different concentrations of TRC210258 for 5 hours. The cell lysates were then analyzed for luciferase and β-galactosidase activity. Normalized luciferase activity values were used to determine the EC50 value in nM.
TGR5-mediated GLP1 secretion
GLP1 secretion was monitored after TGR5 activation in a human enteroendocrine cell line, ie, NCI-H716 (American Type Culture Collection, catalog number CCL-251).13 The cells were treated with vehicle, TRC210258, or lithocholic acid for 1 hour in the presence of a 0.05% dipeptidyl peptidase-4 inhibitor (sitagliptin). Lithocholic acid was used as the positive control. The supernatant from the culture medium was analyzed for active GLP1 levels by enzyme-linked immunosorbent assay (ELISA; Millipore, Billerica, MA, USA). The results are expressed as the amount of active GLP1 released.
In vivo studies
In vivo efficacy studies in the present investigation comprised three sets of experimental protocols as shown schematically in Figure 2. Experiment A was conducted to assess the effect of treatment with TRC210258 on energy expenditure in a mouse model of DIO (Figure 2A). Experiment B was conducted to assess the effect of treatment with TRC210258 on glycemic control in a mouse model of DIO (Figure 2B). Experiment C was conducted to study the effect of treatment with TRC210258 on dyslipidemic parameters and triglyceride clearance in a hamster model (Figure 2C).
Figure 2.

(A) Schematic representation of study design for in vivo efficacy experiment A to evaluate the effect of treatment with TRC210258 on energy expenditure in DIO mice. High-fat DIO C57bl/6J mice were randomized into two treatment groups. Mice in group 1 (n=5) received vehicle (0.5% w/v Tween-80 + 0.5% w/v Na-CMC solution in water) throughout the study. Group 2 (n=5) received TRC210258 2.3 mg/kg for the first 4 days. On day 5, the dose of TRC210258 was increased to 4.5 mg/kg, and on day 9 dose was increased further to 9.0 mg/kg. All treatments were administered once a day via the oral route. Oxygen consumption was recorded using the Oxymax® system (Columbus Instruments, Columbus, OH, USA) on day 4 of the respective incremental dose. (B) Schematic representation of study design for in vivo efficacy experiment B, which investigated the effect of TRC210258 on glycemic control in DIO mice. High-fat DIO C57bl/6J mice were randomized into three treatment groups. Animals in group 1 (n=10) received vehicle (0.5% w/v Tween-80 + 0.5% w/v Na-CMC solution in water), those in group 2 (n=9) received TRC210258 9 mg/kg, and those in group 3 (n=9) received sitagliptin 1 mg/kg. All treatments were administered once a day via the oral route. (C) Schematic representation of the design for the in vivo efficacy experiment studying the effect of TRC210258 on dyslipidemic parameters and triglyceride clearance in the high fat-fed DIO hamster. Male Golden Syrian hamsters with DIO were randomized into three treatment groups. Hamsters in group 1 (n=10) received vehicle (0.9% Na-CMC + 0.1% Tween-80 in phosphate-buffered saline, pH 7.4) of TRC210258 by the intraperitoneal route, and the vehicle (0.5% w/v Tween-80 + 0.5% w/v Na-CMC in water) for sitagliptin by the oral route; group 2 (n=10) received TRC210258 6 mg/kg by the intraperitoneal route and vehicle for sitagliptin by the oral route; group 3 (n=9) received sitagliptin 3 mg/kg by the oral route and vehicle for TRC210258 by the intraperitoneal route.
Abbreviation: DIO, diet-induced obesity; HFD, high-fat diet; FTT, fat tolerance test; OGTT, oral glucose tolerance test; wks, weeks; w/V, weight by volume; Na-CMC, sodium carboxy methyl cellulose; TRC210258 N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide.
Animals
Male C57bl/6J mice and male Golden Syrian hamsters were housed under controlled conditions (a 12:12 light-dark cycle, 22°C±3°C, and 30%–70% relative humidity) with free access to food in an individually ventilated caging system (Citizen Enterprise, Ahmedabad, India). For induction of DIO, the animals were fed a high-fat diet (HFD) containing 45% kCal from fat (D12451; Research Diets, Brunswick, NJ, USA) for 8±2 weeks before inclusion in the studies, with feeding continued until the end of experiment. Animal care and all experimental procedures involving the animals were carried out in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals, Government of India. The study protocols were approved by the institutional animal ethics committee of Torrent Pharmaceuticals Ltd, Gujarat, India.
Groups and treatments
Experiment A
For determination of the effect on energy expenditure, DIO mice were randomized into two treatment groups. Group I received vehicle (0.5% w/v Tween-80 + 0.5% w/v Na-Carboxy Methyl Cellulose solution in water) by the oral route throughout the study and group II received a formulation of TRC210258 2.3 mg/kg by the oral route. On day 5 of the study, the TRC210258 dose was increased to 4.5 mg/kg and on day 9 was increased to 9.0 mg/kg. All treatments were administered once daily.
Experiment B
To assess the effect of oral administration of TRC210258 on glycemic control in the mouse model of DIO, mice were randomized into three treatment groups. Animals in group I received vehicle (0.5% w/v Tween-80 + 0.5% w/v Na-CMC solution in water), those in group II received a 9 mg/kg dose of TRC210258, and those in group III received a 1 mg/kg dose of sitagliptin. All the treatments were administered once a day by the oral route for 4 weeks.
Experiment C
For the efficacy study in high fat-fed male Golden Syrian hamsters, the animals were randomized into three groups after measurement of their pretreatment glycemic and lipid parameters. Animals in group I received vehicle of TRC210258 (0.9% Na-CMC + 0.1% Tween-80 in phosphate-buffered saline, pH 7.4) as well as vehicle of sitagliptin (0.5% w/v Tween-80 + 0.5% Na-CMC in water); group II received a 6 mg/kg dose of TRC210258 by the intraperitoneal route, and group III received a 3 mg/kg dose of sitagliptin by the oral route.
Experimental methodology for in vivo study
Experiment A
For determination of the effect of TRC210258 on energy expenditure, the animals were housed at a thermoneutral temperature (30°C±0.5°C). The temperature inside the cage was regulated by attaching heating units along with thermostats to the customized individualized ventilated caging system. After acclimatization to these conditions for 7 days, pretreatment oxygen consumption (VO2), carbon dioxide (CO2) production, and respiratory quotient (RQ: ratio of volume of CO2 produced to volume of O2 consumed) measurements were recorded using an open circuit indirect calorimeter, ie, an Oxymax® system (Columbus Instruments, Columbus, OH, USA). Post-treatment, oxygen consumption was recorded on day 4 of administration of each respective incremental dose of TRC210258.
Experiment B
For the oral efficacy study to assess the effect of TRC210258 on glycemic control in mice with DIO, an oral glucose tolerance test was carried out at the end of weeks 2 and 4 of treatment (Figure 2B). For this test, the animals were administered active drug or vehicle after 6 hours of fasting. Fifteen minutes after drug administration, a preglucose (0 minute) sample collection was done. The glucose load was administered at a dose of 2 g/kg to each mouse at 0 minutes post-blood collection. Thereafter, blood samples were collected by the tail snip method at 15, 30, 60, and 120 minutes post-glucose loading.
Experiment C
Pretreatment basal fasting blood samples were collected from hamsters with DIO to assess basal plasma lipid parameters, including triglycerides, total cholesterol, LDL cholesterol, and HDL cholesterol. These parameters were also estimated after 2, 4, and 5 weeks of treatment. At each time point, remnant cholesterol was estimated by subtracting the LDL and HDL cholesterol values from the total cholesterol value.9 To assess the effect on triglyceride clearance, an oral fat tolerance test was carried out at the end of treatment week 4. The hamsters were fasted for 6 hours before the fat tolerance test. A pre-oil load blood sample was collected, and an oral olive oil load at a dose of 5 mL/kg was then administered to each hamster. Next, blood samples were collected at 60, 90, 120, 150, 180, and 240 minutes. An oral glucose tolerance test was carried out at the end of treatment week 2 to assess the effect on glucose tolerance. In the hamster studies, blood samples were collected by the retro-orbital route under mild anesthesia.
All blood samples were collected in microcentrifuge tubes containing 10% potassium ethylene diamine tetra acetic acid (K-EDTA), and centrifuged at 6,000 rpm and 4°C for 10 minutes for plasma separation. Plasma glucose, triglyceride, total cholesterol, LDL cholesterol, and HDL cholesterol determinations were done using a biochemical autoanalyzer (AU-400; Beckman Coulter, Fullerton, CA, USA). Fasting insulin determination was done using a rat/mouse insulin ELISA kit (Millipore) for the mouse samples, and a hamster-specific insulin ELISA kit (Crystal Chem Inc., Downers Grove, IL, USA) was used for the hamster samples.
Data compilation and statistical analysis
The data were expressed as the mean ± standard error of the mean (unless otherwise mentioned). Statistical analysis was performed using SAS version 9.1 (SAS Institute Inc., Cary, NC, USA) software and GraphPad Prism software version 3.2 (San Diego, CA, USA). For comparison of parameters where data were generated at multiple time points during treatment, repeated-measures analysis of variance was used to test for significant differences between the intervention and control groups over time. For other parameters, where a point comparison was made between two groups, the unpaired Student’s t-test was used. Values exceeding 95% critical limits (P<0.05) were considered to be statistically significant.
Results
TRC210258-activated TGR5 under in vitro conditions
Treatment with TRC210258 resulted in a dose-dependent increase in cAMP levels in CHO-K1 cells overexpressing the TGR5 receptor, with an EC50 of 354 nM (Figure 3A). Lithocholic acid, a natural ligand of TGR5, showed an EC50 of 571.5 nM in the same experiment. Treatment with TRC210258 also resulted in dose-dependent activation of cAMP-responsive element-driven luciferase reporter in TGR5-overexpressing CHO-K1 cells, with an EC50 of 221 nM (Figure 3B). Lithocholic acid showed an EC50 of 446 nM in the same experiment. Treatment with TRC210258 resulted in significant induction of GLP1 secretion from human enteroendocrine cells (NCI-H716), and this effect was found to be superior to that of lithocholic acid (Figure 3C).
Figure 3.

Activation of TGR5 by TRC210258. (A) Effect of TRC210258 on TGR5-mediated cAMP formation. EC50 value expressed in nM. (B) Effect of TRC210258 on CRE luciferase reporter. EC50 value expressed in nM. (C) Effect of TRC210258 on GLP1 secretion from NCI-H716, a human enteroendocrine cell line. GLP1 levels expressed in pM. Each value is the average of two experiments, each performed in duplicate. *P<0.05, TRC210258 versus vehicle control.
Abbreviations: DMSO, dimethyl sulfoxide; TRC210258, N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide; GLP, glucagon like peptide 1; CRE, cyclic AMP responsive element; pM, picomole; conc, concentration.
TRC210258 enhanced energy expenditure in DIO mice
Stimulation of TGR5 enhances energy expenditure. Therefore, the increase in energy expenditure by TRC210258 was used as a functional measure of TGR5 activation in vivo. Oral treatment of DIO mice with incremental doses of TRC210258 resulted in enhanced energy expenditure in a dose-dependent manner. A prominent effect on energy expenditure was observed during the first 5 hours after treatment with TRC210258. The 9 mg/kg/day oral dose was found to be the most effective (Figure 4A and B). Hence, further efficacy studies in DIO mice were done using the 9 mg/kg/day dose of TRC210258.
Figure 4.

Effect of TRC210258 on energy expenditure in DIO mice. (A) The oxygen consumption profile was measured for ~22 hours (started at 9.0 am) on day 4 of treatment with different doses of TRC210258 or vehicle. #P<0.05, 4.5 mg/kg group versus respective vehicle group; *P<0.05, 9 mg/kg group versus respective vehicle group. (B) Average oxygen consumption during initial 5 hours post-drug administration (shaded area, right Y-axis scale) and the total 22 hours of recording (nonshaded area, left Y-axis scale) on treatment with different doses of TRC210258 or vehicle. *P<0.05 for TRC210258 9 mg/kg versus vehicle control group; **P<0.01 for TRC210258 9 mg/kg group versus vehicle control group & #P<0.05 for TRC210258 4.5 mg/kg group versus vehicle control group.
Abbreviation: DIO, diet-induced obesity; TRC210258, N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide; h, hour.
TRC210258 improved glycemic control in DIO mice
Four weeks of treatment with TRC210258 9 mg/kg achieved a marked reduction in glucose excursion in mice with DIO (Figure 5A and B). After 4 weeks of treatment, the mean area under the curve (AUC) for glucose was reduced by 16.6% in the TRC210258 group when compared with the vehicle control group, while a 24% reduction in the mean AUC for glucose was observed in the sitagliptin group.
Figure 5.

Effect of TRC210258 on glycemic parameters in DIO mice. (A) Bar graph represents the average area under the curve (AUC0–120 min) for plasma glucose during an oral glucose tolerance test at pretreatment basal level and after 2 and 4 weeks of treatment. (B) Plasma glucose profile during oral glucose tolerance test after 4 weeks of treatment. (C) Fasting plasma glucose (left y axis) and insulin (right y axis) after 4 weeks of treatment. (D) HOMA-IR after 4 weeks of treatment. *P<0.05 for treatment group versus vehicle control group; **P<0.01 for treatment group versus vehicle control group.
Abbreviations: AUC, area under curve; DIO, diet-induced obesity; m, minutes; TRC 210258, N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide; HOMA-IR, homeostasis model assessment –insulin resistance.
When the oral glucose tolerance test results were compared at weeks 2 and 4 of treatment, there was an improvement in glucose excursion in sitagliptin-treated DIO mice of 29% and 24%, respectively. However, treatment with TRC210258 resulted in a gradual and incremental improvement in glucose excursion after week 4 as compared with week 2 (reduction in plasma glucose AUC(0–120 min) by 9% at week 2 and by 16.6% at week 4) in DIO mice.
Additionally, 4 weeks of treatment with TRC210258 in DIO mice reduced fasting plasma insulin by 40.3%, while in the sitagliptin group the reduction was 22.7% as compared with the vehicle control group (Figure 5C). Correspondingly, fasting plasma glucose reduced by 6.5% in the TRC210258 group and by 4.4% in the sitagliptin group (Figure 5C). Treatment with TRC210258 for 4 weeks in DIO mice also showed a trend toward amelioration of Homeostasis Model of Assessment-Insulin Resistance, an index of insulin resistance (Figure 5D).
TRC210258 improved dyslipidemic parameters in the DIO hamster
HFD feeding induces features of diabesity, including insulin resistance, dysglycemia, and dyslipidemia in hamsters.21 Our data indicate that with respect to normal chow-fed hamsters, HFD-fed hamsters gained significant amounts of weight and developed dyslipidemia, glucose intolerance, fasting hyperinsulinemia, and significant impairment in triglyceride clearance (see Table S1).
In this model of diabesity, treatment with TRC210258 achieved significant improvement in various dyslipidemic parameters (Figure 6A–D) and plasma triglyceride clearance (Figure 7) during treatment week 5. Treatment with TRC210258 improved various lipid parameters, ie, total cholesterol by 25.4%, LDL cholesterol by 26.2%, non-HDL cholesterol by 32%, and triglycerides by 28.3% as compared with the vehicle control group at the end of treatment week 5. TRC210258 also achieved a reduction in remnant cholesterol levels of 41.6% as compared with the vehicle control group. Sitagliptin has no significant effect on any of these lipid parameters.
Figure 6.

Effect of TRC210258 on plasma lipid parameters in DIO hamsters. (A) Total cholesterol, (B) LDL cholesterol, (C) non-HDL cholesterol, and (D) remnant cholesterol, in TRC210258, sitagliptin, and vehicle control groups at pretreatment basal level and after weeks 2, 4, and 5 of treatment. *P<0.05, **P<0.01, and ***P<0.001 for TRC210258 group versus vehicle group; #P<0.05 and ##P<0.01 for TRC210258 group versus sitagliptin group.
Abbreviations: DIO, diet-induced obesity; TC, total cholesterol; nHDL-C, non high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Rem-C, remnant cholesterol; TC, total cholesterol; Wk, week; TRC210258, N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide.
Figure 7.

Effect of TRC210258 on plasma triglyceride clearance in DIO hamsters. (A) Incremental plasma triglyceride profile after an olive oil load in the TRC210258, sitagliptin, and vehicle control groups during fat tolerance testing after 4 weeks of treatment. *P<0.05, for TRC210258 group compared at multiple time points with the vehicle group. (B) Bar graph represents the mean AUC for plasma triglycerides during the fat tolerance test. *P<0.05 for TRC210258 group versus vehicle control group.
Abbreviations: DIO, diet-induced obesity; AUC, area under the curve; m, minutes; TG, triglyceride; TRC210258, N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide.
During the fat tolerance test, mean plasma triglyceride levels in the TRC210258 group remained lower at all time points post-fat loading as compared with those in the vehicle control and sitagliptin groups. The AUC for plasma triglycerides was significantly reduced in the TRC210258 group compared with the vehicle control group, and this reduction was better than in the sitagliptin group. On oral fat loading, the mean peak concentration of incremental plasma triglyceride levels was 48.5% lower in the TRC210258 group, while the sitagliptin group showed levels that were 28.6% lower than in the vehicle control group. After oral fat loading, the time taken to reach peak plasma triglyceride levels in the TRC210258 group was 120 minutes and triglyceride levels declined thereafter, whereas in the vehicle control group, the time taken to reach peak plasma levels was 156±12.5 minutes, and triglyceride levels remained high until 240 minutes.
TRC210258 improved glycemic parameters in DIO hamsters
The favorable effect of TRC210258 on plasma glucose and insulin release in response to a glucose load during an oral glucose tolerance test was also observed in DIO hamsters (Figure 8). After 2 weeks of treatment, the mean AUC(0–120 min) for glucose was reduced by 19.7% and 16.8% in the TRC210258 and sitagliptin groups, respectively, when compared with the vehicle control group. The TRC210258 and sitagliptin groups also showed a higher AUC(0–120 min) for insulin. Therefore, the doses of TRC210258 and sitagliptin selected for the DIO hamster study were effective in terms of improving glycemic excursion, and the effect of TRC210258 and sitagliptin was comparable at these dose levels.
Figure 8.

Effect of TRC210258 on glycemic parameters in DIO hamsters: (A) Plasma glucose profile during oral glucose tolerance test after 4 weeks of treatment (B) AUC(0–120 min) plasma glucose (left y axis) and insulin (right y axis) after 4 weeks of treatment in DIO hamsters. **P<0.05 versus vehicle control group.
Abbreviation: DIO, diet-induced obesity; AUC, area under the curve; m, minutes.
Discussion
In the present study, we first demonstrated that treatment with TRC210258 resulted in activation of TGR5 under in vitro conditions. TRC210258 was found to be a potent TGR5 agonist, resulting in release of GLP1 from human enteroendocrine cells under in vitro conditions. We then carried out an efficacy study of TRC210258 in DIO mice. The DIO mouse is a clinically relevant model of diabesity, with key features of visceral fat accumulation, glucose intolerance, fasting hyperglycemia, and hyperinsulinemia. In preliminary studies using DIO mice, we demonstrated that treatment with TRC210258 resulted in a dose-related increase in energy expenditure. A similar effect on energy expenditure via TGR5 activation, by bile acid and a semisynthetic TGR5 agonist, INT-777, has been reported previously.13,14 This increase in energy expenditure is predicted to be due to D2 activation mediated by the TGR5 receptor.11,17,18
In further chronic efficacy studies in DIO mice, treatment with TRC210258 resulted in a gradual improvement in glucose tolerance. The improved glucose tolerance observed in this study could involve two mechanisms, ie, TGR5-mediated improvement in insulin secretion as well as enhanced substrate metabolism during increased energy expenditure. A gradual improvement in glucose tolerance was observed in the TRC210258 group, while a constant effect on glucose tolerance was exerted by sitagliptin throughout the study period. Such a differential effect of TRC210258 as compared with sitagliptin could be due to TGR5-mediated enhancement of energy expenditure which can concurrently improve insulin sensitivity.
We then demonstrated the efficacy of TRC210258 on dyslipidemic parameters using the DIO hamster model. Hamsters mimic the human lipid metabolism and lipid profile better than rodents.20 The disease characteristics of the DIO hamster model include moderate visceral fat accumulation, glucose intolerance, hyperinsulinemia, impaired triglyceride clearance, and increased LDL and non-HDL cholesterol.21
We administered TRC210258 in the DIO hamster via the intraperitoneal route because of the lower oral bioavailability of this compound in hamsters compared with mice. The observed lower oral bioavailability of TRC210258 in the hamster seems to be species-specific because, in comparison with the hamster, multiple folds of plasma exposure of TRC210258 has been observed on oral administration in other species, including the mouse, rat, and dog (data not shown). In the present study, although we used different doses and routes of administration in the mouse and hamster, we ensured that the doses selected provided similar exposure of TRC210258 based on pharmacokinetic studies in these species (Table S2).
Treatment with TRC210258 resulted in significantly improved parameters of dyslipidemia, including plasma triglycerides, LDL cholesterol, non-HDL cholesterol, and remnant cholesterol in DIO hamsters. Dyslipidemia, an established risk factor for cardiovascular disease, is common in patients with type 2 diabetes, affecting almost 50% of this population. In addition to hyperglycemia and hypertension, dyslipidemia is a modifiable cardiovascular risk factor that remains largely uncontrolled in patients with type 2 diabetes. Hyperglycemia and dyslipidemia both augment the risk of microvascular complications in this group of patients.2 The dyslipidemia associated with insulin resistance is characterized by moderately increased triglyceride levels (carried in very low-density lipoprotein and chylomicron remnant particles) and reduced HDL cholesterol levels. Thus, the observed efficacy of TRC210258 with regard to various dyslipidemic parameters in DIO hamsters highlights the potential of TRC210258 to improve the atherogenic dyslipidemic parameters that are concurrently present in diabesity.
We also evaluated the effect of TRC210258 on triglyceride clearance after oral fat loading in DIO hamsters. Such studies are highly relevant given that a positive association between insulin resistance and impaired postprandial triglyceride clearance is well supported in the literature.22–24 For example, postprandial triglyceride levels have been shown to be an independent predictor of coronary heart disease.25,26 Similarly, postprandial accumulation of triglyceride-rich lipoproteins in plasma has been reported to be a significant risk factor for coronary artery disease.27 Plasma triglyceride-rich lipoprotein clearance is a function of lipoprotein lipase.23 Lipoprotein lipase protein amount and activity in preheparin plasma correlate negatively with insulin resistance and fasting insulin.24 Human studies have also shown significantly delayed triglyceride-rich lipoprotein metabolism after fat loading in obese young men with impaired glucose tolerance as compared with lean young men.22 In accordance with these findings supporting an association between insulin resistance and impaired triglyceride clearance, the observed effect of TRC210258 on triglyceride clearance has great clinical significance. Moreover, the observed effect of TRC210258 on triglyceride clearance was found to be significantly better than that of sitagliptin under these experimental conditions. This better effect of TRC210258 on triglyceride clearance when compared with sitagliptin could be due to the comparatively better effect of TRC210258 on energy expenditure and consequent gradual improvement in glucose tolerance. TRC210258 has also shown a significant ability to attenuate remnant cholesterol (elevated cholesterol content in triglyceride-rich lipoprotein particles). A recent study has demonstrated a causal link between elevated remnant cholesterol and ischemic heart disease.9 Similarly, other studies have highlighted a relationship between atherogenic dyslipidemia, a combination of elevated triglycerides and low HDL cholesterol, and microvascular risk in type 2 diabetes.28,29 All these studies taken together highlight the need for interventions aimed at reducing levels of remnant cholesterol and triglyceride-rich lipoproteins. In view of this, the findings of the present study are pertinent in that they reveal the potential of TGR5 agonists to impact cardiovascular risk significantly in patients with type 2 diabetes.
Summarizing the present investigation, a novel TGR5 agonist, TRC210258, showed improvement in glycemic as well as dyslipidemic parameters in animal models of diabesity (Figure 9). Additionally, the study demonstrated the efficacy of TRC210258 in reducing elevated triglycerides and remnant cholesterol, which are being considered to contribute toward residual cardiovascular risk in type 2 diabetes. The improvement in lipid parameters was superior to that achieved by the DPP-4 inhibitor sitagliptin. These findings with TRC210258 further strengthen the potential of TGR5 agonists as emerging therapy for diabesity.
Figure 9.

Schematic representation summarizing TRC210258-mediated TGR5 activation and improvement of glycemic and dyslipidemic cardiovascular risk.
Abbreviations: GLP1, glucagon like peptide-1; D2, deiodinase 2; wt, weight; TG, triglyceride; CV, cardiovascular; TRC210258, N-(4-chlorophenyl)-2-(4-fluorophenoxy)-N-methylimidazo [1, 2-a] pyrimidine-3-carboxamide.
Supplementary material
Characteristics of the high-fat DIO hamster model
Male Golden Syrian hamsters (aged 6–8 weeks) were housed under controlled conditions (a 12:12 light-dark cycle, 22°C±3°C, and 30%–70% relative humidity) with free access to food in an individually ventilated caging system. After recording of basal parameters, the animals were divided into two groups. One group was fed with a normal growing chow diet (D10012G, Research Diet, Inc., New Brunswick, NJ) and the other was fed on a high-fat diet containing 45% kCal from fat (D12451, Research Diet, Inc., New Brunswick, NJ) for 8 weeks. After 8 weeks of dietary intervention, oral glucose and fat tolerance tests were carried out. The effects of dietary intervention on various parameters are enumerated in Supplemental Table 1 and the relevant pharmacokinetic parameters are shown in Supplemental Table 2.
Table S1.
Characteristics of the high-fat DIO hamster model
| Parameters | Time point | Normal chow-fed hamster | HFD-fed hamster |
|---|---|---|---|
| Body weight (g) | Week 0 | 97.8±2.9 | 97.9±2.5 |
| Week 8 | 117.8±2.6 | 135.4±3.9*** | |
| Cumulative food intake (g) | Week 8 | 257.5±13.8 | 283.8±8.8 |
| Total cholesterol (mg/dL) | Week 0 | 109.6±6.8 | 100.5±7.4 |
| Week 8 | 117.0±6.2 | 136.0±3.1* | |
| LDL cholesterol (mg/dL) | Week 0 | 32.0±4.0 | 28.6±2.3 |
| Week 8 | 36.0±2.5 | 43.5±1.4* | |
| Non-HDL cholesterol (mg/dL) | Week 0 | 36.1±4.1 | 36.2±4.5 |
| Week 8 | 50.4±2.2 | 61.1±1.7** | |
| Remnant cholesterol (mg/dL) | Week 0 | 4.0±0.9 | 4.7±0.7 |
| Week 8 | 14.3±0.6 | 17.0±0.2** | |
| Triglycerides (mg/dL) | Week 0 | 120.3±10.8 | 116.5±9.3 |
| Week 8 | 199.8±14.5 | 220.4±7.8 | |
| Fasting glucose (mg/dL) | Week 0 | 103.5±6.4 | 94.5±3.5 |
| Week 8 | 104.0±6.3 | 106.2±4.0 | |
| Fasting insulin (ng/mL) | Week 8 | 2.7±0.4 | 5.8±1.2* |
| Glucose AUC(0–120 min) (mg/dL×min) | Week 8 | 20,680.6±577.1 | 28,183.0±2,566.5* |
| Triglyceride AUC(0–360 min) (mg/dL×min) | Week 8 | 17,204.4±3,784.9 | 25,449±986.6* |
Notes: Values are shown as the mean ± standard error of mean
P<0.05
P<0.01
P<0.001, compared with respective normal chow group.
Abbreviations: AUC, area under the curve; DIO, diet-induced obesity; HDL, high-density lipoprotein; HFD, high-fat diet; LDL, low-density lipoprotein.
Table S1.
Pharmacokinetic data for TRC210258 in DIO mice (9 mg/kg orally) and DIO hamsters (6 mg/kg, intraperitoneally)
| PK parameter | Units | DIO mice | DIO hamsters |
|---|---|---|---|
| Tmax | Hours | 2.25±1.25 | 1.5±0.71 |
| Cmax | ng/mL | 59.4±22.2 | 78.7±31.8 |
| AUClast | Hours×ng/mL | 525.7±127.0 | 726.2±223.8 |
Note: Values are shown as the mean ± standard deviation.
Abbreviations: PK, pharmacokinetic; DIO, diet-induced obesity; Cmax, peak plasma concentration; Tmax, time taken to reach peak plasma concentration; AUC, area under the curve.
Acknowledgments
The authors are thankful to Dr Ram Gupta, Mr Umang Patel, and Mr Nilesh Badgujar for support and assistance during conduct of the experimental work. We are also thankful to Mr. Ajay Shiwalkar for support with the statistical analysis and to Mrs Sneha Moss for her kind help with English language copy editing.
Footnotes
Disclosure
The authors are employees of Torrent Pharmaceuticals Ltd. The authors report no other conflicts of interest in this work.
References
- 1.Farag YM, Gaballa MR. Diabesity: an overview of a rising epidemic. Nephrol Dial Transplant. 2011;26:28–35. doi: 10.1093/ndt/gfq576. [DOI] [PubMed] [Google Scholar]
- 2.Laakso M. Cardiovascular disease in type 2 diabetes from population to man to mechanisms: the Kelly West Award Lecture 2008. Diabetes Care. 2010;33:442–449. doi: 10.2337/dc09-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tschop MH, DiMarchi RD. Outstanding Scientific Achievement Award Lecture 2011: defeating diabesity. The case for personalized combinatorial therapies. Diabetes. 2012;61:1309–1314. doi: 10.2337/db12-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haslam D. Obesity and diabetes: the links and common approaches. Prim Care Diabetes. 2010;4:105–112. doi: 10.1016/j.pcd.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Meigs JB. Epidemiology of type 2 diabetes and cardiovascular disease: translation from population to prevention: the Kelly West award lecture 2009. Diabetes Care. 2010;33:1865–1871. doi: 10.2337/dc10-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fruchart JC, Sacks FM, Hermans MP. Implications of the ACCORD lipid study: perspective from the Residual Risk Reduction Initiative (R(3)i) Curr Med Res Opin. 2010;26:1793–1797. doi: 10.1185/03007995.2010.489341. [DOI] [PubMed] [Google Scholar]
- 7.Hermans MP, Fruchart JC. Reducing vascular events risk in patients with dyslipidaemia: an update for clinicians. Ther Adv Chronic Dis. 2011;2:307–323. doi: 10.1177/2040622311413952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nesto RW. Beyond low-density lipoprotein: addressing the atherogenic lipid triad in type 2 diabetes mellitus and the metabolic syndrome. Am J Cardiovasc Drugs. 2005;5:379–387. doi: 10.2165/00129784-200505060-00005. [DOI] [PubMed] [Google Scholar]
- 9.Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–436. doi: 10.1016/j.jacc.2012.08.1026. [DOI] [PubMed] [Google Scholar]
- 10.Fruchart JC, Sacks FM, Hermans MP, et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in dyslipidaemic patient. Diab Vasc Dis Res. 2008;5:319–335. doi: 10.3132/dvdr.2008.046. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 12.Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 13.Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svensson PA, Olsson M, Andersson-Assarsson JC, et al. The TGR5 gene is expressed in human subcutaneous adipose tissue and is associated with obesity, weight loss and resting metabolic rate. Biochem Biophys Res Commun. 2013;433:563–566. doi: 10.1016/j.bbrc.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pols TW, Nomura M, Harach T, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 17.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 18.Wu T, Bound MJ, Standfield SD, et al. Effects of rectal administration of taurocholic acid on glucagon-like peptide-1 and peptide YY secretion in healthy humans. Diabetes Obes Metab. 2013;15:474–477. doi: 10.1111/dom.12043. [DOI] [PubMed] [Google Scholar]
- 19.Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- 20.Ugawa T, Kakuta H, Moritani H, Shikama H. Experimental model of escape phenomenon in hamsters and the effectiveness of YM-53601 in the model. Br J Pharmacol. 2002;135:1572–1578. doi: 10.1038/sj.bjp.0704595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G, Liu X, Zhu H, et al. Insulin resistance in insulin-resistant and diabetic hamsters (Mesocricetus auratus) is associated with abnormal hepatic expression of genes involved in lipid and glucose metabolism. Comp Med. 2009;59:449–458. [PMC free article] [PubMed] [Google Scholar]
- 22.Nabeno-Kaeriyama Y, Fukuchi Y, Hayashi S, Kimura T, Tanaka A, Naito M. Delayed postprandial metabolism of triglyceride-rich lipoproteins in obese young men compared to lean young men. Clin Chim Acta. 2010;411:1694–1699. doi: 10.1016/j.cca.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Pruneta-Deloche V, Sassolas A, Linga-Thie GM, Berthezene F, Ponsin G, Moulin P. Alteration in lipoprotein lipase activity bound to triglyceride-rich lipoproteins in the postprandial state in type 2 diabetes. J Lipid Res. 2004;45:859–865. doi: 10.1194/jlr.M300435-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Tornvall P, Olivecrona G, Karpe F, Hamsten A, Olivecrona T. Lipoprotein lipase mass and activity in plasma and their increase after heparin are separate parameters with different relations to plasma lipoproteins. Arterioscler Thromb Vasc Biol. 1995;15:1086–1093. doi: 10.1161/01.atv.15.8.1086. [DOI] [PubMed] [Google Scholar]
- 25.Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–316. doi: 10.1001/jama.298.3.309. [DOI] [PubMed] [Google Scholar]
- 26.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 27.Havel RJ. Postprandial hyperlipidemia and remnant lipoproteins. Curr Opin Lipidol. 1994;5:102–109. doi: 10.1097/00041433-199404000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Gupta R, Guptha S, Agrawal A, Kaul V, Gaur K, Gupta VP. Secular trends in cholesterol lipoproteins and triglycerides and prevalence of dyslipidemias in an urban Indian population. Lipids Health Dis. 2008;7:40. doi: 10.1186/1476-511X-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zoppini G, Negri C, Stoico V, Casati S, Pichiri I, Bonora E. Triglyceride-high-density lipoprotein cholesterol is associated with microvascular complications in type 2 diabetes mellitus. Metabolism. 2012;61:22–29. doi: 10.1016/j.metabol.2011.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Characteristics of the high-fat DIO hamster model
| Parameters | Time point | Normal chow-fed hamster | HFD-fed hamster |
|---|---|---|---|
| Body weight (g) | Week 0 | 97.8±2.9 | 97.9±2.5 |
| Week 8 | 117.8±2.6 | 135.4±3.9*** | |
| Cumulative food intake (g) | Week 8 | 257.5±13.8 | 283.8±8.8 |
| Total cholesterol (mg/dL) | Week 0 | 109.6±6.8 | 100.5±7.4 |
| Week 8 | 117.0±6.2 | 136.0±3.1* | |
| LDL cholesterol (mg/dL) | Week 0 | 32.0±4.0 | 28.6±2.3 |
| Week 8 | 36.0±2.5 | 43.5±1.4* | |
| Non-HDL cholesterol (mg/dL) | Week 0 | 36.1±4.1 | 36.2±4.5 |
| Week 8 | 50.4±2.2 | 61.1±1.7** | |
| Remnant cholesterol (mg/dL) | Week 0 | 4.0±0.9 | 4.7±0.7 |
| Week 8 | 14.3±0.6 | 17.0±0.2** | |
| Triglycerides (mg/dL) | Week 0 | 120.3±10.8 | 116.5±9.3 |
| Week 8 | 199.8±14.5 | 220.4±7.8 | |
| Fasting glucose (mg/dL) | Week 0 | 103.5±6.4 | 94.5±3.5 |
| Week 8 | 104.0±6.3 | 106.2±4.0 | |
| Fasting insulin (ng/mL) | Week 8 | 2.7±0.4 | 5.8±1.2* |
| Glucose AUC(0–120 min) (mg/dL×min) | Week 8 | 20,680.6±577.1 | 28,183.0±2,566.5* |
| Triglyceride AUC(0–360 min) (mg/dL×min) | Week 8 | 17,204.4±3,784.9 | 25,449±986.6* |
Notes: Values are shown as the mean ± standard error of mean
P<0.05
P<0.01
P<0.001, compared with respective normal chow group.
Abbreviations: AUC, area under the curve; DIO, diet-induced obesity; HDL, high-density lipoprotein; HFD, high-fat diet; LDL, low-density lipoprotein.
Table S1.
Pharmacokinetic data for TRC210258 in DIO mice (9 mg/kg orally) and DIO hamsters (6 mg/kg, intraperitoneally)
| PK parameter | Units | DIO mice | DIO hamsters |
|---|---|---|---|
| Tmax | Hours | 2.25±1.25 | 1.5±0.71 |
| Cmax | ng/mL | 59.4±22.2 | 78.7±31.8 |
| AUClast | Hours×ng/mL | 525.7±127.0 | 726.2±223.8 |
Note: Values are shown as the mean ± standard deviation.
Abbreviations: PK, pharmacokinetic; DIO, diet-induced obesity; Cmax, peak plasma concentration; Tmax, time taken to reach peak plasma concentration; AUC, area under the curve.