

Abstract
It is hypothesized that differential AKT phosphorylation between sexes is important in abdominal aortic aneurysm (AAA) formation. Male C57BL/6 mice undergoing elastase treatment showed a typical AAA phenotype (80% over baseline, P < 0.001) and significantly increased phosphorylated AKT-308 (p308) and total-AKT (T-AKT) at day 14 compared with female mice. Elastase-treated Raw cells produced increased p308 and significant amounts of matrix metalloproteinase 9 (MMP-9), and these effects were suppressed by LY294002 treatment, a known AKT inhibitor. Male and female rat aortic smooth muscle cells treated with elastase for 1, 6, or 24 hours demonstrated that the p308/T-AKT and AKT-Ser-473/T-AKT ratios peaked at 6 hours and were significantly higher in the elastase-treated cells compared with controls. Similarly, male cells had higher phosphorylated AKT/T-AKT levels than female cells. LY294002 also inhibited elastase-induced p308 formation more in female smooth muscle cells than in males, and the corresponding cell media had less pro–MMP-9. AKT siRNA significantly decreased secretion of pro–MMP-9, as well as pro–MMP-2 and active MMP-2 from elastase-treated male rat aortic smooth muscle cells. IHC of male mice AAA aortas showed increased p308, AKT-Ser-473, and T-AKT compared with female mice. Aortas from male AAA patients had a significantly higher p308/T-AKT ratio than female AAA tissues. These data suggest that AKT phosphorylation is important in the upstream regulation of MMP activity, and that differential phosphorylation may be important in sex differences in AAA.
Abdominal aortic aneurysm (AAA) formation is accompanied by chronic inflammation of the aorta wall, with males four times more likely to develop the disease than females.1 The activation of matrix metalloproteinases (MMPs), especially MMP-2 and MMP-9, has long been known to play a vital role in the destruction of the aortic wall, leading to AAA development.2–6 However, little is known about upstream modulation of the MMPs during AAA formation. Activation of the molecules of the mitogen-activated protein (MAP) kinase pathways is often observed in inflammatory diseases, such as in AAAs. Two MAP kinases in particular, c-Jun N-terminal kinase (JNK) and extracellular signal–regulated kinase (ERK), have been shown to be important in the activation of MMP-2 and MMP-9 in the aorta.7–10 In addition to the MAP kinase cascade, the molecules of the phosphoinositide 3-kinase pathway are also known to be important for the modulation of MMPs.9,11–15 Moreover, AKT, a key molecule in the phosphoinositide 3-kinase pathway, has been shown to play an important role in sex-specific differences of various diseases.16–20 The role of AKT in the modulation of MMPs has not been studied in AAA formation. Therefore, using an in vivo mouse model of AAA and human AAA tissue, as well as in vitro experiments with rat aortic smooth muscle cells (RASMCs) and Raw cells, we show increased phosphorylation of AKT in males compared with females.
Materials and Methods
Experimental AAA Formation
Male and female C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME), aged 8 to 10 weeks, who weighed 20 to 27 g, were used in this study. The experiments were approved by the University of Michigan (Ann Arbor) Universal Committee on the Use and Care of Animals (number 8593).
Mice were anesthetized under 2% isoflurane inhalational anesthetic, a midline laparotomy was performed, and the abdominal aorta from just below the left renal vein to the bifurcation was isolated.21,22 The aortic diameter (AD) was measured with a Spot Insight Color Optical Camera (Diagnostic Instruments, Sterling Heights, MI) attached to an operating microscope (SMX-800; Nikon, Melville, NY) using NIS-Elements software version D.3.10 (Media Cybernetics, Rockville, MD). The aorta was then perfused for 5 minutes with porcine pancreatic elastase (specific activity 6.6 U/mg protein; E1250; Sigma, St. Louis, MO) at a concentration of 0.332 U/mL in 1 mL isotonic saline (experimental or control). After aortic perfusion, AD measurements were obtained. Saline- and elastase-perfused aortas were measured on 1, 3, and 14 days after perfusion (N = 5 or 6 per treatment group per day). Segments of the infrarenal aorta were studied for Western blot analysis, immunohistochemistry (IHC), and zymography. AD increases were reported as a percentage increase from baseline measurements, and an AAA was defined as a 50% increase in AD compared with baseline.
To investigate whether inhibition of phosphorylation of AKT with LY294002 could prevent the formation of AAA in the elastase model, a separate set of experiments was performed, in which mice were divided into four groups, each with N = 8 mice. The groups of mice were as follows: i) mice only treated with dimethyl sulfoxide (DMSO) and LY294002 at a dose of 3 μg/g body weight of mice (No Surgery group), ii) mice that received elastase perfusion as described before and also received DMSO treatment (Surgery + DMSO group but no LY294002), iii) mice that received elastase perfusion as described before and treated with LY294002 and DMSO (Surgery + DMSO + LY group), and iv) mice that underwent elastase perfusion as described earlier (Surgery only group) but received no LY294002 or DMSO treatment. Treatment of mice consisted of i.p. injection of LY294002 (dissolved in 25% DMSO in PBS) at a dose of 3 μg/g body weight of mice every day from day 4 after elastase perfusion to day 14 (the day the mice were sacrificed and tissue was harvested). DMSO treatment consisted of i.p. injection of 25% DMSO in PBS from day 4 after elastase perfusion to day 14 to investigate whether DMSO alone had any effect on AAA formation.
Cell Culture
Male and female RASMCs were isolated, as described previously,7 and were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 10% fetal calf serum (Invitrogen) and 1% penicillin, streptomycin, and glutamine mixture (Invitrogen) at 37°C in a humidified, 5% carbon dioxide atmosphere. Cells (passages 5 to 8) were treated with elastase at 5 μg/mL for 24 hours in serum-free Dulbecco’s modified Eagle’s medium containing the antibiotics and 1% bovine serum albumin. For the in vitro inhibition studies, the cells were pretreated for 2 hours with 50 μmol/L LY294002, an inhibitor of AKT phosphorylation, before treatment with elastase. Media were collected at 1, 6, and 24 hours and assayed for MMP-2 and MMP-9 activities by zymography. RASMCs were lysed, and total AKT (T-AKT), phosphorylated AKT-Thr-308 (p308), and AKT-Ser-473 (p473) levels were assessed by using Western blot analysis (N = 3 per time point per sample, repeated in triplicate).
siRNA Transfection
Cells were plated in 6-well tissue-culture plates at 60% to 80% confluency. One day later, the cells were transfected with 1 μmol/L of siRNA per well for AKT1 (Thermo Scientific, Pittsburgh, PA) or 200 pmol per well for control siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) using the Accell siRNA delivery medium for AKT siRNA (Thermo Scientific). For control siRNA, siRNA transfection reagent and medium from Santa Cruz Biotechnology, according to the manufacturer’s instructions, were used. Seventy-two hours after transfection, cells were washed with PBS, starved in serum-free medium overnight, washed in PBS, and then treated with elastase-containing media or media alone for a further 24 hours. The media and cells were then collected for zymography and Western blot analysis, respectively.
Western Blot Analysis
Proteins were isolated from aortic segments (N = 3 aortas per time point per sample, repeated in triplicate) using radioimmunoprecipitation assay buffer (Thermo Scientific), containing 1% SDS by sonication, and overnight incubation on a shaker. Protein concentration was measured by a bicinchoninic acid kit (Thermo Scientific), and equal amounts of proteins were loaded in each lane for gel electrophoresis using 10% Bis-Tris Nupage gels (Invitrogen). The electrophoretically separated proteins were transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Billerica, MA) by using a semidry transfer apparatus (Bio-Rad Laboratories, Hercules, CA), as per the manufacturer’s guidelines. Non-specific binding was blocked by incubating the membrane for 1 hour in StartBlock TBS (Thermo Scientific). The membranes were incubated with primary antibodies in blocking buffer at 4°C overnight, washed with 25 mmol/L Tris, 150 mmol/L NaCl, and 0.05% Tween-20, pH 7.4, and then incubated with horseradish peroxidase–conjugated secondary antibodies in blocking buffer for 1 hour, washed in 25 mmol/L Tris, 150 mmol/L NaCl, and 0.05% Tween-20, pH 7.4, and developed with the West-Pico electrochemiluminescence kit (Thermo Scientific). The primary antibodies used were phosphorylated Akt (Ser473) (D9E) XP Rabbit monoclonal antibody (mAb) 4060, lot 9, phosphorylated Akt (Thr308) (C31E5E) Rabbit mAb 2965, lot 3, Akt (pan) (C67E7) Rabbit mAb 4691, lot 11 (all from Cell Signaling Technology, Danvers, MA; dilution 1:500), actin antibody sc-4778 horseradish peroxidase, lot L0908, and anti-rabbit secondary antibodies (Santa Cruz Biotechnology, dilution 1:2000). Mac-2 CL8942AP, lot 4223 (dilution 1:200) (Cedarlane Laboratories Limited, Burlington, ON, Canada), anti-rabbit fluorescein isothiocyanate sc-2012, lot D1009 (dilution 1:200) (Santa Cruz Biotechnology), and anti-rat Texas Red TI-9400, lot W0322 (dilution 1:200) (Vector Laboratories, Burlingame, CA), were also used. β-Actin staining was used to check for equal loading of proteins.
Substrate Gel Zymography
Gelatin substrate zymograms were run in precast 10% SDS-PAGE gels containing 1 mg/mL of gelatin (Invitrogen). Equal volumes of experimental media or equal amounts of proteins were diluted into 2× Tris-glycine SDS sample buffer and electrophoretically separated under nonreducing conditions. Proteins were renatured for 30 minutes in Renaturing Buffer (Invitrogen) and then the gels were incubated in the Developing Buffer (Invitrogen) for 30 minutes and again in the same buffer overnight at 37°C. The gel was stained in SimplyBlue SafeStain (Invitrogen), and the gelatinase activity was observed by clear bands against the blue background.
IHC Data
Harvested aortas were fixed in fresh 4% paraformaldehyde for 16 to 24 hours, followed by 70% ethanol. Segments were then paraffin embedded, and sections (5 μm thick) were mounted onto slides. The sections were deparaffinized and rehydrated according to standard protocols, and those prepared for IHC studies were treated for 10 minutes with 3% hydrogen peroxide to block endogenous peroxidase activity. The sections were blocked and then incubated with the primary antibodies overnight at a dilution of 1:100 in blocking solution and stained according to the instructions in the Vectastain ABC Elite Kit (Vector Laboratories). The immunolocalized proteins were then visualized using the Novared kit (Vector Laboratories) and imaged using a Nikon Eclipse Ti microscope (Nikon, Melville, NY).
Human Tissue Preparation
Tissues were obtained from patients undergoing treatment for AAA (N = 18) and cadavers (N = 4) following the Institutional Review Board guidelines of the University of Michigan (HUM1999-0413). Proteins were extracted from aortas using radioimmunoprecipitation assay buffer (Thermo Scientific) supplemented with 1% SDS, and protein concentration was measured by a bicinchoninic acid kit (Thermo Scientific).
Densitometric Analysis
Densitometric analysis of protein bands in the Western blots and zymograms were done using ImageJ software version 1.45s (NIH, Bethesda, MD).
Data Analysis
Data are expressed as means ± SE. The means of two groups were compared using a Student’s t-test. P < 0.05 was considered statistically significant.
Results
Increased p308 in Mouse AAA Model
The mouse AAA model was used to evaluate the status of AKT phosphorylation on different days (1, 3, and 14).7,23,24 Western blot data showed that on day 1 after elastase perfusion, there was no difference in the levels of p308 (P = 0.43) and T-AKT (P = 0.35) between male and female mice (Figure 1A). However, by day 3 after elastase infusion, there was a significant increase in the level of p308 (P = 0.0002) in male compared with female mice (Figure 1, A and B), although female mice had 147% more T-AKT than male mice (Figure 1, A and C). The level of p308 and T-AKT in male mice at 14 days after elastase infusion was significantly more (P = 0.0009 and P = 0.0162, respectively) than in female mice (Figure 1, D and E). The level of p473 in elastase-treated mice at all time points did not show any specific bands (data not shown). Increased phosphorylation of AKT in the elastase-perfused aorta in male mice correlated with increased AAA formation in male compared with female mice (Figure 2).
Figure 1.

A: Western blot analysis for p308, T-AKT, and β-actin on proteins isolated from aortas of elastase-treated male (ME) and female (FE) mice at days 1, 3, and 14, respectively. B–E: Densitometric analysis of the Western blot in A for p308 and T-AKT at days 3 and 14.
Figure 2.
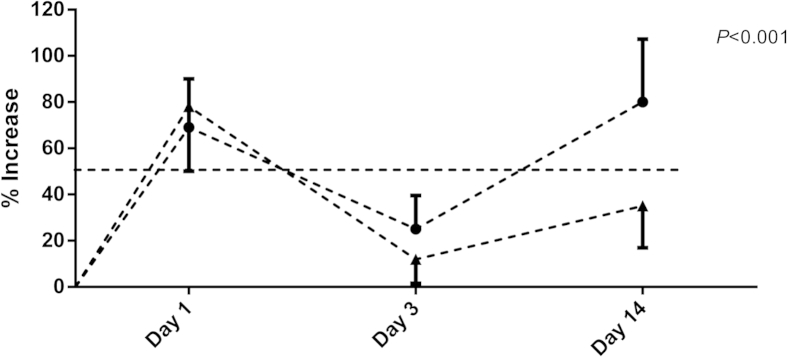
Increase in aortic diameter in male (circles) versus female (triangles) mice at days 1, 3, and 14. P < 0.001. WT, wild type.
Increased Phosphorylation of AKT in Human AAA
Human tissues from AAA and control aortas were analyzed for their AKT content by using Western blot analysis (Figure 3A). AAA patients had significantly higher levels of p308 (P = 0.0027) (Figure 3B), p473 (P = 0.0290) (Figure 3C), and T-AKT (P = 0.0014) (Figure 3D) than control tissues (N = 4). We also observed that the ratio of p308/T-AKT was significantly higher in male (N = 12) human AAA tissues compared with female (N = 6) AAA tissues (P = 0.0252) (Figure 3E).
Figure 3.

A: Western blot analysis for p308, p473, T-AKT, and β-actin on proteins extracted from control and abdominal aortic aneurysm (AAA) human aortas. B–E: Densitometric analysis of the Western blot in A.
Effect of Inhibiting AKT Phosphorylation on AAA Formation in Mice
Western blot experiments demonstrate that formation of AAA in mice and humans was accompanied with an increase in phosphorylation of AKT. We, therefore, wanted to investigate whether inhibiting AKT phosphorylation in vivo would have any effect on AAA formation in mice. We injected the mice with LY294002 (an inhibitor of AKT phosphorylation) at only a single dose of 3 μg/g, before subjecting the mice to elastase perfusion, as described earlier. We observed that treating mice with LY294002 after elastase perfusion did not completely inhibit AAA formation (decrease of 5.5%) (data not shown). However, Verhoff’s–van Gieson staining, as well as H&E counterstaining, showed that the elastin fibers were less disrupted in the LY294002-treated mice compared with those mice that received DMSO (used to dissolve LY294002) treatment or no drug treatment (surgery only) (Figure 4).
Figure 4.

Verhoff’s–van Gieson and H&E staining on male mouse aorta showing elastin fibers. A: Aorta from control mice that were only treated with LY294002 dissolved in DMSO (no surgery, only DMSO + LY). B: Elastase-perfused mouse aorta (surgery only). C: Mice that underwent elastase perfusion but received only DMSO as treatment (surgery + DMSO). D: Mice that underwent elastase perfusion and were then treated with LY294002 that was dissolved in DMSO (surgery + DMSO + LY). Arrows show sites of intact and broken elastin fibers.
We next performed IHC to visualize the presence of p308, p473, and T-AKT in male mice that were treated with LY294002 after perfusing their aorta with elastase. As seen in Figure 5, mice that were treated with LY294002 after elastase perfusion (surgery + DMSO + LY) exhibited reduced staining for p308 and p473 compared with the mice that received no drug treatments (surgery only and surgery + DMSO groups). The staining for T-AKT between these groups of mice did not show any discernible differences. We also observed less macrophage infiltration in the LY294002-treated mice than those mice that did not receive the drug treatment, as demonstrated by less Mac-2 staining, a known marker for macrophages (Figure 5). Our IHC data were corroborated with Western blot experiments in which we observed significant differences in p308 and p308/T-AKT levels in the aortas isolated from the LY294002-treated mice compared with the DMSO + surgery and surgery-only groups of mice (Supplemental Figure S1, B and D). Moreover, Western blot data showed no differences in the levels of T-AKT between the different treatment groups of mice (Supplemental Figure S1C).
Figure 5.

IHC of the mouse aorta from Figure 4. Arrows show areas of decreased levels of p308, p473, and macrophages (Mac-2) in the LY294002-treated group of mice and steady levels of T-AKT in the three groups of mice.
Immunofluorescence Analysis of p308 and Macrophages in Human and Murine AAA Tissues
Western blot data showed that, in both mice and humans, there was an up-regulation of p308 in the AAA tissues. It is known that macrophage infiltration occurs at the site of AAA formation.24 We, therefore, used immunofluorescence to localize p308 and Mac-2 staining in the aorta of the mice that were either treated or not treated with LY294002 after elastase perfusion. As seen in Figure 6, both in the drug-treated and untreated groups, there were areas of colocalization of p308-producing cells and Mac-2 cells, as seen in the merged immunofluorescence, which exhibited the characteristic yellow fluorescence. However, the LY294002-treated mice showed less yellow fluorescence, indicating less p308 production and macrophage infiltration in the tissue.
Figure 6.

Immunofluorescence microscopy of the aorta from control (only surgery) and LY294002-treated (surgery + DMSO + LY) group of mice showing the levels of p308 and presence of macrophages (Mac-2). Arrows show areas for p308 (green) and Mac-2 (red) stains and the areas of their colocalization, which exhibited yellow fluorescence. Original magnification, ×100.
We performed similar experiments on tissues from human patients with AAA and compared p308 and Mac-2 staining with those of the control tissues. We observed similar patterns of colocalization of p308 and macrophages in human AAA tissues (Figure 7) as in mouse experiments. Moreover, colocalization was observed in both male and female AAA patients. In control human tissues, we did not observe colocalization of p308-producing cells and macrophages.
Figure 7.

Immunofluorescence microscopy of male and female AAA patients and control tissues for p308 (green) and macrophages (Mac-2) (red). The images for p308 and Mac-2 were merged with their nuclear stain (DAPI, blue) to visualize regions where p308 and Mac-2 colocalize and, thereby, emit yellow fluorescence. Arrows show areas of p308 and macrophages (Mac-2) and also the sites of their costaining as yellow fluorescence. Original magnification, ×400.
Elastase-Induced p308 Formation in RAW Cells Is Attenuated by LY294002 Treatment
Immunofluorescence studies showed that p308-producing cells and macrophages were colocalized (Figures 6 and 7) in the AAA tissues. We, therefore, used in vitro cell culture assays to determine whether mouse macrophages were capable of producing p308 on stimulation with elastase. Elastase, 5 μg/mL, was used for treating the cells. The dose is nontoxic and was chosen based on published literature.25–29 As seen in the Western blot analysis in Figure 8A, when male mouse macrophages (RAW 264.7 cells) were incubated for 24 hours with elastase, an agent used to induce AAA in our mouse model, p308, was increased (Figure 8A) compared with media-treated control cells. This p308 induction was subsequently abolished when the cells were pretreated with LY294002 (Figure 8A). Treatment of macrophages with elastase for shorter periods of 6 hours (Figure 8A) did not elicit p308 formation. Western blot analysis also showed that elastase treatment did not alter T-AKT and actin levels in these macrophages.
Figure 8.

Western blot analysis (A) and zymogram (B) show that LY294002 inhibits the formation of p308 and MMP-9 secretion in mouse macrophages (Raw cells) treated with elastase. A: Western blot analysis for p308, T-AKT, and actin. Lane 1, untreated control cells; 2, cells treated with LY294002 for 6 hours; 3, cells treated with elastase and LY294002 for 6 hours; 4, cells treated with elastase for 24 hours; and 5, cells treated with elastase and LY294002 for 24 hours. B: Zymogram with growth medium from cells as in A. Untreated control cells (control), cells treated with LY294002 for 24 hours (Inh), cells treated with elastase for 24 hours (Elast), and cells treated for 24 hours with elastase and LY294002 (Elast + Inh). All lanes were run in triplicate. C: Densitometric analysis of the zymogram in B. ∗P < 0.005, ∗∗∗∗P < 0.0001.
We next investigated whether these macrophages were capable of secreting MMPs when stimulated with elastase. As shown by zymography in Figure 8B and densitometric analysis in Figure 8C, elastase induced significant amounts of MMP-9 secretion from macrophages under in vitro conditions. However, when the macrophages were pretreated with LY294002, an inhibitor for AKT phosphorylation, there was a significant decrease in MMP-9 secretion from the macrophages in the presence of elastase.
Elastase-Induced Activation of AKT in Male and Female RASMCs
Smooth muscle cells (SMCs) are the most abundant cell type in the aorta. Our immunofluorescence experiments with mouse and human tissues and a subsequent in vitro experiment with mouse macrophages had shown that the macrophages were able to produce p308 and secrete MMP-9 (Figures 6, 7, and 8). Therefore, in conjunction with macrophages, we wanted to investigate AKT phosphorylation and MMP-2 and MMP-9 production by aortic smooth muscle cells when they were treated with elastase and also to observe sex-dependent AKT phosphorylation in the SMCs in response to elastase. Male and female RASMCs were treated for 1, 6, and 24 hours with elastase.23 We observed a temporal response to elastase in that there was an increase in p308 and p473 production in both male and female RASMCs, which peaked at 6 hours (Supplemental Figure S2). There was a significant increase in production of p308 (P < 0.0001) (Supplemental Figures S3, A and B, and S4A) and p473 (P < 0.002) (Supplemental Figures S3, C and D, and S4C) at 6 and 24 hours. At all time points, male RASMCs had more p308 and p473 than female RASMCs (Table 1).
Table 1.
Significance (P Values) of Levels of p308 and p473 with Respect to T-AKT in Elastase-Treated Male and Female RASMCs at Various Times
| Time (hours) | p308/T-AKT P values (male vs female) | p473/T-AKT P values (male vs female) |
|---|---|---|
| 1 | 0.001 | 0.005 |
| 6 | 0.022 | 0.030 |
| 24 | 0.002 | 0.010 |
Pretreatment of RASMCs with LY294002 Decreases Elastase-Induced p308 and p473
LY294002 is a known inhibitor of AKT phosphorylation.30 Therefore, to confirm the phosphorylation of AKT in RASMCs in the presence of elastase, we pretreated RASMCs with LY294002 for 2 hours and then treated the cells with elastase in the presence of the inhibitor. As seen in the Western blot in Supplemental Figure S2, there was a significant reduction in p308 and p473 in female RASMCs compared with male RASMCs at 6 hours (Table 2).
Table 2.
Significance (P Values) of Levels of p308 and p473 with Respect to T-AKT in LY294002-Pretreated Male and Female RASMCs at 6 Hours
| Variable | P value (male) | P value (female) |
|---|---|---|
| p308/T-AKT ratio | 0.016 | 0.002 |
| p473/T-AKT ratio | 0.117 | 0.004 |
Elastase Induced Higher Pro–MMP-2 Activities in RASMCs and Attenuation of Pro–MMP-9 Induction by AKT Inhibitor
To determine whether inhibiting AKT phosphorylation had any effect on MMP-2 and MMP-9 activity, zymography was performed on the media from the cells treated with either elastase or elastase in the presence of LY294002. Zymography showed that male RASMCs had higher levels of pro–MMP-2 than female RASMCs at all time points after elastase treatment (Figure 9 and Supplemental Table S1). Moreover, pro–MMP-9 activity at 24 hours was completely attenuated in female RASMCs by the AKT inhibitor, LY294002, compared with the male RASMCs (Figure 9).
Figure 9.

Zymogram shows that elastase induces higher pro–MMP-2 activities in male RASMCs than female RASMCs. In addition, inhibition of AKT phosphorylation attenuated pro–MMP-9 induction at 24 hours in both male and female RASMCs.
Decreased Pro–MMP-2, Pro–MMP-9, and Active MMP-2 in Male RASMCs Transfected with AKT siRNA
In initial experiments, we had shown that use of AKT inhibitor, LY294002, prevented male RASMCs from secreting MMPs into the medium when cells were treated with elastase (Figure 9). The mechanism of action of pharmacological inhibitors is often global and may not act solely on one particular molecule. To better define this, siRNA technology was used to selectively inhibit a particular molecule without affecting any other molecule in a non-specific manner. We, thus, transfected male and female RASMCs with siRNA for AKT and then treated them with elastase for 24 hours. We observed that AKT siRNA-transfected male RASMCs secreted less pro–MMP-9, pro–MMP-2, and active MMP-2 in the presence of elastase than control siRNA-transfected cells (Figure 10A). Densitometric analysis of the zymogram revealed that AKT siRNA-transfected male RASMCs secreted significantly reduced pro–MMP-9 (E versus AKT-si + E; P = 0.0089) (Figure 10B), pro–MMP-2 (E versus AKT-si + E; P = 0.0097) (Figure 10C), and active MMP-2 (E versus AKT-si + E; P = 0.0020) (Figure 10D). Transfection of female RASMCs with AKT-siRNA did not have any effect on MMP production in the presence of elastase (data not shown).
Figure 10.

A: Zymogram depicting decreased MMP-2 and MMP-9 in male rat aortic smooth muscle cells (RASMCs) transfected with AKT siRNA. B–D: Densitometric analysis of the zymogram in A. E, cells that were treated with elastase (5 μg/mL for 24 hours in serum-free DMEM); M, cells that were treated with serum free DMEM only.
Discussion
AKT, a serine/threonine kinase, was first discovered in 1977 as an oncogene in the transforming retrovirus AKT8.31 It is a critical mediator of extracellular information into intracellular biological responses. AKT regulates a vast array of cellular responses, such as cell motility, growth, proliferation, apoptosis, survival, transcription, protein synthesis, and nutrient metabolism. There are three different AKT isoforms (AKT1, AKT2, and AKT3), all of which have similar structural homologies.32 AKT-mutant mice have shown that mutations in AKT1 may cause schizophrenia.33 AKT2 alterations may result in familial diabetes,34 whereas changes in AKT3 result in reduced size of the brain.35 AKT has been shown to be highly activated by low concentrations of insulin in female intra-abdominal adipose tissue compared with males.36 Cortical phosphorylated AKT was increased by isoflurane in male mice compared with female mice, and this resulted in better neuroprotection in the male mice.37 Again, hearts from female mice had greater recovery of left ventricular function and higher phosphorylated AKT levels compared with males.38 There are reports showing that young women have higher levels of nuclear-localized phosphorylated AKT in cardiomyocytes than comparably aged men or postmenopausal women.17
Studies have shown that the hormones, estrogen and testosterone, mediate some of the effects of the AKT. In male C57BL/6J mice, estrogen confers cardioprotection after myocardial infarction by decreasing MMP-9 activity and increasing the AKT signaling pathway.39 Administration of testosterone, on the other hand, down-regulated the AKT pathway in male rat hearts after ischemia/reperfusion.40 Estrogen also activates the AKT pathway and induces vascular endothelial growth factor-A expression in the epithelial cells of the rat uterus.41 Estrogen also induces endothelial nitric oxide synthase via an AKT-dependent pathway in vascular endothelial cells.42 All of these studies suggest that AKT might play a role in sex-mediated disease differences.
By using the elastase perfusion AAA model, it was shown that male rats developed larger AAAs than female rats in association with increased macrophage infiltration and MMP-9 RNA expression in male rats compared with female rats.23 It was also shown that female rat aortas transplanted into male rats lost their resistance toward aneurysm formation and were similar to their male counterparts. Treatment with 17-β-estradiol reduced the growth of aneurysm, elastin disintegration, macrophage infiltration, and MMP-9 levels in male mice. By using the same elastase perfusion model, it was demonstrated that oophorectomized female rats exhibited an increased size of AAAs compared with oophorectomized female and male rats that received 17-β-estradiol treatment.43 This increased aneurysm formation correlated with increased levels of MMP-2 and MMP-9 expression in these oophorectomized and 17-β-estradiol–treated male and female mice.44 Moreover, in earlier research articles, we showed that male mice have more JNK-1 than female mice, which could account for a higher incidence of AAA in males.45 We also have shown that female mice have higher amounts of PAI-1 than male mice and, thus, PAI-1 could impart resistance to AAA formation in female mice.46 In another set of in vitro studies, we have shown that p-ERK signaling is up-regulated in male rat aortic smooth muscle cells.7 We have also shown that treatment of male rats with 17-β-estradiol greatly reduced the male aortic MMP-2 production.47 Our studies have also documented that there is decreased expression of multiple cytokines, chemokines, and leukocyte infiltration in female rat aortas compared with male rats in the elastase perfusion model of AAA.1 Therefore, from all of these studies, we conclude that female hormones regulate certain cytokines, chemokines, and proteins, such as JNK and ERK, having a protective role in aneurysm formation. The present study suggests that the sex-specific differences in AKT phosphorylation and MMP-2 and MMP-9 secretions that we observed in RASMCs in response to elastase treatment may be another component of the complex regulatory mechanisms that contribute to the differences that are observed in male and female animal models of AAA, as well as probably in humans.
AKT is a known modulator of MMPs.32 Tumor necrosis factor-α induces MMP-9 expression via activation of the AKT pathway in vascular smooth muscle cells and in mouse epidermal cells.11,12,48 The migration of vascular smooth muscle cells requires phosphorylation of AKT for the activation of MMP-2 and MMP-9.49 Cigarette smoke extract has been shown to induce MMP-9 expression in rat alveolar macrophages via phosphorylation of AKT.50 IL-1–induced MMP-1 and MMP-13 expressions in human chondrocytes were blocked by LY294002, suggesting that activation of AKT is important in cartilage collagenolysis.51
Recently, it was demonstrated that AKT2 confers protection against aortic aneurysms and dissection.52 Using AKT2 knockout mice and infusing them with angiotensin II resulted in the knockout mice forming larger aortic aneurysms compared with the wild-type control. The said study also demonstrated that there is reduced expression of AKT2 in the degenerative medial layer of human aortic aneurysm dissection tissues. The angiotensin model of mice that was used is different from the elastase model of AAA disease in mice. In the elastase model, we observe aneurysms by day 14, whereas in the angiotensin II model, the mice develop aneurysm at approximately 28 days, with a more profound inflammatory response. Therefore, the intracellular events in the angiotensin and elastase model of AAA may not be comparable. Moreover, the authors did not look into the expression levels of AKT1 and AKT3 in their study to see whether the expression levels of these AKT isoforms differed in the AKT2 knockout mice that underwent angiotensin II treatment and also in the human AAA tissues. We agree with the authors of the previous studies that the literature documents that a link between AKT isoforms and inflammation is complex and often one isoform compensates for the other.
The present study is the first to implicate the importance of AKT during AAA formation in the elastase model of the disease in mice. In the present study, AKT phosphorylation was increased in males compared with females, both in rodents and in humans. In addition, we document that elastase treatment of the aortic smooth muscle cells differentially turns on the AKT signaling pathway in the male and female RASMCs. These cells showed increases in phosphorylated AKT levels with increasing incubation times with elastase, which peaked at 6 hours. Male SMCs were more susceptible to increased phosphorylation of AKT than female cells. Furthermore, at all time points, phosphorylated AKT levels were higher in male cells than female cells (Table 1).
Interestingly, inhibiting phosphorylation of AKT with LY294002 resulted in reduction of pro–MMP-9 secretion by both male and female RASMCs at 24 hours of elastase treatment. However, treatment of cells with LY294002 did not have any effect on active MMP-2, as seen in Figure 9. We next treated the cells with siRNA for AKT, incubated them with elastase, and performed zymography. Similar to LY294002, transfection of male RASMCs with siRNA for AKT and treatment with elastase resulted in a significant reduction in pro–MMP-9 activity. However, unlike LY294002, transfection of male RASMCs with AKT siRNA also resulted in significant decreases in pro– and active MMP-2 secretion. Again, such as LY294002, transfection of female RASMCs with AKT siRNA had no effect on secretion of pro– and active MMP-2 into the medium (data not shown).
In this series of experiments, we first used an in vivo mouse model of AAA to determine the status of AKT in male and female mice. We observed that, as the disease progressed, there was a significant increase in phosphorylated AKT in male compared with female mice. In addition, tissues from male human patients with AAA had much higher levels of phosphorylated AKT than tissues from male cadavers. Our immunofluorescence experiments showed that in both mice with AAA and human tissues from patients with AAA, p308 levels were higher than in controls and p308 costained with macrophages. Based on the in vitro experiments, we also speculate that most of the p308-producing cells were mainly the smooth muscle cells because they are the most abundant type of cells in the aorta and, to a lesser extent, by the macrophages that get recruited to the site. Likewise, we think that MMP-2 and MMP-9 are mostly secreted by SMCs, and some MMP-9 is also secreted by the macrophages at the site of AAA.
Treatment of mice with LY294002, a known AKT phosphorylation inhibitor, did not completely abolish AAA formation after elastase perfusion in mice. We speculate that LY294002 treatment did not inhibit the progression of AAA formation as the result of either the low dose of LY294002 that was used or the timing of its delivery started 4 days after AAA induction. Probably, an increased dose of LY294002 or delivery maintaining a continuous sustained release might have a greater effect on the progression of AAA in the elastase model of the disease. Also, pretreatment of mice with LY294002 for a few days before elastase perfusion may be able to arrest AAA formation. It is also possible that phosphorylation of AKT could be involved in AAA initiation but not critical for AAA progression. However, we did observe that LY294002 was able to preserve the elastin fibers in the aorta, as seen in the Verhoff’s–van Gieson staining (Figure 4). Also, LY294002 treatment resulted in less p308 and p473 staining in mouse AAA tissues (Figure 5). When we treated mouse macrophages with elastase under in vitro conditions, we observed increased levels of p308 and MMP-9 secretion from these cells (Figure 8), which was similar to our experimental observations with vascular aortic smooth muscle cells (Figure 9 and Supplemental Figure S2). This increased production of MMP-9 from the macrophages was inhibited when the cells were treated with LY294002.
In conclusion, this study shows that, under in vitro conditions, elastase induces phosphorylation of AKT in RASMCs and there are increased levels of phosphorylated AKT in male RASMCs compared with female RASMCs. Elastase-induced differential phosphorylation of AKT in male and female RASMCs parallels differential activation of MMPs in RASMCs. Induction of MMP-9 from both male and female RASMCs by elastase is inhibited by pharmacological inhibitors of AKT phosphorylation, whereas the use of AKT siRNA resulted in inhibition of both MMP-2 and MMP-9 by male RASMCs. This study perhaps at least partially explains why AAA is more common in men than women. Although more studies are necessary, the observation that inhibition of the AKT pathway inhibits activation of MMPs may lead to the development of new drugs for the treatment of AAA.
Footnotes
Supported by NIH grants R01 HL081629-01 and 3R01 HL081629-03S1 (G.R.U.).
Supplemental Data
Western blot and densitometric analyses of the levels of p308 and T-AKT of the tissues from the three groups of mice used in Figure 5. A: Western blot analysis for p308, T-AKT, and actin done in triplicate lanes. B–D: Densitometric analysis of the Western blot from A. Actin staining was used to determine equal loading of proteins in the lanes. B:∗P = 0.4389, ∗∗P = 0.0006, and ∗∗∗P = 0.0006. C:∗P = 0.9265, ∗∗P = 0.2428, and ∗∗∗P = 0.13029. D:∗P = 0.0316, ∗∗P = 0.00009, and ∗∗∗P = 0.0013.
Western blot analysis showing the levels of p308, p473, and T-AKT in male and female RASMCs treated with medium, elastase, and phosphorylated AKT inhibitor (LY294002) or a combination of elastase and LY294002 for 1, 6, and 24 hours, respectively.
A–D: Densitometric analysis of the Western blot in Supplemental Figure S2 for 1 and 6 hours. ∗P, ∗∗P < 0.0001, ††P, ††P < 0.002.
A–D: Densitometric analysis of the Western blot in Supplemental Figure S2 for 24 hours. ∗P < 0.0001, †P < 0.002.
References
- 1.Sinha I., Cho B.S., Roelofs K.J., Stanley J.C., Henke P.K., Upchurch G.R., Jr. Female gender attenuates cytokine and chemokine expression and leukocyte recruitment in experimental rodent abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:367–379. doi: 10.1196/annals.1383.027. [DOI] [PubMed] [Google Scholar]
- 2.Eagleton M.J., Ballard N., Lynch E., Srivastava S.D., Upchurch G.R., Jr., Stanley J.C. Early increased MT1-MMP expression and late MMP-2 and MMP-9 activity during angiotensin II induced aneurysm formation. J Surg Res. 2006;135:345–351. doi: 10.1016/j.jss.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 3.Goodall S., Crowther M., Hemingway D.M., Bell P.R., Thompson M.M. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001;104:304–309. doi: 10.1161/01.cir.104.3.304. [DOI] [PubMed] [Google Scholar]
- 4.Longo G.M., Xiong W., Greiner T.C., Zhao Y., Fiotti N., Baxter B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen E., Gineitis A., Wagberg F., Angquist K.A. Activity of matrix metalloproteinase-2 and -9 in abdominal aortic aneurysms: relation to size and rupture. Eur J Vasc Endovasc Surg. 2000;20:457–461. doi: 10.1053/ejvs.2000.1211. [DOI] [PubMed] [Google Scholar]
- 6.Wilson W.R., Anderton M., Schwalbe E.C., Jones J.L., Furness P.N., Bell P.R., Thompson M.M. Matrix metalloproteinase-8 and -9 are increased at the site of abdominal aortic aneurysm rupture. Circulation. 2006;113:438–445. doi: 10.1161/CIRCULATIONAHA.105.551572. [DOI] [PubMed] [Google Scholar]
- 7.Ehrlichman L.K., Ford J.W., Roelofs K.J., Tedeschi-Filho W., Futchko J.S., Ramacciotti E., Eliason J.L., Henke P.K., Upchurch G.R., Jr. Gender-dependent differential phosphorylation in the ERK signaling pathway is associated with increased MMP2 activity in rat aortic smooth muscle cells. J Surg Res. 2010;160:18–24. doi: 10.1016/j.jss.2009.03.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshimura K., Aoki H., Ikeda Y., Fujii K., Akiyama N., Furutani A., Hoshii Y., Tanaka N., Ricci R., Ishihara T., Esato K., Hamano K., Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y., Naggar J.C., Welzig C.M., Beasley D., Moulton K.S., Park H.J., Galper J.B. Simvastatin inhibits angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-knockout mice: possible role of ERK. Arterioscler Thromb Vasc Biol. 2009;29:1764–1771. doi: 10.1161/ATVBAHA.109.192609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh A., DiMusto P.D., Ehrlichman L.K., Sadiq O., McEvoy B., Futchko J.S., Henke P.K., Eliason J.L., Upchurch G.R., Jr. The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. J Am Coll Surg. 2012;215:668–680. doi: 10.1016/j.jamcollsurg.2012.06.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee C.W., Lin C.C., Lin W.N., Liang K.C., Luo S.F., Wu C.B., Wang S.W., Yang C.M. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L799–L812. doi: 10.1152/ajplung.00311.2006. [DOI] [PubMed] [Google Scholar]
- 12.Lee S.J., Seo K.W., Yun M.R., Bae S.S., Lee W.S., Hong K.W., Kim C.D. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-kappaB signaling pathways. Free Radic Biol Med. 2008;45:1487–1492. doi: 10.1016/j.freeradbiomed.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 13.Ruhul Amin A.R., Senga T., Oo M.L., Thant A.A., Hamaguchi M. Secretion of matrix metalloproteinase-9 by the proinflammatory cytokine, IL-1beta: a role for the dual signalling pathways, Akt and Erk. Genes Cells. 2003;8:515–523. doi: 10.1046/j.1365-2443.2003.00652.x. [DOI] [PubMed] [Google Scholar]
- 14.Yao J.S., Chen Y., Zhai W., Xu K., Young W.L., Yang G.Y. Minocycline exerts multiple inhibitory effects on vascular endothelial growth factor-induced smooth muscle cell migration: the role of ERK1/2, PI3K, and matrix metalloproteinases. Circ Res. 2004;95:364–371. doi: 10.1161/01.RES.0000138581.04174.2f. [DOI] [PubMed] [Google Scholar]
- 15.Yin W., Park J.I., Loeser R.F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-kinase-Akt and MEK-ERK MAPK signaling pathways. J Biol Chem. 2009;284:31972–31981. doi: 10.1074/jbc.M109.056838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bae S., Zhang L. Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. J Pharmacol Exp Ther. 2005;315:1125–1135. doi: 10.1124/jpet.105.090803. [DOI] [PubMed] [Google Scholar]
- 17.Camper-Kirby D., Welch S., Walker A., Shiraishi I., Setchell K.D., Schaefer E., Kajstura J., Anversa P., Sussman M.A. Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ Res. 2001;88:1020–1027. doi: 10.1161/hh1001.090858. [DOI] [PubMed] [Google Scholar]
- 18.Cao Z., Liu L., Packwood W., Merkel M., Hurn P.D., Van Winkle D.M. Sex differences in the mechanism of Met5-enkephalin-induced cardioprotection: role of PI3K/Akt. Am J Physiol Heart Circ Physiol. 2008;294:H302–H310. doi: 10.1152/ajpheart.00845.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceylan-Isik A.F., LaCour K.H., Ren J. Gender disparity of streptozotocin-induced intrinsic contractile dysfunction in murine ventricular myocytes: role of chronic activation of Akt. Clin Exp Pharmacol Physiol. 2006;33:102–108. doi: 10.1111/j.1440-1681.2006.04331.x. [DOI] [PubMed] [Google Scholar]
- 20.Sugden P.H., Clerk A. Akt like a woman: gender differences in susceptibility to cardiovascular disease. Circ Res. 2001;88:975–977. doi: 10.1161/hh1001.091864. [DOI] [PubMed] [Google Scholar]
- 21.Anidjar S., Salzmann J.L., Gentric D., Lagneau P., Camilleri J.P., Michel J.B. Elastase-induced experimental aneurysms in rats. Circulation. 1990;82:973–981. doi: 10.1161/01.cir.82.3.973. [DOI] [PubMed] [Google Scholar]
- 22.Pyo R., Lee J.K., Shipley J.M., Curci J.A., Mao D., Ziporin S.J., Ennis T.L., Shapiro S.D., Senior R.M., Thompson R.W. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ailawadi G., Eliason J.L., Roelofs K.J., Sinha I., Hannawa K.K., Kaldjian E.P., Lu G., Henke P.K., Stanley J.C., Weiss S.J., Thompson R.W., Upchurch G.R., Jr. Gender differences in experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2004;24:2116–2122. doi: 10.1161/01.ATV.0000143386.26399.84. [DOI] [PubMed] [Google Scholar]
- 24.Cho B.S., Roelofs K.J., Ford J.W., Henke P.K., Upchurch G.R., Jr. Decreased collagen and increased matrix metalloproteinase-13 in experimental abdominal aortic aneurysms in males compared with females. Surgery. 2010;147:258–267. doi: 10.1016/j.surg.2009.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buczek-Thomas J.A., Nugent M.A. Elastase-mediated release of heparan sulfate proteoglycans from pulmonary fibroblast cultures: a mechanism for basic fibroblast growth factor (bFGF) release and attenuation of bFGF binding following elastase-induced injury. J Biol Chem. 1999;274:25167–25172. doi: 10.1074/jbc.274.35.25167. [DOI] [PubMed] [Google Scholar]
- 26.Gasparini C., Menegazzi R., Patriarca P., Dri P. Evidence that elastase is the TNF-R75 shedding enzyme in resting human polymorphonuclear leukocytes. FEBS Lett. 2003;553:360–364. doi: 10.1016/s0014-5793(03)01046-9. [DOI] [PubMed] [Google Scholar]
- 27.Nemoto E., Kanaya S., Minamibuchi M., Shimauchi H. Cleavage of PDGF receptor on periodontal ligament cells by elastase. J Dent Res. 2005;84:629–633. doi: 10.1177/154405910508400709. [DOI] [PubMed] [Google Scholar]
- 28.Ortiz-Munoz G., Houard X., Martin-Ventura J.L., Ishida B.Y., Loyau S., Rossignol P., Moreno J.A., Kane J.P., Chalkley R.J., Burlingame A.L., Michel J.B., Meilhac O. HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 2009;23:3129–3139. doi: 10.1096/fj.08-127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tull S.P., Bevins A., Kuravi S.J., Satchell S.C., Al-Ani B., Young S.P., Harper L., Williams J.M., Rainger G.E., Savage C.O. PR3 and elastase alter PAR1 signaling and trigger vWF release via a calcium-independent mechanism from glomerular endothelial cells. PLoS One. 2012;7:e43916. doi: 10.1371/journal.pone.0043916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vlahos C.J., Matter W.F., Hui K.Y., Brown R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 31.Staal S.P., Hartley J.W., Rowe W.P. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc Natl Acad Sci U S A. 1977;74:3065–3067. doi: 10.1073/pnas.74.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balu D.T., Carlson G.C., Talbot K., Kazi H., Hill-Smith T.E., Easton R.M., Birnbaum M.J., Lucki I. Akt1 deficiency in schizophrenia and impairment of hippocampal plasticity and function. Hippocampus. 2012;22:230–240. doi: 10.1002/hipo.20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan K., Kimber W.A., Luan J., Soos M.A., Semple R.K., Wareham N.J., O’Rahilly S., Barroso I. Analysis of genetic variation in Akt2/PKB-beta in severe insulin resistance, lipodystrophy, type 2 diabetes, and related metabolic phenotypes. Diabetes. 2007;56:714–719. doi: 10.2337/db06-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Easton R.M., Cho H., Roovers K., Shineman D.W., Mizrahi M., Forman M.S., Lee V.M., Szabolcs M., de Jong R., Oltersdorf T., Ludwig T., Efstratiadis A., Birnbaum M.J. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macotela Y., Boucher J., Tran T.T., Kahn C.R. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009;58:803–812. doi: 10.2337/db08-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitano H., Young J.M., Cheng J., Wang L., Hurn P.D., Murphy S.J. Gender-specific response to isoflurane preconditioning in focal cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:1377–1386. doi: 10.1038/sj.jcbfm.9600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turcato S., Turnbull L., Wang G.Y., Honbo N., Simpson P.C., Karliner J.S., Baker A.J. Ischemic preconditioning depends on age and gender. Basic Res Cardiol. 2006;101:235–243. doi: 10.1007/s00395-006-0585-4. [DOI] [PubMed] [Google Scholar]
- 39.Cao J., Zhu T., Lu L., Geng L., Wang L., Zhang Q., Yang K., Wang H., Shen W. Estrogen induces cardioprotection in male C57BL/6J mice after acute myocardial infarction via decreased activity of matrix metalloproteinase-9 and increased Akt-Bcl-2 anti-apoptotic signaling. Int J Mol Med. 2011;28:231–237. doi: 10.3892/ijmm.2011.681. [DOI] [PubMed] [Google Scholar]
- 40.Huang C., Gu H., Zhang W., Herrmann J.L., Wang M. Testosterone-down-regulated Akt pathway during cardiac ischemia/reperfusion: a mechanism involving BAD, Bcl-2 and FOXO3a. J Surg Res. 2010;164:e1–e11. doi: 10.1016/j.jss.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kazi A.A., Molitoris K.H., Koos R.D. Estrogen rapidly activates the PI3K/AKT pathway and hypoxia-inducible factor 1 and induces vascular endothelial growth factor A expression in luminal epithelial cells of the rat uterus. Biol Reprod. 2009;81:378–387. doi: 10.1095/biolreprod.109.076117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hisamoto K., Ohmichi M., Kurachi H., Hayakawa J., Kanda Y., Nishio Y., Adachi K., Tasaka K., Miyoshi E., Fujiwara N., Taniguchi N., Murata Y. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2001;276:3459–3467. doi: 10.1074/jbc.M005036200. [DOI] [PubMed] [Google Scholar]
- 43.Wu X.F., Zhang J., Paskauskas S., Xin S.J., Duan Z.Q. The role of estrogen in the formation of experimental abdominal aortic aneurysm. Am J Surg. 2009;197:49–54. doi: 10.1016/j.amjsurg.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 44.Starr J.E., Halpern V. Abdominal aortic aneurysms in women. J Vasc Surg. 2013;57:3S–10S. doi: 10.1016/j.jvs.2012.08.125. [DOI] [PubMed] [Google Scholar]
- 45.DiMusto P.D., Lu G., Ghosh A., Roelofs K.J., Sadiq O., McEvoy B., Su G., Laser A., Bhamidipati C.M., Ailawadi G., Henke P.K., Eliason J.L., Upchurch G.R., Jr. Increased JNK in males compared with females in a rodent model of abdominal aortic aneurysm. J Surg Res. 2012;176:687–695. doi: 10.1016/j.jss.2011.11.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiMusto P.D., Lu G., Ghosh A., Roelofs K.J., Su G., Zhao Y., Lau C.L., Sadiq O., McEvoy B., Laser A., Diaz J.A., Wakefield T.W., Henke P.K., Eliason J.L., Upchurch G.R., Jr. Increased PAI-1 in females compared with males is protective for abdominal aortic aneurysm formation in a rodent model. Am J Physiol Heart Circ Physiol. 2012;302:H1378–H1386. doi: 10.1152/ajpheart.00620.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woodrum D.T., Ford J.W., Cho B.S., Hannawa K.K., Stanley J.C., Henke P.K., Upchurch G.R., Jr. Differential effect of 17-beta-estradiol on smooth muscle cell and aortic explant MMP2. J Surg Res. 2009;155:48–53. doi: 10.1016/j.jss.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee E.J., Kim D.I., Kim W.J., Moon S.K. Naringin inhibits matrix metalloproteinase-9 expression and AKT phosphorylation in tumor necrosis factor-alpha-induced vascular smooth muscle cells. Mol Nutr Food Res. 2009;53:1582–1591. doi: 10.1002/mnfr.200800210. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez-Hernando C., Jozsef L., Jenkins D., Di Lorenzo A., Sessa W.C. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:2033–2040. doi: 10.1161/ATVBAHA.109.196394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S.E., Thanh Thuy T.T., Lee J.H., Ro J.Y., Bae Y.A., Kong Y., Ahn J.Y., Lee D.S., Oh Y.M., Lee S.D., Lee Y.S. Simvastatin inhibits induction of matrix metalloproteinase-9 in rat alveolar macrophages exposed to cigarette smoke extract. Exp Mol Med. 2009;41:277–287. doi: 10.3858/emm.2009.41.4.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Litherland G.J., Dixon C., Lakey R.L., Robson T., Jones D., Young D.A., Cawston T.E., Rowan A.D. Synergistic collagenase expression and cartilage collagenolysis are phosphatidylinositol 3-kinase/Akt signaling-dependent. J Biol Chem. 2008;283:14221–14229. doi: 10.1074/jbc.M710136200. [DOI] [PubMed] [Google Scholar]
- 52.Shen Y.H., Zhang L., Ren P., Nguyen M.T., Zou S., Wu D., Wang X.L., Coselli J.S., LeMaire S.A. AKT2 confers protection against aortic aneurysms and dissections. Circ Res. 2013;112:618–632. doi: 10.1161/CIRCRESAHA.112.300735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blot and densitometric analyses of the levels of p308 and T-AKT of the tissues from the three groups of mice used in Figure 5. A: Western blot analysis for p308, T-AKT, and actin done in triplicate lanes. B–D: Densitometric analysis of the Western blot from A. Actin staining was used to determine equal loading of proteins in the lanes. B:∗P = 0.4389, ∗∗P = 0.0006, and ∗∗∗P = 0.0006. C:∗P = 0.9265, ∗∗P = 0.2428, and ∗∗∗P = 0.13029. D:∗P = 0.0316, ∗∗P = 0.00009, and ∗∗∗P = 0.0013.
Western blot analysis showing the levels of p308, p473, and T-AKT in male and female RASMCs treated with medium, elastase, and phosphorylated AKT inhibitor (LY294002) or a combination of elastase and LY294002 for 1, 6, and 24 hours, respectively.
A–D: Densitometric analysis of the Western blot in Supplemental Figure S2 for 1 and 6 hours. ∗P, ∗∗P < 0.0001, ††P, ††P < 0.002.
A–D: Densitometric analysis of the Western blot in Supplemental Figure S2 for 24 hours. ∗P < 0.0001, †P < 0.002.